Potential Roles of Antiphospholipid Antibodies in Generating Platelet-C4d in Systemic Lupus Erythematosus

Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Participants and Blood Specimens
2.2. Plasma, Serum, and Immunoglobulin Preparation
2.3. Flow Cytometry of P-C4d Measurement
2.4. aPL Antibody Immunoassays
2.5. In Vitro P-C4d Induction Assay
2.6. Statistical Analysis
3. Results
3.1. Patient Characteristics
3.2. Relationships between P-C4d and aPL Antibodies
3.3. Involvement of aPL Antibodies in P-C4d Generation
3.4. Potential Involvement of Other Platelet-Reactive Autoantibodies in P-C4d Generation
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Manzi, S.; Meilahn, E.N.; Rairie, J.E.; Conte, C.G.; Medsger, T.A., Jr.; Jansen-McWilliams, L.; D’Agostino, R.B.; Kuller, L.H. Age-specific incidence rates of myocardial infarction and angina in women with systemic lupus erythematosus: Comparison with the framingham study. Am. J. Epidemiol. 1997, 145, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Ward, M.M. Premature morbidity from cardiovascular and cerebrovascular diseases in women with systemic lupus erythematosus. Arthritis Rheum. 1999, 42, 338–346. [Google Scholar] [CrossRef]
- Nikpour, M.; Urowitz, M.B.; Gladman, D.D. Premature atherosclerosis in systemic lupus erythematosus. Rheum. Dis. Clin. N. Am. 2005, 31, 329–354. [Google Scholar] [CrossRef] [PubMed]
- Haque, S.; Bruce, I.N. Therapy insight: Systemic lupus erythematosus as a risk factor for cardiovascular disease. Nat. Clin. Pract. Cardiovasc. Med. 2005, 2, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Yaniv, G.; Twig, G.; Shor, D.B.; Furer, A.; Sherer, Y.; Mozes, O.; Komisar, O.; Slonimsky, E.; Klang, E.; Lotan, E.; et al. A volcanic explosion of autoantibodies in systemic lupus erythematosus: A diversity of 180 different antibodies found in sle patients. Autoimmun. Rev. 2015, 14, 75–79. [Google Scholar] [CrossRef] [PubMed]
- McNeil, H.P.; Simpson, R.J.; Chesterman, C.N.; Krilis, S.A. Anti-phospholipid antibodies are directed against a complex antigen that includes a lipid-binding inhibitor of coagulation: β2-glycoprotein I (apolipoprotein H). Proc. Natl. Acad. Sci. USA 1990, 87, 4120–4124. [Google Scholar] [CrossRef] [PubMed]
- Bevers, E.M.; Galli, M. β2-glycoprotein I for binding of anticardiolipin antibodies to cardiolipin. Lancet 1990, 336, 952–953. [Google Scholar] [CrossRef]
- Galli, M.; Comfurius, P.; Maassen, C.; Hemker, H.C.; de Baets, M.H.; van Breda-Vriesman, P.J.; Barbui, T.; Zwaal, R.F.; Bevers, E.M. Anticardiolipin antibodies (ACA) directed not to cardiolipin but to a plasma protein cofactor. Lancet 1990, 335, 1544–1547. [Google Scholar] [CrossRef]
- Bevers, E.M.; Galli, M.; Barbui, T.; Comfurius, P.; Zwaal, R.F. Lupus anticoagulant igg’s (LA) are not directed to phospholipids only, but to a complex of lipid-bound human prothrombin. Thromb. Haemost. 1991, 66, 629–632. [Google Scholar] [PubMed]
- Bertolaccini, M.L.; Hughes, G.R.; Khamashta, M.A. Revisiting antiphospholipid antibodies: From targeting phospholipids to phospholipid binding proteins. Clin. Lab. 2004, 50, 653–665. [Google Scholar] [PubMed]
- Petri, M. Epidemiology of the antiphospholipid antibody syndrome. J. Autoimmun. 2000, 15, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Horbach, D.A.; van Oort, E.; Donders, R.C.; Derksen, R.H.; de Groot, P.G. Lupus anticoagulant is the strongest risk factor for both venous and arterial thrombosis in patients with systemic lupus erythematosus. Comparison between different assays for the detection of antiphospholipid antibodies. Thromb. Haemost. 1996, 76, 916–924. [Google Scholar] [PubMed]
- Greaves, M. Antiphospholipid antibodies and thrombosis. Lancet 1999, 353, 1348–1353. [Google Scholar] [CrossRef]
- Galli, M.; Luciani, D.; Bertolini, G.; Barbui, T. Lupus anticoagulants are stronger risk factors for thrombosis than anticardiolipin antibodies in the antiphospholipid syndrome: A systematic review of the literature. Blood 2003, 101, 1827–1832. [Google Scholar] [CrossRef] [PubMed]
- Nojima, J.; Kuratsune, H.; Suehisa, E.; Kitani, T.; Iwatani, Y.; Kanakura, Y. Strong correlation between the prevalence of cerebral infarction and the presence of anti-cardiolipin/β2-glycoprotein I and anti-phosphatidylserine/prothrombin antibodies—Co-existence of these antibodies enhances ADP-induced platelet activation in vitro. Thromb. Haemost. 2004, 91, 967–976. [Google Scholar] [PubMed]
- Lim, W. Thrombotic risk in the antiphospholipid syndrome. Semin. Thromb. Hemost. 2014, 40, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Sorice, M.; Longo, A.; Capozzi, A.; Garofalo, T.; Misasi, R.; Alessandri, C.; Conti, F.; Buttari, B.; Rigano, R.; Ortona, E.; et al. Anti-β2-glycoprotein I antibodies induce monocyte release of tumor necrosis factor alpha and tissue factor by signal transduction pathways involving lipid rafts. Arthritis Rheum. 2007, 56, 2687–2697. [Google Scholar] [CrossRef] [PubMed]
- Salmon, J.E.; de Groot, P.G. Pathogenic role of antiphospholipid antibodies. Lupus 2008, 17, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Rand, J.H.; Wu, X.X.; Quinn, A.S.; Taatjes, D.J. The annexin A5-mediated pathogenic mechanism in the antiphospholipid syndrome: Role in pregnancy losses and thrombosis. Lupus 2010, 19, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Satta, N.; Kruithof, E.K.; Fickentscher, C.; Dunoyer-Geindre, S.; Boehlen, F.; Reber, G.; Burger, D.; de Moerloose, P. Toll-like receptor 2 mediates the activation of human monocytes and endothelial cells by antiphospholipid antibodies. Blood 2011, 117, 5523–5531. [Google Scholar] [CrossRef] [PubMed]
- Perez-Sanchez, C.; Ruiz-Limon, P.; Aguirre, M.A.; Bertolaccini, M.L.; Khamashta, M.A.; Rodriguez-Ariza, A.; Segui, P.; Collantes-Estevez, E.; Barbarroja, N.; Khraiwesh, H.; et al. Mitochondrial dysfunction in antiphospholipid syndrome: Implications in the pathogenesis of the disease and effects of coenzyme Q10 treatment. Blood 2012, 119, 5859–5870. [Google Scholar] [CrossRef] [PubMed]
- Giannakopoulos, B.; Krilis, S.A. The pathogenesis of the antiphospholipid syndrome. N. Engl. J. Med. 2013, 368, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Martinuzzo, M.E.; Maclouf, J.; Carreras, L.O.; Levy-Toledano, S. Antiphospholipid antibodies enhance thrombin-induced platelet activation and thromboxane formation. Thromb. Haemost. 1993, 70, 667–671. [Google Scholar] [PubMed]
- Galli, M.; Bevers, E.M.; Comfurius, P.; Barbui, T.; Zwaal, R.F. Effect of antiphospholipid antibodies on procoagulant activity of activated platelets and platelet-derived microvesicles. Br. J. Haematol. 1993, 83, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Nojima, J.; Suehisa, E.; Kuratsune, H.; Machii, T.; Koike, T.; Kitani, T.; Kanakura, Y.; Amino, N. Platelet activation induced by combined effects of anticardiolipin and lupus anticoagulant IGg antibodies in patients with systemic lupus erythematosus—Possible association with thrombotic and thrombocytopenic complications. Thromb. Haemost. 1999, 81, 436–441. [Google Scholar] [PubMed]
- Shi, T.; Giannakopoulos, B.; Yan, X.; Yu, P.; Berndt, M.C.; Andrews, R.K.; Rivera, J.; Iverson, G.M.; Cockerill, K.A.; Linnik, M.D.; et al. Anti-β2-glycoprotein I antibodies in complex with β2-glycoprotein I can activate platelets in a dysregulated manner via glycoprotein Ib-IX-V. Arthritis Rheum. 2006, 54, 2558–2567. [Google Scholar] [CrossRef] [PubMed]
- Urbanus, R.T.; Pennings, M.T.; Derksen, R.H.; de Groot, P.G. Platelet activation by dimeric β2-glycoprotein I requires signaling via both glycoprotein Ibα and apolipoprotein E receptor 2’. J. Thromb. Haemost. 2008, 6, 1405–1412. [Google Scholar] [CrossRef] [PubMed]
- Arad, A.; Proulle, V.; Furie, R.A.; Furie, B.C.; Furie, B. β2-glycoprotein-1 autoantibodies from patients with antiphospholipid syndrome are sufficient to potentiate arterial thrombus formation in a mouse model. Blood 2011, 117, 3453–3459. [Google Scholar] [CrossRef] [PubMed]
- Meroni, P.L.; Borghi, M.O.; Raschi, E.; Tedesco, F. Pathogenesis of antiphospholipid syndrome: Understanding the antibodies. Nat. Rev. Rheumatol. 2011, 7, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Holers, V.M.; Girardi, G.; Mo, L.; Guthridge, J.M.; Molina, H.; Pierangeli, S.S.; Espinola, R.; Xiaowei, L.E.; Mao, D.; Vialpando, C.G.; et al. Complement C3 activation is required for antiphospholipid antibody-induced fetal loss. J. Exp. Med. 2002, 195, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Girardi, G.; Redecha, P.; Salmon, J.E. Heparin prevents antiphospholipid antibody-induced fetal loss by inhibiting complement activation. Nat. Med. 2004, 10, 1222–1226. [Google Scholar] [CrossRef] [PubMed]
- Fischetti, F.; Durigutto, P.; Pellis, V.; Debeus, A.; Macor, P.; Bulla, R.; Bossi, F.; Ziller, F.; Sblattero, D.; Meroni, P.; et al. Thrombus formation induced by antibodies to β2-glycoprotein i is complement dependent and requires a priming factor. Blood 2005, 106, 2340–2346. [Google Scholar] [CrossRef] [PubMed]
- Davis, W.D.; Brey, R.L. Antiphospholipid antibodies and complement activation in patients with cerebral ischemia. Clin. Exp. Rheumatol. 1992, 10, 455–460. [Google Scholar] [PubMed]
- Navratil, J.S.; Manzi, S.; Kao, A.H.; Krishnaswami, S.; Liu, C.C.; Ruffing, M.J.; Shaw, P.S.; Nilson, A.C.; Dryden, E.R.; Johnson, J.J.; et al. Platelet C4d is highly specific for systemic lupus erythematosus. Arthritis Rheum. 2006, 54, 670–674. [Google Scholar] [CrossRef] [PubMed]
- Kao, A.H.; McBurney, C.A.; Sattar, A.; Lertratanakul, A.; Wilson, N.L.; Rutman, S.; Paul, B.; Navratil, J.S.; Scioscia, A.; Ahearn, J.M.; et al. Relation of platelet c4d with all-cause mortality and ischemic stroke in patients with systemic lupus erythematosus. Transl. Stroke Res. 2014, 5, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Peerschke, E.I.; Yin, W.; Alpert, D.R.; Roubey, R.A.; Salmon, J.E.; Ghebrehiwet, B. Serum complement activation on heterologous platelets is associated with arterial thrombosis in patients with systemic lupus erythematosus and antiphospholipid antibodies. Lupus 2009, 18, 530–538. [Google Scholar] [CrossRef] [PubMed]
- Lood, C.; Eriksson, S.; Gullstrand, B.; Jonsen, A.; Sturfelt, G.; Truedsson, L.; Bengtsson, A.A. Increased c1q, c4 and c3 deposition on platelets in patients with systemic lupus erythematosus—A possible link to venous thrombosis? Lupus 2012, 21, 1423–1432. [Google Scholar] [CrossRef] [PubMed]
- Lood, C.; Tyden, H.; Gullstrand, B.; Sturfelt, G.; Jonsen, A.; Truedsson, L.; Bengtsson, A.A. Platelet activation and anti-phospholipid antibodies collaborate in the activation of the complement system on platelets in systemic lupus erythematosus. PLoS ONE 2014, 9, e99386. [Google Scholar] [CrossRef] [PubMed]
- Mehta, N.; Uchino, K.; Fakhran, S.; Sattar, M.A.; Branstetter, B.F.T.; Au, K.; Navratil, J.S.; Paul, B.; Lee, M.; Gallagher, K.M.; et al. Platelet C4d is associated with acute ischemic stroke and stroke severity. Stroke 2008, 39, 3236–3241. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.M.; Cohen, A.S.; Fries, J.F.; Masi, A.T.; McShane, D.J.; Rothfield, N.F.; Schaller, J.G.; Talal, N.; Winchester, R.J. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982, 25, 1271–1277. [Google Scholar] [CrossRef] [PubMed]
- Hochberg, M.C. Updating the american college of rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997, 40, 1725. [Google Scholar] [CrossRef] [PubMed]
- Burchett, W.W.; Ellis, A.R.; Harrar, S.W.; Bathke, A.C. Nonparametric inference of multivariate data: The R package npmv. J. Stat. Softw. 2017, 76, 18. [Google Scholar] [CrossRef]
- Machin, S.J. Platelets and antiphospholipid antibodies. Lupus 1996, 5, 386–387. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Manzi, S.; Kao, A.H.; Navratil, J.S.; Ahearn, J.M. Cell-bound complement biomarkers for systemic lupus erythematosus: From benchtop to bedside. Rheum. Dis. Clin. N. Am. 2010, 36, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Ahearn, J.M.; Manzi, S.; Liu, C.C. The lupus biomarker odyssey: One experience. Methods Mol. Biol. 2014, 1134, 17–35. [Google Scholar] [PubMed]
- Liu, C.C.; Manzi, S.; Ahearn, J.M. Antilymphocyte autoantibodies generate T cell-C4d signatures in systemic lupus erythematosus. Transl. Res. 2014, 164, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, K.; Tani, P.; O’Toole, T.E.; Ginsberg, M.H.; McMillan, R. Different specificities of platelet-associated and plasma autoantibodies to platelet GPIIb-IIIa in patients with chronic immune thrombocytopenic purpura. Blood 1992, 79, 1441–1446. [Google Scholar] [PubMed]
- Macchi, L.; Rispal, P.; Clofent-Sanchez, G.; Pellegrin, J.L.; Nurden, P.; Leng, B.; Nurden, A.T. Anti-platelet antibodies in patients with systemic lupus erythematosus and the primary antiphospholipid antibody syndrome: Their relationship with the observed thrombocytopenia. Br. J. Haematol. 1997, 98, 336–341. [Google Scholar] [CrossRef] [PubMed]
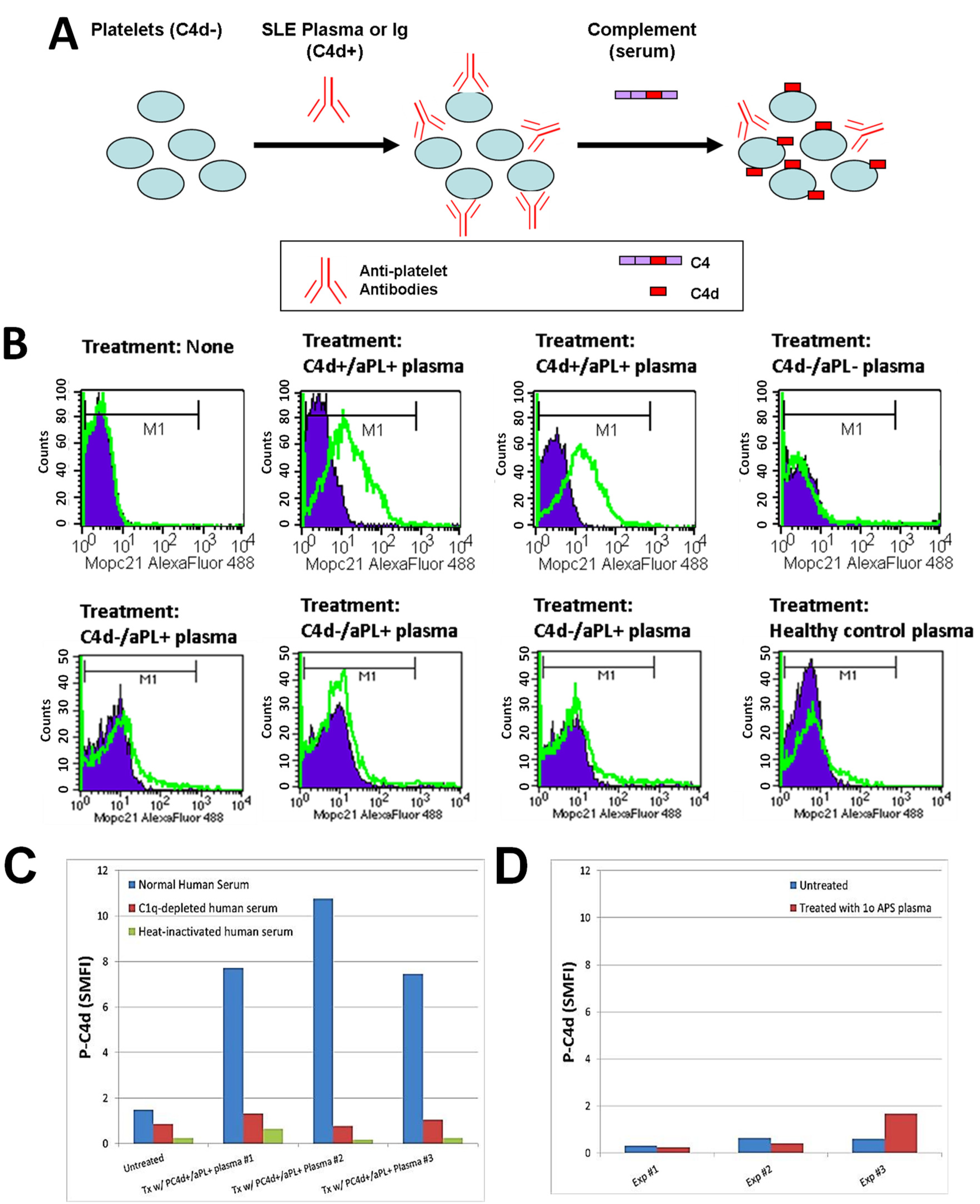
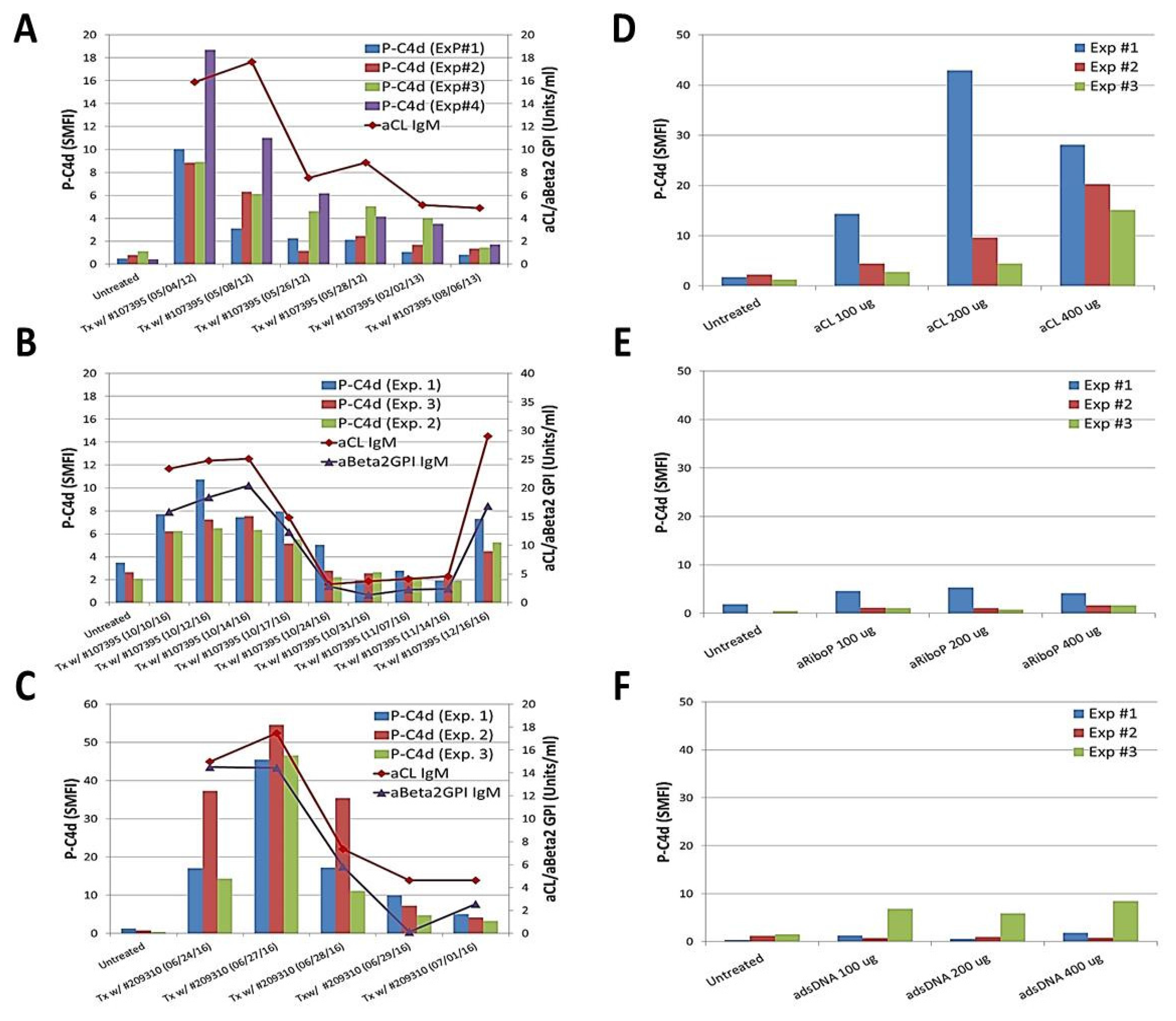
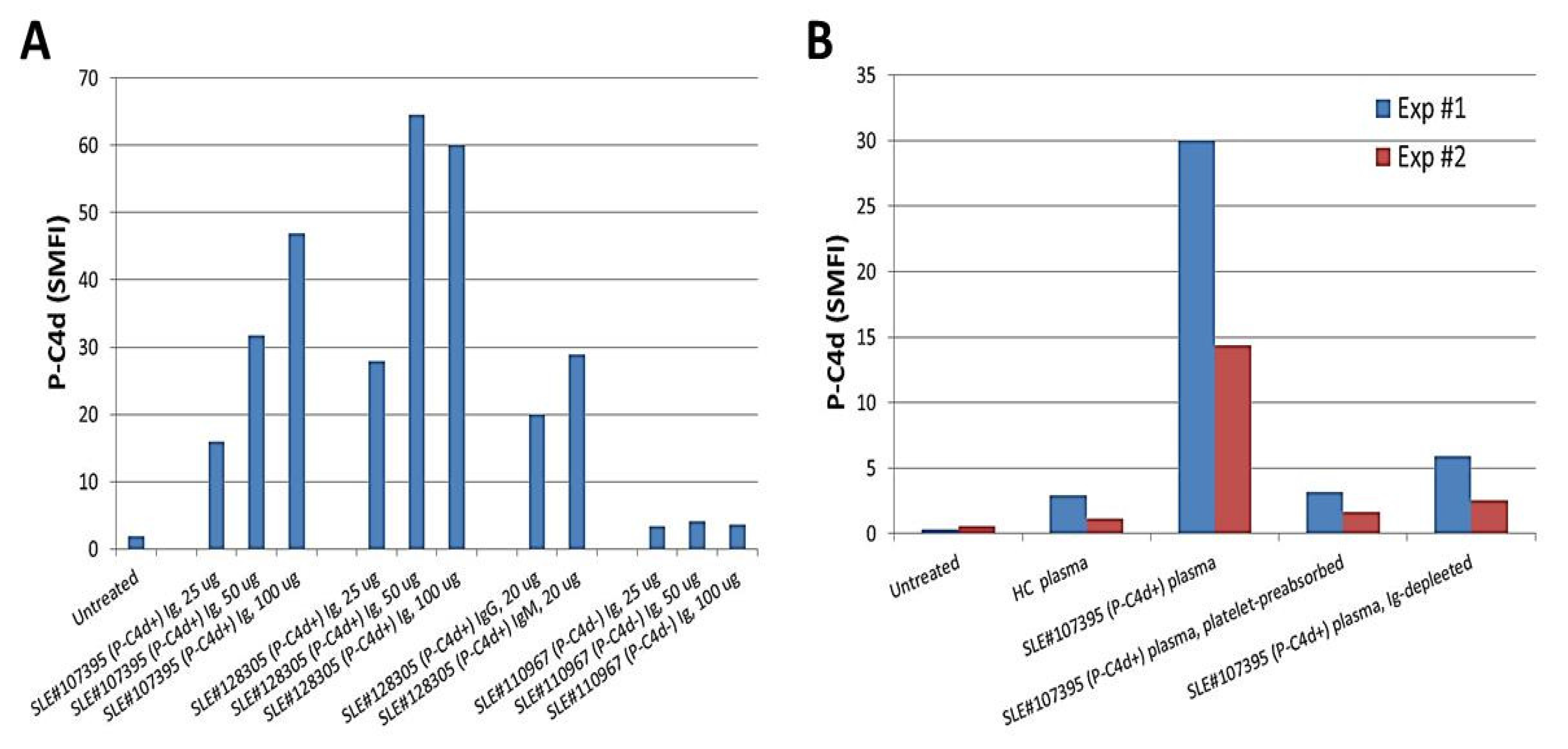

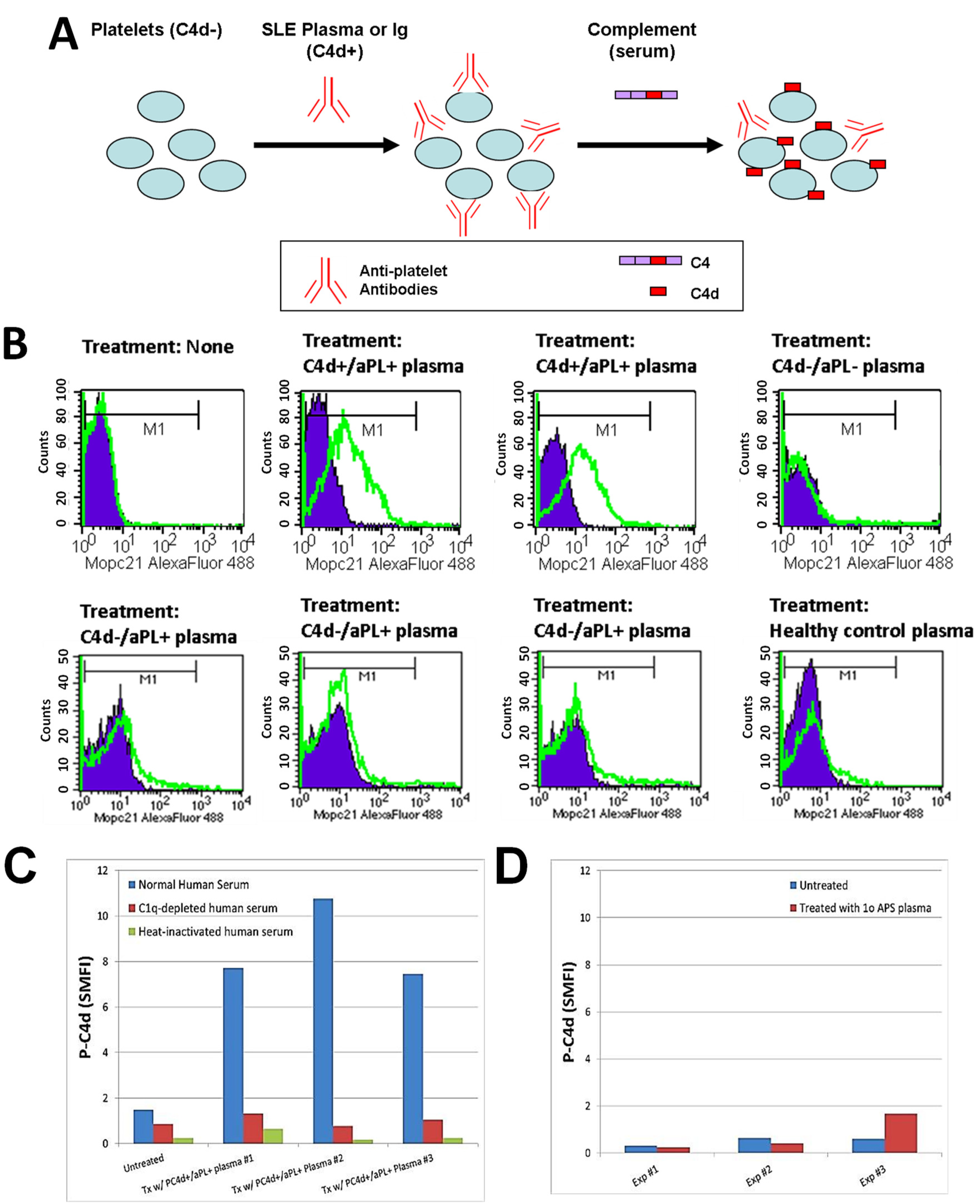{kind=link}
{kind=link}
{kind=link}
| Platelet-C4d | ||||
|---|---|---|---|---|
| All (n = 180) | Positive (n = 34) | Negative (n = 146) | p-Value | |
| Age, year, mean (SD) | 47.2 (11.9) | 46.9 (10.7) | 47.3 (12.2) | 0.802 |
| Duration of SLE, year, mean (SD) | 15.2 (9.7) | 16.5 (9.6) | 14.8 (9.7) | 0.276 |
| Sex, n (%) | ||||
| Women | 167 (92.8) | 31 (91.2) | 136 (93.2) | 0.714 |
| Race, n (%) | ||||
| White | 156 (86.7) | 28 (82.4) | 128 (87.7) | 0.240 |
| Black | 21 (11.7) | 6 (18.6) | 15 (10.3) | |
| Others | 3 (1.7) | 3 (2.0) | ||
| ACR & criteria (ever), n (%) | ||||
| Malar rash | 83 (46.1) | 13 (38.2) | 70 (47.9) | 0.344 |
| Discoid rash | 14 (7.8) | 4 (11.8) | 10 (6.8) | 0.306 |
| Photosensitivity | 111 (61.7) | 17 (50.0) | 94 (64.4) | 0.170 |
| Oral ulcers | 122 (67.8) | 19 (55.9) | 103 (70.3) | 0.107 |
| Arthritis | 172 (95.6) | 32 (94.1) | 140 (95.9) | 0.647 |
| Serositis | 64 (35.6) | 14 (41.2) | 50 (34.2) | 0.551 |
| Renal disease | 54 (30.0) | 8 (23.5) | 46 (31.5) | 0.412 |
| Neurological | 20 (11.1) | 4 (11.8) | 16 (10.9) | 1.000 |
| Seizure | 15 (8.3) | 1 (2.9) | 14 (9.6) | 0.310 |
| Psychosis | 9 (5.0) | 3 (8.8) | 6 (4.1) | 0.402 |
| Hematological | 96 (53.3) | 25 (73.5) | 71 (48.6) | 0.012 |
| Hemolytic anemia | 10 (5.6) | 6 (17.6) | 4 (2.7) | 0.004 |
| Leukopenia | 46 (25.6) | 11 (32.4) | 35 (24.0) | 0.382 |
| Lymphopenia | 61 (33.9) | 19 (55.9) | 42 (22.8) | 0.004 |
| Thrombocytopenia | 32 (17.8) | 10 (29.4) | 22 (15.1) | 0.047 |
| Antinuclear antibody | 177 (98.3) | 33 (97.1) | 144 (98.6) | 0.468 |
| Serological | 142 (78.9) | 33 (97.1) | 109 (74.7) | 0.002 |
| Anti-Phospholipid #,^ | 83 (46.1) | 24 (70.6) | 59 (40.4) | 0.002 |
| Anti-cardiolipin | 60 (33.3) | 17 (50.0) | 43 (29.5) | 0.028 |
| Lupus anticoagulant | 45 (25.0) | 16 (47.1) | 29 (19.9) | 0.002 |
| Anti-dsDNA | 97 (53.9) | 23 (67.6) | 74 (50.7) | 0.087 |
| Anti-Smith | 24 (13.3) | 4 (11.8) | 20 (13.7) | 1.000 |
| Medication use, current **, n (%) | ||||
| Steroid | 109 (60.6) | 25 (73.5) | 84 (57.5) | 0.074 |
| Antimalarial | 128 (71.1) | 23 (67.6) | 105 (71.9) | 0.676 |
| Anticoagulant | ||||
| Warfarin | 24 (13.3) | 10 (29.4) | 14 (9.6) | 0.005 |
| Heparin | 7 (3.9) | 4 (11.8) | 3 (2.1) | 0.025 |
| FXa inhibitor | 2 (1.1) | 1 (2.9) | 1 (0.7) | 0.343 |
| Antiplatelet | ||||
| Aspirin | 69 (38.3) | 11 (32.4) | 58 (39.7) | 0.557 |
| P2Y12 antagonist | 10 (5.6) | 4 (11.8) | 6 (4.1) | 0.096 |
| Statins | 37 (20.6) | 7 (20.6) | 30 (20.5) | 1.000 |
| Immunosuppressant | 101 (56.1) | 21 (61.8) | 80 (50.8) | 0.566 |
| Biologicals | 18 (10.0) | 4 (11.8) | 14 (9.6) | 0.751 |
| Antihypertensive | 77 (42.8) | 13 (38.2) | 64 (43.8) | 0.571 |
| Diuretics | 31 (17.2) | 3 (8.8) | 28 (19.2) | 0.208 |
| Insulin | 6 (3.3) | 0 (0.0) | 6 (4.1) | 0.592 |
| Antidiabetic | 4 (2.2) | 0 (0.0) | 4 (2.7) | 1.000 |
| P-C4d level, current, median (IQR) | 0.6 (0.2–1.4) | 5.6 (3.4–11.1) | 0.4 (0.1–0.8) | <0.001 |
| Platelet-C4d | ||||
|---|---|---|---|---|
| All (n = 180) | Positive (n = 34) | Negative (n = 146) | p-Value (P-C4d+ vs. P-C4d−) | |
| aPL (aCL and/or aβ2GPI) antibodies positivity, n (%) | 48 (26.7) | 20 (58.5) | 28 (19.2) | <0.001 |
| aCL IgM positivity, n (%) † | 19 (10.6) | 8 (23.5) | 11 (7.5) | 0.012 |
| aCL IgM level (U/mL) median (IQR) * | 3.1 (2.0–6.5) | 4.2 (2.2–11.6) | 2.8 (2.0–6.0) | 0.068 |
| aCL IgG positivity, n (%) † | 17 (9.4) | 11 (32.4) | 6 (4.1) | <0.001 |
| aCL IgG level (U/mL) median (IQR) * | 5.0 (2.8–9.6) | 12.5 (5.6–37.9) | 4.6 (2.7–7.7) | <0.001 |
| aCL IgA positivity, n (%) † | 6 (3.3) | 4 (11.8) | 2 (1.4) | 0.012 |
| aCL IgA level (U/mL) median (IQR) * | 1.2 (0.6–2.3) | 1.3 (1.0–2.9) | 1.0 (0.5–2.0) | 0.053 |
| Any aCL positivity, n (%) | 32 (17.8) | 13 (38.2) | 19 (13.0) | 0.002 |
| aβ2GPI IgM positivity, n (%) | 10 (5.6) | 5 (14.7) | 5 (3.4) | 0.022 |
| aβ2GPI IgM level (U/mL) median (IQR) | 0.4 (0.0–1.3) | 1.0 (0.4–3.6) | 0.3 (0.0–0.9) | <0.001 |
| aβ2GPI IgG positivity, n (%) | 14 (7.8) | 11 (32.4) | 8 (5.5) | <0.001 |
| aβ2GPI IgG level (U/mL) median (IQR) | 1.6 (0.0–3.7) | 4.6 (1.1–76.1) | 1.3 (0.0–3.1) | <0.001 |
| aβ2GPI IgA positivity, n (%) | 21 (11.7) | 13 (38.2) | 16 (11.0) | <0.001 |
| aβ2GPI IgA level (U/mL) median (IQR) | 0.4 (0.0–2.6) | 3.9 (0.8–12.8) | 0.4 (0.0–1.2) | <0.001 |
| Any aβ2GPI positivity, n (%) | 34 (18.9) | 18 (52.9) | 16 (11.0) | <0.001 |
| Number of Positive aCL and aβ2GPI Antibodies | Number of Patients | Isotypes of Positive aCL and aβ2GPI Antibodies |
|---|---|---|
| 6 | 1 | aCL IgM/IgG/IgA; aβ2GPI IgM/IgG/IgA |
| 5 | 1 | aCL IgM/IgG/IgA; aβ2GPI IgG/IgA |
| 1 | aCL IgM/IgG/IgA; aβ2GPI IgM/IgG | |
| 4 | 1 | aCL IgG/IgA; aβ2GPI IgG/IgA |
| 1 | aCL IgM/IgG; aβ2GPI IgG/IgA | |
| 3 | 2 | aCL IgG; aβ2GPI IgG/IgA |
| 1 | aCL IgM/IgG; aβ2GPI IgG | |
| 1 | aβ2GPI IgM/IgG/IgA | |
| 1 | aCL IgM/IgG/IgA | |
| 2 | 3 | aCL IgM; aβ2GPI IgM |
| 5 | aCL IgG; aβ2GPI IgG | |
| 2 | aCL IgM; aβ2GPI IgA | |
| 1 | 8 | aCL IgM |
| 3 | aCL IgG | |
| 2 | aCLIgA | |
| 2 | aβ2GPI IgM | |
| 1 | aβ2GPI IgG | |
| 12 | aβ2GPI IgA |
| P-C4d Median (IQR) * | p-Value (vs. aPL−) | |
|---|---|---|
| aPL− (n = 132) | 0.52 (0.13–0.91) | |
| aPL+ (n = 48) ** | 1.18 (0.25–4.55) | 0.002 |
| aCL+ alone (n = 14) | 0.48 (0.20–1.51) | 0.586 |
| aβ2GPI+ alone (n = 16) | 1.44 (0.25–3.24) | 0.042 |
| aCL+/aβ2GPI+ (n = 18) | 4.61 (0.53–11.44) | 0.001 |
| Plasma Source a | Plasma aPL Positivity b | Ex vivo P-C4d Positivity c | Platelet Source d | In Vitro P-C4d (SMFI) e |
|---|---|---|---|---|
| SLE patient #1 | + | + | SLE patient #4 | 34.21 |
| SLE patient #1 | SLE patient #5 | 39.17 | ||
| SLE patient #1 | SLE patient #6 | 19.31 | ||
| SLE patient #1 | SLE patient #7 | 14.70 | ||
| SLE patient #1 | SLE patient #8 | 6.83 | ||
| SLE patient #1 | SLE patient #9 | 27.34 | ||
| SLE patient #1 | SLE patient #10 | 39.29 | ||
| SLE patient #1 | SLE patient #11 | 31.52 | ||
| SLE patient #1 | Healthy Control #1 | 27.50 | ||
| SLE patient #1 | Healthy Control #2 | 5.38 | ||
| SLE patient #2 | + | + | SLE patient #4 | 4.93 |
| SLE patient #2 | SLE patient #7 | 5.20 | ||
| SLE patient #2 | SLE patient #8 | 0.44 | ||
| SLE patient #2 | SLE patient #9 | 6.20 | ||
| SLE patient #2 | SLE patient #10 | 9.37 | ||
| SLE patient #2 | SLE patient #11 | 9.53 | ||
| SLE patient #2 | SLE patient #12 | 3.36 | ||
| SLE patient #2 | Healthy Control #1 | 1.15 | ||
| SLE patient #2 | Healthy Control #2 | 0.78 | ||
| SLE patient #3 | + | + | SLE patient #12 | 20.48 |
| SLE patient #3 | Healthy Control #1 | 24.92 | ||
| SLE patient #3 | Healthy Control #2 | 14.30 | ||
| SLE patient #4 | − | + | SLE patient #5 | 3.07 |
| SLE patient #4 | SLE patient #6 | 0.80 | ||
| SLE patient #4 | SLE patient #7 | 2.08 | ||
| SLE patient #4 | SLE patient #8 | 0.26 | ||
| SLE patient #4 | SLE patient #9 | 3.96 | ||
| SLE patient #4 | SLE patient #10 | 7.20 | ||
| SLE patient #4 | SLE patient #11 | 8.70 | ||
| SLE patient #4 | Healthy Control #1 | 0.10 | ||
| SLE patient #4 | Healthy Control #2 | 1.14 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, C.-C.; Schofield, T.; Tang, A.; Manzi, S.; Ahearn, J.M. Potential Roles of Antiphospholipid Antibodies in Generating Platelet-C4d in Systemic Lupus Erythematosus. Antibodies 2017, 6, 9. https://doi.org/10.3390/antib6030009
Liu C-C, Schofield T, Tang A, Manzi S, Ahearn JM. Potential Roles of Antiphospholipid Antibodies in Generating Platelet-C4d in Systemic Lupus Erythematosus. Antibodies. 2017; 6(3):9. https://doi.org/10.3390/antib6030009
Chicago/Turabian StyleLiu, Chau-Ching, Travis Schofield, Amy Tang, Susan Manzi, and Joseph M. Ahearn. 2017. "Potential Roles of Antiphospholipid Antibodies in Generating Platelet-C4d in Systemic Lupus Erythematosus" Antibodies 6, no. 3: 9. https://doi.org/10.3390/antib6030009
APA StyleLiu, C.-C., Schofield, T., Tang, A., Manzi, S., & Ahearn, J. M. (2017). Potential Roles of Antiphospholipid Antibodies in Generating Platelet-C4d in Systemic Lupus Erythematosus. Antibodies, 6(3), 9. https://doi.org/10.3390/antib6030009