The Ligands for Human IgG and Their Effector Functions



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Immunoglobulin G (IgG)
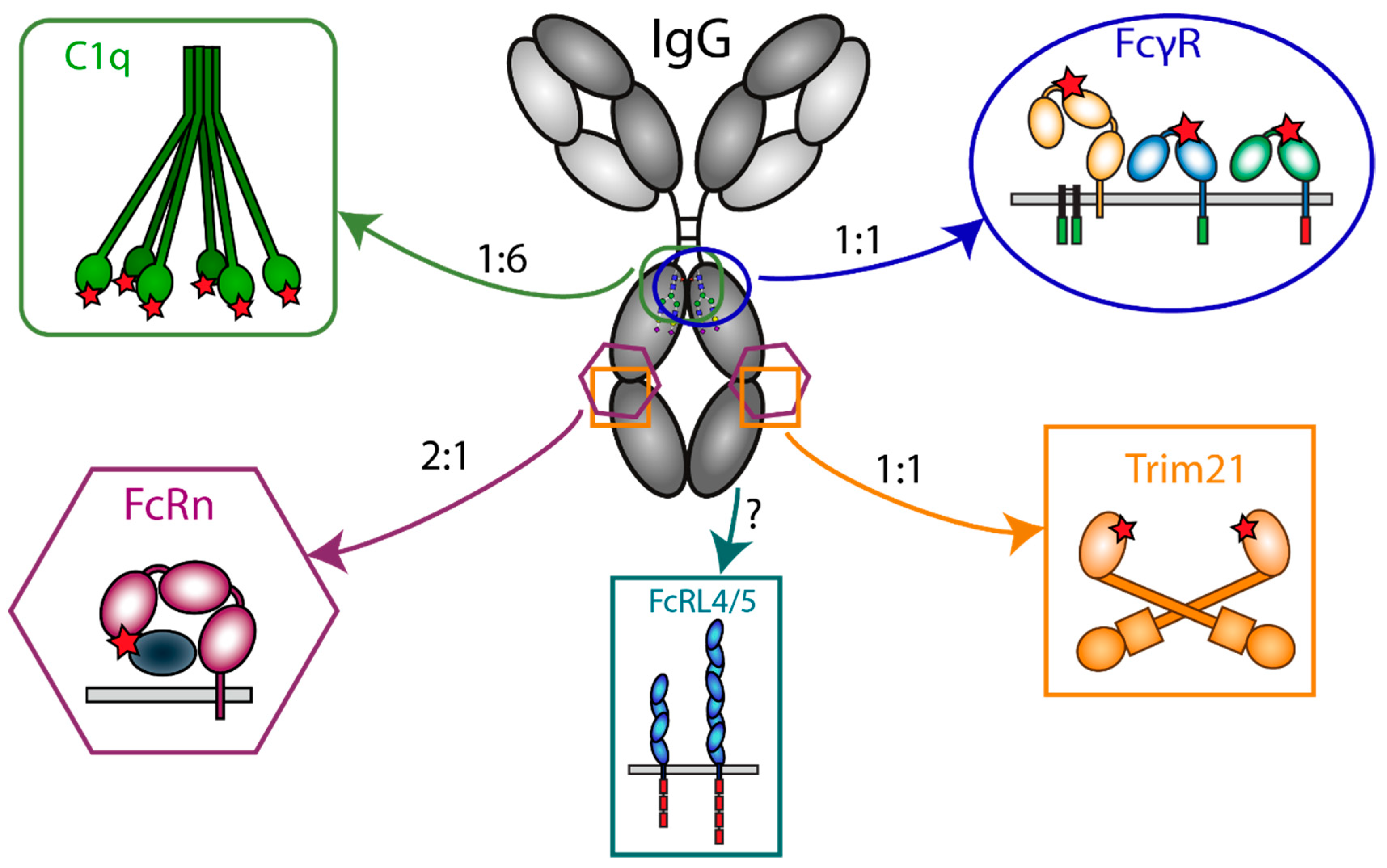
3. IgG-Fc-Engaging Effector Molecules
3.1. Fc-Receptors
3.2. DC-SIGN and CD23
3.3. FcRn
3.4. TRIM21
3.5. FcRL
3.6. Complement (C1q)
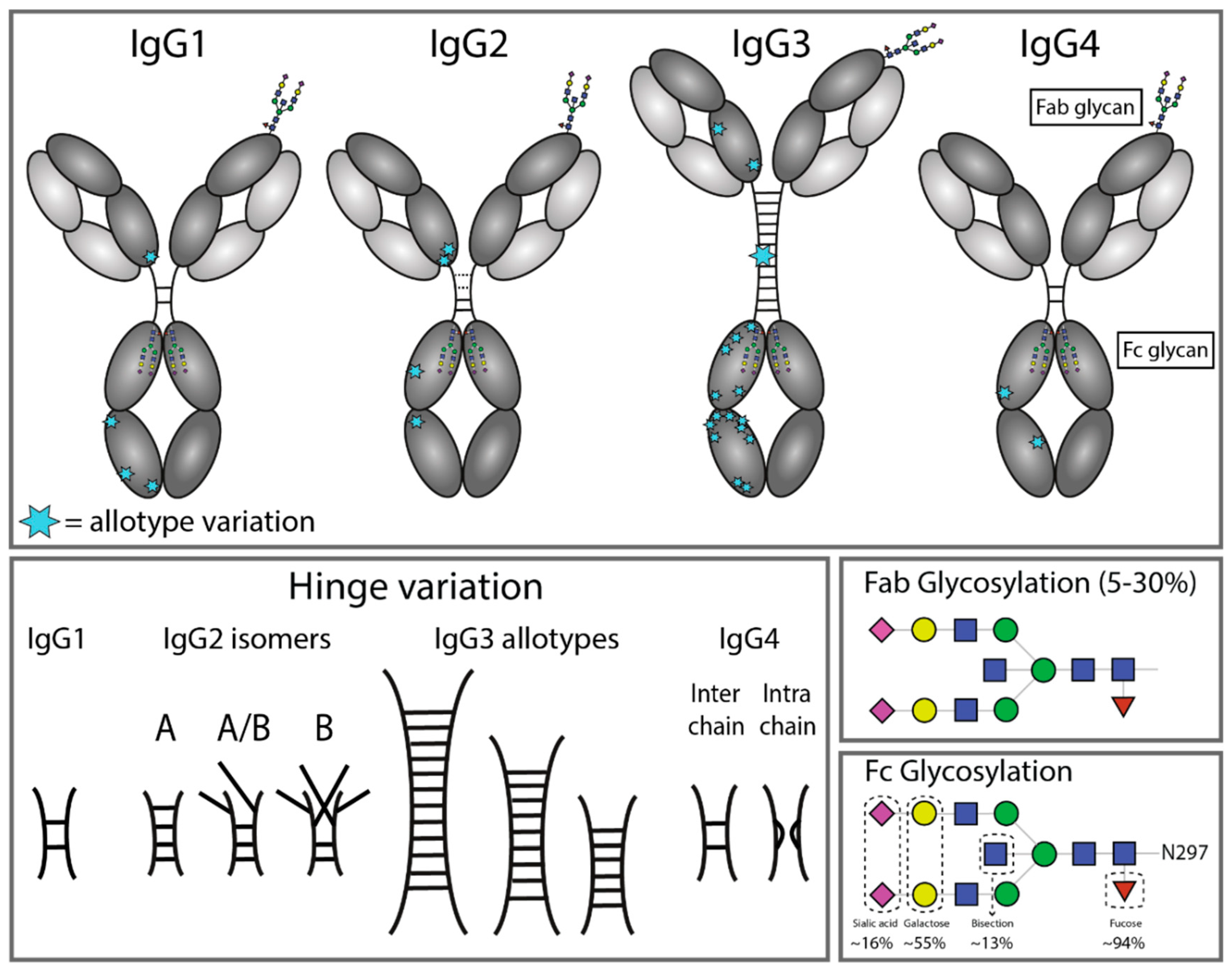
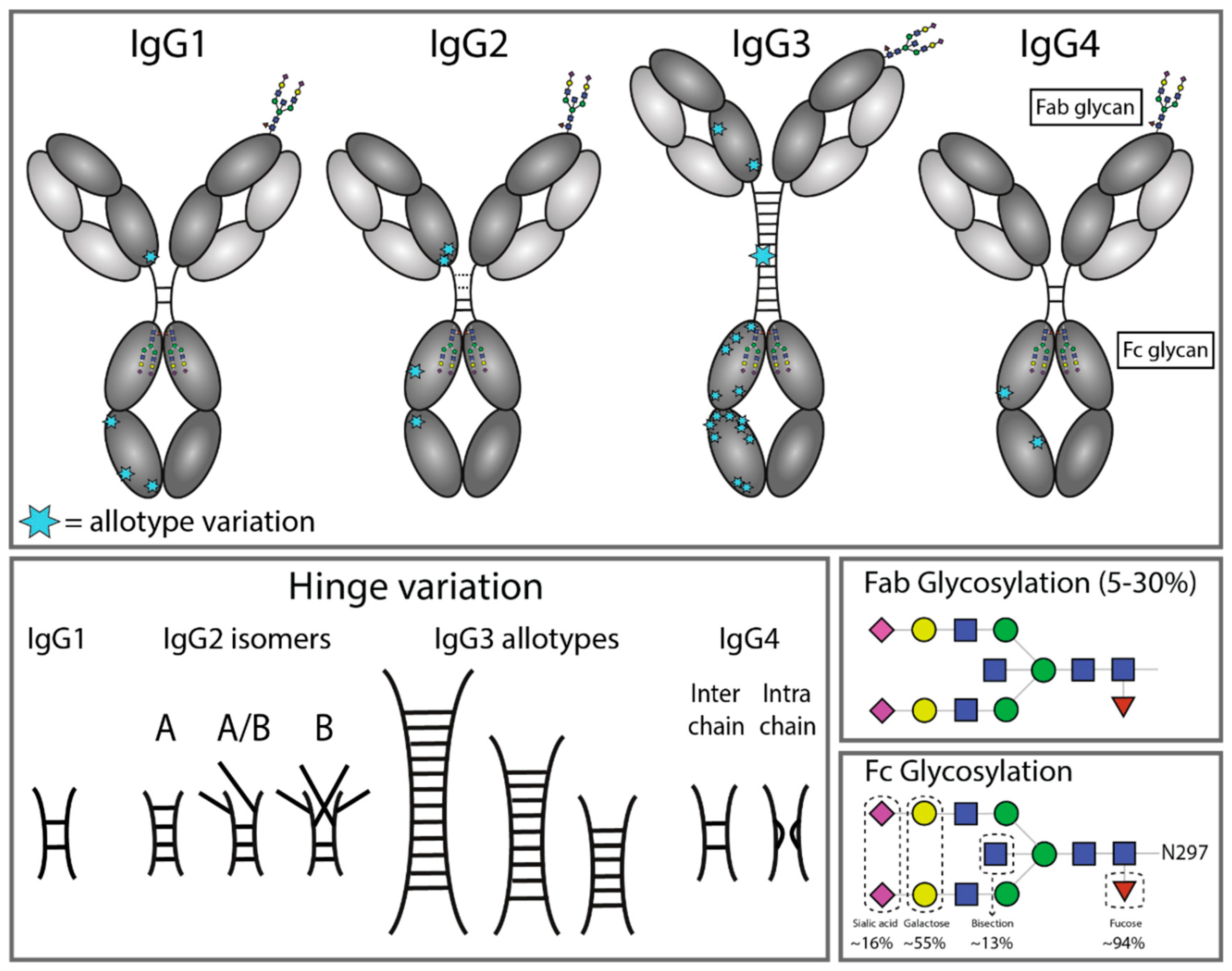
4. IgG Allotypes
5. IgG Glycosylation
6. Antibody Fc engineering
Author Contributions
Funding
Conflicts of Interest
References
- Hiramoto, E.; Tsutsumi, A.; Suzuki, R.; Matsuoka, S.; Arai, S.; Kikkawa, M.; Miyazaki, T. The IgM pentamer is an asymmetric pentagon with an open groove that binds the AIM protein. Sci. Adv. 2018, 4, eaau1199. [Google Scholar] [CrossRef]
- Berkowska, M.A.; Driessen, G.J.A.; Bikos, V.; Grosserichter-Wagener, C.; Stamatopoulos, K.; Cerutti, A.; He, B.; Biermann, K.; Lange, J.F.; van der Burg, M.; et al. Human memory B cells originate from three distinct germinal center-dependent and -independent maturation pathways. Blood 2011, 118, 2150–2158. [Google Scholar] [CrossRef] [Green Version]
- Methot, S.P.; Di Noia, J.M. Molecular Mechanisms of Somatic Hypermutation and Class Switch Recombination. Adv. Immunol. 2017, 133, 37–87. [Google Scholar] [PubMed]
- Shan, M.; Carrillo, J.; Yeste, A.; Gutzeit, C.; Segura-Garzón, D.; Walland, A.C.; Pybus, M.; Grasset, E.K.; Yeiser, J.R.; Matthews, D.B.; et al. Secreted IgD Amplifies Humoral T Helper 2 Cell Responses by Binding Basophils via Galectin-9 and CD44. Immunity 2018, 49, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Gutzeit, C.; Chen, K.; Cerutti, A. The enigmatic function of IgD: some answers at last. Eur. J. Immunol. 2018, 48, 1101–1113. [Google Scholar] [CrossRef] [Green Version]
- Horton, R.E.; Vidarsson, G. Antibodies and Their Receptors: Different Potential Roles in Mucosal Defense. Front. Immunol. 2013, 4, 200. [Google Scholar] [CrossRef] [PubMed]
- Herr, A.B.; Ballister, E.R.; Bjorkman, P.J. Insights into IgA-mediated immune responses from the crystal structures of human FcαRI and its complex with IgA1-Fc. Nature 2003, 423, 614–620. [Google Scholar] [CrossRef]
- van Egmond, M.; van Garderen, E.; van Spriel, A.B.; Damen, C.A.; van Amersfoort, E.S.; van Zandbergen, G.; van Hattum, J.; Kuiper, J.; van de Winkel, J.G.J. FcαRI-positive liver Kupffer cells: Reappraisal of the function of immunoglobulin A in immunity. Nat. Med. 2000, 6, 680–685. [Google Scholar] [CrossRef]
- Vidarsson, G.; van Der Pol, W.L.; van Den Elsen, J.M.; Vilé, H.; Jansen, M.; Duijs, J.; Morton, H.C.; Boel, E.; Daha, M.R.; Corthésy, B.; et al. Activity of human IgG and IgA subclasses in immune defense against Neisseria meningitidis serogroup B. J. Immunol. 2001, 166, 6250–6256. [Google Scholar] [CrossRef] [PubMed]
- Sutton, B.J.; Davies, A.M. Structure and dynamics of IgE-receptor interactions: FcεRI and CD23/FcεRII. Immunol. Rev. 2015, 268, 222–235. [Google Scholar] [CrossRef] [PubMed]
- Bunker, J.J.; Bendelac, A. IgA Responses to Microbiota. Immunity 2018, 49, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG subclasses and allotypes: from structure to effector functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, A.; Beard, L.J.; Feldman, R.G. IgG subclass distribution of antibodies to bacterial and viral antigens. Pediatr. Infect. Dis. J. 1990, 9, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Lighaam, L.; Rispens, T. The Immunobiology of Immunoglobulin G4. Semin. Liver Dis. 2016, 36, 200–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Neut Kolfschoten, M.; Schuurman, J.; Losen, M.; Bleeker, W.K.; Martínez-Martínez, P.; Vermeulen, E.; den Bleker, T.H.; Wiegman, L.; Vink, T.; Aarden, L.A.; et al. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science 2007, 313, 670–673. [Google Scholar]
- Rispens, T.; Davies, A.M.; Ooijevaar-de Heer, P.; Absalah, S.; Bende, O.; Sutton, B.J.; Vidarsson, G.; Aalberse, R.C. Dynamics of inter-heavy chain interactions in human immunoglobulin G (IgG) subclasses studied by kinetic Fab arm exchange. J. Biol. Chem. 2014, 289, 6098–6109. [Google Scholar] [CrossRef]
- Wypych, J.; Li, M.; Guo, A.; Zhang, Z.; Martinez, T.; Allen, M.J.; Fodor, S.; Kelner, D.N.; Flynn, G.C.; Liu, Y.D.; et al. Human IgG2 antibodies display disulfide-mediated structural isoforms. J. Biol. Chem. 2008, 283, 16194–16205. [Google Scholar] [CrossRef]
- Barrett, D.J.; Ayoub, E.M. IgG2 subclass restriction of antibody to pneumococcal polysaccharides. Clin. Exp. Immunol. 1986, 63, 127–134. [Google Scholar]
- Saeland, E.; Vidarsson, G.; Leusen, J.H.W.; Van Garderen, E.; Nahm, M.H.; Vile-Weekhout, H.; Walraven, V.; Stemerding, A.M.; Verbeek, J.S.; Rijkers, G.T.; et al. Central role of complement in passive protection by human IgG1 and IgG2 anti-pneumococcal antibodies in mice. J. Immunol. 2003, 170, 6158–6164. [Google Scholar] [CrossRef]
- White, A.L.; Chan, H.T.C.; French, R.R.; Willoughby, J.; Mockridge, C.I.; Roghanian, A.; Penfold, C.A.; Booth, S.G.; Dodhy, A.; Polak, M.E.; et al. Conformation of the Human Immunoglobulin G2 Hinge Imparts Superagonistic Properties to Immunostimulatory Anticancer Antibodies. Cancer Cell 2015, 27, 138–148. [Google Scholar] [CrossRef] [Green Version]
- Hargreaves, C.E.; Rose-Zerilli, M.J.J.; Machado, L.R.; Iriyama, C.; Hollox, E.J.; Cragg, M.S.; Strefford, J.C. Fcγ receptors: genetic variation, function, and disease. Immunol. Rev. 2015, 268, 6–24. [Google Scholar] [CrossRef] [PubMed]
- Li, F.J.; Won, W.J.; Becker, E.J.; Easlick, J.L.; Tabengwa, E.M.; Li, R.; Shakhmatov, M.; Honjo, K.; Burrows, P.D.; Davis, R.S. Emerging Roles for the FCRL Family Members in Lymphocyte Biology and Disease. In Curr. Top. Microbiol. Immunol. 2014, 382, 29–50. [Google Scholar]
- Lu, J.; Kishore, U. C1 Complex: An Adaptable Proteolytic Module for Complement and Non-Complement Functions. Front. Immunol. 2017, 8, 592. [Google Scholar] [CrossRef] [PubMed]
- Stapleton, N.M.; Einarsdóttir, H.K.; Stemerding, A.M.; Vidarsson, G. The multiple facets of FcRn in immunity. Immunol. Rev. 2015, 268, 253–268. [Google Scholar] [CrossRef]
- Foss, S.; Watkinson, R.; Sandlie, I.; James, L.C.; Andersen, J.T. TRIM21: a cytosolic Fc receptor with broad antibody isotype specificity. Immunol. Rev. 2015, 268, 328–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillis, C.; Gouel-Chéron, A.; Jönsson, F.; Bruhns, P. Contribution of Human FcγRs to Disease with Evidence from Human Polymorphisms and Transgenic Animal Studies. Front. Immunol. 2014, 5, 254. [Google Scholar] [CrossRef] [PubMed]
- Daëron, M. F c RECEPTOR BIOLOGY. Annu. Rev. Immunol. 1997, 15, 203–234. [Google Scholar] [CrossRef]
- Bruhns, P.; Iannascoli, B.; England, P.; Mancardi, D.A.; Fernandez, N.; Jorieux, S.; Daëron, M. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood 2009, 113, 3716–3725. [Google Scholar] [CrossRef]
- Vidarsson, G.; van de Winkel, J.G. Fc receptor and complement receptor-mediated phagocytosis in host defence. Curr. Opin. Infect. Dis. 1998, 11, 271–278. [Google Scholar] [CrossRef]
- Veri, M.-C.; Gorlatov, S.; Li, H.; Burke, S.; Johnson, S.; Stavenhagen, J.; Stein, K.E.; Bonvini, E.; Koenig, S. Monoclonal antibodies capable of discriminating the human inhibitory Fc?-receptor IIB (CD32B) from the activating Fc?-receptor IIA (CD32A): biochemical, biological and functional characterization. Immunology 2007, 121, 392–404. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J. V Divergent Immunoglobulin G Subclass Activity Through Selective Fc Receptor Binding. Science 2005, 310, 1510–1512. [Google Scholar] [CrossRef]
- Caaveiro, J.M.M.; Kiyoshi, M.; Tsumoto, K. Structural analysis of Fc/FcγR complexes: a blueprint for antibody design. Immunol. Rev. 2015, 268, 201–221. [Google Scholar] [CrossRef]
- Bruggeman, C.W.; Dekkers, G.; Bentlage, A.E.H.; Treffers, L.W.; Nagelkerke, S.Q.; Lissenberg-Thunnissen, S.; Koeleman, C.A.M.; Wuhrer, M.; van den Berg, T.K.; Rispens, T.; et al. Enhanced effector functions due to antibody defucosylation depend on the effector cell Fcγ receptor profile. J. Immunol. 2017, 199, 204–211. [Google Scholar] [CrossRef]
- Treffers, L.W.; van Houdt, M.; Bruggeman, C.W.; Heineke, M.H.; Zhao, X.W.; van der Heijden, J.; Nagelkerke, S.Q.; Verkuijlen, P.J.J.H.; Geissler, J.; Lissenberg-Thunnissen, S.; et al. FcγRIIIb Restricts Antibody-Dependent Destruction of Cancer Cells by Human Neutrophils. Front. Immunol. 2019, 9, 3124. [Google Scholar] [CrossRef]
- Meinderts, S.M.; Sins, J.W.R.; Fijnvandraat, K.; Nagelkerke, S.Q.; Geissler, J.; Tanck, M.W.; Bruggeman, C.; Biemond, B.J.; Rijneveld, A.W.; Kerkhoffs, J.-L.H.; et al. Nonclassical FCGR2C haplotype is associated with protection from red blood cell alloimmunization in sickle cell disease. Blood 2017, 130, 2121–2130. [Google Scholar] [CrossRef]
- van der Heijden, J.; Breunis, W.B.; Geissler, J.; de Boer, M.; van den Berg, T.K.; Kuijpers, T.W. Phenotypic Variation in IgG Receptors by Nonclassical FCGR2C Alleles. J. Immunol. 2012, 188, 1318–1324. [Google Scholar] [CrossRef]
- Li, X.; Wu, J.; Ptacek, T.; Redden, D.T.; Brown, E.E.; Alarcón, G.S.; Ramsey-Goldman, R.; Petri, M.A.; Reveille, J.D.; Kaslow, R.A.; et al. Allelic-Dependent Expression of an Activating Fc Receptor on B Cells Enhances Humoral Immune Responses. Sci. Transl. Med. 2013, 5, 216ra175. [Google Scholar] [CrossRef]
- Metes, D.; Ernst, L.K.; Chambers, W.H.; Sulica, A.; Herberman, R.B.; Morel, P.A. Expression of functional CD32 molecules on human NK cells is determined by an allelic polymorphism of the FcgammaRIIC gene. Blood 1998, 91, 2369–2380. [Google Scholar]
- Breunis, W.B.; van Mirre, E.; Bruin, M.; Geissler, J.; de Boer, M.; Peters, M.; Roos, D.; de Haas, M.; Koene, H.R.; Kuijpers, T.W. Copy number variation of the activating FCGR2C gene predisposes to idiopathic thrombocytopenic purpura. Blood 2008, 111, 1029–1038. [Google Scholar] [CrossRef]
- Stegmann, T.C.; Veldhuisen, B.; Nagelkerke, S.Q.; Winkelhorst, D.; Schonewille, H.; Verduin, E.P.; Kuijpers, T.W.; de Haas, M.; Vidarsson, G.; van der Schoot, C.E. RhIg-prophylaxis is not influenced by FCGR2/3 polymorphisms involved in red blood cell clearance. Blood 2017, 129, 1045–1048. [Google Scholar] [CrossRef]
- Miescher, S.; Spycher, M.O.; Amstutz, H.; de Haas, M.; Kleijer, M.; Kalus, U.J.; Radtke, H.; Hubsch, A.; Andresen, I.; Martin, R.M.; et al. A single recombinant anti-RhD IgG prevents RhD immunization: association of RhD-positive red blood cell clearance rate with polymorphisms in the FcγRIIA and FcγIIIA genes. Blood 2004, 103, 4028–4035. [Google Scholar] [CrossRef]
- Ruyssen-Witrand, A.; Rouanet, S.; Combe, B.; Dougados, M.; Le Loët, X.; Sibilia, J.; Tebib, J.; Mariette, X.; Constantin, A. Fcγ receptor type IIIA polymorphism influences treatment outcomes in patients with rheumatoid arthritis treated with rituximab. Ann. Rheum. Dis. 2012, 71, 875–877. [Google Scholar] [CrossRef]
- Lee, Y.H.; Bae, S.-C.; Song, G.G. Functional FCGR3A 158 V/F and IL-6 −174 C/G polymorphisms predict response to biologic therapy in patients with rheumatoid arthritis: a meta-analysis. Rheumatol. Int. 2014, 34, 1409–1415. [Google Scholar] [CrossRef]
- Liu, D.; Tian, Y.; Sun, D.; Sun, H.; Jin, Y.; Dong, M. The FCGR3A polymorphism predicts the response to rituximab-based therapy in patients with non-Hodgkin lymphoma: a meta-analysis. Ann. Hematol. 2016, 95, 1483–1490. [Google Scholar] [CrossRef]
- Carlotti, E.; Palumbo, G.A.; Oldani, E.; Tibullo, D.; Salmoiraghi, S.; Rossi, A.; Golay, J.; Pulsoni, A.; Foà, R.; Rambaldi, A. FcgammaRIIIA and FcgammaRIIA polymorphisms do not predict clinical outcome of follicular non-Hodgkin’s lymphoma patients treated with sequential CHOP and rituximab. Haematologica 2007, 92, 1127–1130. [Google Scholar] [CrossRef]
- van der Heijden, J.; Nagelkerke, S.; Zhao, X.; Geissler, J.; Rispens, T.; van den Berg, T.K.; Kuijpers, T.W. Haplotypes of Fc RIIa and Fc RIIIb Polymorphic Variants Influence IgG-Mediated Responses in Neutrophils. J. Immunol. 2014, 192, 2715–2721. [Google Scholar] [CrossRef] [Green Version]
- Treffers, L.W.; Zhao, X.W.; van der Heijden, J.; Nagelkerke, S.Q.; van Rees, D.J.; Gonzalez, P.; Geissler, J.; Verkuijlen, P.; van Houdt, M.; de Boer, M.; et al. Genetic variation of human neutrophil Fcγ receptors and SIRPα in antibody-dependent cellular cytotoxicity towards cancer cells. Eur. J. Immunol. 2018, 48, 344–354. [Google Scholar] [CrossRef]
- Sondermann, P.; Pincetic, A.; Maamary, J.; Lammens, K.; Ravetch, J.V. General mechanism for modulating immunoglobulin effector function. Proc. Natl. Acad. Sci. USA 2013, 110, 9868–9872. [Google Scholar] [CrossRef]
- Anthony, R.M.; Wermeling, F.; Karlsson, M.C.I.; Ravetch, J.V. Identification of a receptor required for the anti-inflammatory activity of IVIG. Proc. Natl. Acad. Sci. USA 2008, 105, 19571–19578. [Google Scholar] [CrossRef] [Green Version]
- Crispin, M.; Yu, X.; Bowden, T.A. Crystal structure of sialylated IgG Fc: Implications for the mechanism of intravenous immunoglobulin therapy. Proc. Natl. Acad. Sci. USA 2013, 110, E3544–E3546. [Google Scholar] [CrossRef]
- Yu, X.; Vasiljevic, S.; Mitchell, D.A.; Crispin, M.; Scanlan, C.N. Dissecting the Molecular Mechanism of IVIg Therapy: The Interaction between Serum IgG and DC-SIGN is Independent of Antibody Glycoform or Fc Domain. J. Mol. Biol. 2013, 425, 1253–1258. [Google Scholar] [CrossRef]
- Temming, A.R.; Dekkers, G.; van de Bovenkamp, F.S.; Plomp, H.R.; Bentlage, A.E.H.; Szittner, Z.; Derksen, N.I.L.; Wuhrer, M.; Rispens, T.; Vidarsson, G. Human DC-SIGN and CD23 do not interact with human IgG. Submitted.
- Anthony, R.M.; Wermeling, F.; Ravetch, J.V. Novel roles for the IgG Fc glycan. Ann. N. Y. Acad. Sci. 2012, 1253, 170–180. [Google Scholar] [CrossRef]
- Ahmed, A.A.; Giddens, J.; Pincetic, A.; Lomino, J.V.; Ravetch, J.V.; Wang, L.-X.; Bjorkman, P.J. Structural Characterization of Anti-Inflammatory Immunoglobulin G Fc Proteins. J. Mol. Biol. 2014, 426, 3166–3179. [Google Scholar] [CrossRef] [Green Version]
- Guhr, T.; Bloem, J.; Derksen, N.I.L.; Wuhrer, M.; Koenderman, A.H.L.; Aalberse, R.C.; Rispens, T. Enrichment of sialylated IgG by lectin fractionation does not enhance the efficacy of immunoglobulin G in a murine model of immune thrombocytopenia. PLoS One 2011, 6, e21246. [Google Scholar] [CrossRef]
- Vidarsson, G.; Stemerding, A.M.; Stapleton, N.M.; Spliethoff, S.E.; Janssen, H.; Rebers, F.E.; de Haas, M.; van de Winkel, J.G. FcRn: an IgG receptor on phagocytes with a novel role in phagocytosis. Blood 2006, 108, 3573–3579. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, A.; Honzawa, M.; Ito, S.; Miyazaki, T.; Matsumoto, H.; Nakamura, H.; Michaelsen, T.E.; Arata, Y. H NMR studies of the Fc region of human IgG1 and IgG3 immunoglobulins: assignment of histidine resonances in the CH3 domain and identification of IgG3 protein carrying G3m(st) allotypes. Mol. Immunol. 1983, 20, 141–148. [Google Scholar] [CrossRef]
- Vaughn, D.E.; Bjorkman, P.J. Structural basis of pH-dependent antibody binding by the neonatal Fc receptor. Structure 1998, 6, 63–73. [Google Scholar] [CrossRef]
- Stapleton, N.M.; Andersen, J.T.; Stemerding, A.M.; Bjarnarson, S.P.; Verheul, R.C.; Gerritsen, J.; Zhao, Y.; Kleijer, M.; Sandlie, I.; de Haas, M.; et al. Competition for FcRn-mediated transport gives rise to short half-life of human IgG3 and offers therapeutic potential. Nat. Commun. 2011, 2, 599. [Google Scholar] [CrossRef] [Green Version]
- Einarsdottir, H.; Ji, Y.; Visser, R.; Mo, C.; Luo, G.; Scherjon, S.; van der Schoot, C.E.; Vidarsson, G. H435-containing immunoglobulin G3 allotypes are transported efficiently across the human placenta: implications for alloantibody-mediated diseases of the newborn. Transfusion 2014, 54, 665–671. [Google Scholar] [CrossRef]
- Dechavanne, C.; Dechavanne, S.; Sadissou, I.; Lokossou, A.G.; Alvarado, F.; Dambrun, M.; Moutairou, K.; Courtin, D.; Nuel, G.; Garcia, A.; et al. Associations between an IgG3 polymorphism in the binding domain for FcRn, transplacental transfer of malaria-specific IgG3, and protection against Plasmodium falciparum malaria during infancy: A birth cohort study in Benin. PLoS Med. 2017, 14, e1002403. [Google Scholar] [CrossRef]
- Schoch, A.; Kettenberger, H.; Mundigl, O.; Winter, G.; Engert, J.; Heinrich, J.; Emrich, T. Charge-mediated influence of the antibody variable domain on FcRn-dependent pharmacokinetics. Proc. Natl. Acad. Sci. USA 2015, 112, 5997–6002. [Google Scholar] [CrossRef]
- Monnet, C.; Jorieux, S.; Urbain, R.; Fournier, N.; Bouayadi, K.; De Romeuf, C.; Behrens, C.K.; Fontayne, A.; Mondon, P. Selection of IgG Variants with Increased FcRn Binding Using Random and Directed Mutagenesis: Impact on Effector Functions. Front. Immunol. 2015, 6, 39. [Google Scholar] [CrossRef]
- James, L.C.; Keeble, A.H.; Khan, Z.; Rhodes, D.A.; Trowsdale, J. Structural basis for PRYSPRY-mediated tripartite motif (TRIM) protein function. Proc. Natl. Acad. Sci. USA 2007, 104, 6200–6205. [Google Scholar] [CrossRef]
- Keeble, A.H.; Khan, Z.; Forster, A.; James, L.C. TRIM21 is an IgG receptor that is structurally, thermodynamically, and kinetically conserved. Proc. Natl. Acad. Sci. USA 2008, 105, 6045–6050. [Google Scholar] [CrossRef]
- Mallery, D.L.; McEwan, W.A.; Bidgood, S.R.; Towers, G.J.; Johnson, C.M.; James, L.C. Antibodies mediate intracellular immunity through tripartite motif-containing 21 (TRIM21). Proc. Natl. Acad. Sci. USA 2010, 107, 19985–19990. [Google Scholar] [CrossRef] [Green Version]
- Foss, S.; Watkinson, R.E.; Grevys, A.; McAdam, M.B.; Bern, M.; Høydahl, L.S.; Dalhus, B.; Michaelsen, T.E.; Sandlie, I.; James, L.C.; et al. TRIM21 Immune Signaling Is More Sensitive to Antibody Affinity Than Its Neutralization Activity. J. Immunol. 2016, 196, 3452–3459. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, A.J.; Mallery, D.L.; Watkinson, R.E.; Dickson, C.F.; James, L.C. Sequential ubiquitination and deubiquitination enzymes synchronize the dual sensor and effector functions of TRIM21. Proc. Natl. Acad. Sci. USA 2015, 112, 10014–10019. [Google Scholar] [CrossRef] [Green Version]
- Rostamzadeh, D.; Kazemi, T.; Amirghofran, Z.; Shabani, M. Update on Fc receptor-like (FCRL) family: new immunoregulatory players in health and diseases. Expert Opin. Ther. Targets 2018, 22, 487–502. [Google Scholar] [CrossRef]
- Swainson, L.A.; Mold, J.E.; Bajpai, U.D.; McCune, J.M. Expression of the Autoimmune Susceptibility Gene FcRL3 on Human Regulatory T Cells Is Associated with Dysfunction and High Levels of Programmed Cell Death-1. J. Immunol. 2010, 184, 3639–3647. [Google Scholar] [CrossRef]
- Bin Dhuban, K.; d’Hennezel, E.; Nashi, E.; Bar-Or, A.; Rieder, S.; Shevach, E.M.; Nagata, S.; Piccirillo, C.A. Coexpression of TIGIT and FCRL3 Identifies Helios + Human Memory Regulatory T Cells. J. Immunol. 2015, 194, 3687–3696. [Google Scholar] [CrossRef]
- Kulemzin, S.V.; Zamoshnikova, A.Y.; Yurchenko, M.Y.; Vitak, N.Y.; Najakshin, A.M.; Fayngerts, S.A.; Chikaev, N.A.; Reshetnikova, E.S.; Kashirina, N.M.; Peclo, M.M.; et al. FCRL6 receptor: Expression and associated proteins. Immunol. Lett. 2011, 134, 174–182. [Google Scholar] [CrossRef]
- Wilson, T.J.; Fuchs, A.; Colonna, M. Cutting Edge: Human FcRL4 and FcRL5 Are Receptors for IgA and IgG. J. Immunol. 2012, 188, 4741–4745. [Google Scholar] [CrossRef]
- Franco, A.; Damdinsuren, B.; Ise, T.; Dement-Brown, J.; Li, H.; Nagata, S.; Tolnay, M. Human Fc receptor-like 5 binds intact IgG via mechanisms distinct from those of Fc receptors. J. Immunol. 2013, 190, 5739–5746. [Google Scholar] [CrossRef]
- Miller, I.; Hatzivassiliou, G.; Cattoretti, G.; Mendelsohn, C.; Dalla-Favera, R. IRTAs: a new family of immunoglobulinlike receptors differentially expressed in B cells. Blood 2002, 99, 2662–2669. [Google Scholar] [CrossRef] [Green Version]
- Haga, C.L.; Ehrhardt, G.R.A.; Boohaker, R.J.; Davis, R.S.; Cooper, M.D. Fc receptor-like 5 inhibits B cell activation via SHP-1 tyrosine phosphatase recruitment. Proc. Natl. Acad. Sci. USA 2007, 104, 9770–9775. [Google Scholar] [CrossRef] [Green Version]
- Franco, A.; Kraus, Z.; Li, H.; Seibert, N.; Dement-Brown, J.; Tolnay, M. CD21 and FCRL5 form a receptor complex with robust B-cell activating capacity. Int. Immunol. 2018, 30, 569–578. [Google Scholar] [CrossRef]
- Niles, M.J.; Matsuuchi, L.; Koshland, M.E. Polymer IgM assembly and secretion in lymphoid and nonlymphoid cell lines: evidence that J chain is required for pentamer IgM synthesis. Proc. Natl. Acad. Sci. USA 1995, 92, 2884–2888. [Google Scholar] [CrossRef]
- Ugurlar, D.; Howes, S.C.; de Kreuk, B.-J.; Koning, R.I.; de Jong, R.N.; Beurskens, F.J.; Schuurman, J.; Koster, A.J.; Sharp, T.H.; Parren, P.W.H.I.; et al. Structures of C1-IgG1 provide insights into how danger pattern recognition activates complement. Science 2018, 359, 794–797. [Google Scholar] [CrossRef]
- van den Bremer, E.T.; Beurskens, F.J.; Voorhorst, M.; Engelberts, P.J.; de Jong, R.N.; van der Boom, B.G.; Cook, E.M.; Lindorfer, M.A.; Taylor, R.P.; van Berkel, P.H.; et al. Human IgG is produced in a pro-form that requires clipping of C-terminal lysines for maximal complement activation. MAbs 2015, 7, 672–680. [Google Scholar] [CrossRef] [Green Version]
- Dekkers, G.; Treffers, L.; Plomp, R.; Bentlage, A.E.H.; de Boer, M.; Koeleman, C.A.M.; Lissenberg-Thunnissen, S.N.; Visser, R.; Brouwer, M.; Mok, J.Y.; et al. Decoding the human immunoglobulin G-glycan repertoire reveals a spectrum of Fc-receptor- and complement-mediated-effector activities. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef]
- Quast, I.; Keller, C.W.; Maurer, M.A.; Giddens, J.P.; Tackenberg, B.; Wang, L.-X.; Münz, C.; Nimmerjahn, F.; Dalakas, M.C.; Lünemann, J.D. Sialylation of IgG Fc domain impairs complement-dependent cytotoxicity. J. Clin. Invest. 2015, 125, 4160–4170. [Google Scholar] [CrossRef] [Green Version]
- Peschke, B.; Keller, C.W.; Weber, P.; Quast, I.; Lünemann, J.D. Fc-Galactosylation of Human Immunoglobulin Gamma Isotypes Improves C1q Binding and Enhances Complement-Dependent Cytotoxicity. Front. Immunol. 2017, 8, 646. [Google Scholar] [CrossRef]
- Brüggemann, M.; Williams, G.T.; Bindon, C.I.; Clark, M.R.; Walker, M.R.; Jefferis, R.; Waldmann, H.; Neuberger, M.S. Comparison of the effector functions of human immunoglobulins using a matched set of chimeric antibodies. J. Exp. Med. 1987, 166, 1351–1361. [Google Scholar] [CrossRef] [Green Version]
- Natsume, A.; In, M.; Takamura, H.; Nakagawa, T.; Shimizu, Y.; Kitajima, K.; Wakitani, M.; Ohta, S.; Satoh, M.; Shitara, K.; et al. Engineered antibodies of IgG1/IgG3 mixed isotype with enhanced cytotoxic activities. Cancer Res. 2008, 68, 3863–3872. [Google Scholar] [CrossRef]
- Giuntini, S.; Granoff, D.M.; Beernink, P.T.; Ihle, O.; Bratlie, D.; Michaelsen, T.E. Human IgG1, IgG3, and IgG3 Hinge-Truncated Mutants Show Different Protection Capabilities against Meningococci Depending on the Target Antigen and Epitope Specificity. Clin. Vaccine Immunol. 2016, 23, 698–706. [Google Scholar] [CrossRef] [Green Version]
- Oxelius, V.-A.; Pandey, J.P. Human immunoglobulin constant heavy G chain (IGHG) (Fcγ) (GM) genes, defining innate variants of IgG molecules and B cells, have impact on disease and therapy. Clin. Immunol. 2013, 149, 475–486. [Google Scholar] [CrossRef] [Green Version]
- Oxelius, V.-A. Immunoglobulin constant heavy G subclass chain genes in asthma and allergy. Immunol. Res. 2008, 40, 179–191. [Google Scholar] [CrossRef]
- Oxelius, V.A.; Carlsson, A.M.; Aurivillius, M. Alternative G1m, G2m and G3m allotypes of IGHG genes correlate with atopic and nonatopic pathways of immune regulation in children with bronchial asthma. Int. Arch. Allergy Immunol. 1998, 115, 215–219. [Google Scholar] [CrossRef]
- O’Hanlon, T.P.; Rider, L.G.; Schiffenbauer, A.; Targoff, I.N.; Malley, K.; Pandey, J.P.; Miller, F.W. Immunoglobulin gene polymorphisms are susceptibility factors in clinical and autoantibody subgroups of the idiopathic inflammatory myopathies. Arthritis Rheum. 2008, 58, 3239–3246. [Google Scholar] [CrossRef]
- Pandey, J.P.; Namboodiri, A.M. Genetic variants of IgG1 antibodies and FcγRIIIa receptors influence the magnitude of antibody-dependent cell-mediated cytotoxicity against prostate cancer cells. Oncoimmunology 2014, 3, e27317. [Google Scholar] [CrossRef] [Green Version]
- Pandey, J.P.; Luo, Y.; Elston, R.C.; Wu, Y.; Philp, F.H.; Astemborski, J.; Thomas, D.L.; Netski, D.M. Immunoglobulin allotypes influence IgG antibody responses to hepatitis C virus envelope proteins E1 and E2. Hum. Immunol. 2008, 69, 158–164. [Google Scholar] [CrossRef]
- Seppälä, I.J.; Sarvas, H.; Mäkelä, O. Low concentrations of Gm allotypic subsets G3 mg and G1 mf in homozygotes and heterozygotes. J. Immunol. 1993, 151, 2529–2537. [Google Scholar]
- Pan, Q.; Petit-Frére, C.; Hammarström, L. An allotype-associated polymorphism in the γ3 promoter determines the germ-line γ3 transcriptional rate but does not influence switching and subsequent IgG3 production. Eur. J. Immunol. 2000, 30, 2388–2393. [Google Scholar] [CrossRef] [Green Version]
- Jonsson, S.; Sveinbjornsson, G.; de Lapuente Portilla, A.L.; Swaminathan, B.; Plomp, R.; Dekkers, G.; Ajore, R.; Ali, M.; Bentlage, A.E.H.; Elmér, E.; et al. Identification of sequence variants influencing immunoglobulin levels. Nat. Genet. 2017, 49, 1182–1191. [Google Scholar] [CrossRef]
- Pan, Q.; Hammarström, L. Molecular basis of IgG subclass deficiency. Immunol. Rev. 2000, 178, 99–110. [Google Scholar] [CrossRef]
- Kratochvil, S.; McKay, P.F.; Chung, A.W.; Kent, S.J.; Gilmore, J.; Shattock, R.J. Immunoglobulin G1 allotype influences antibody subclass distribution in response to HIV gp140 vaccination. Front. Immunol. 2017, 8, 1883. [Google Scholar] [CrossRef]
- Ternant, D.; Arnoult, C.; Pugnière, M.; Dhommée, C.; Drocourt, D.; Perouzel, E.; Passot, C.; Baroukh, N.; Mulleman, D.; Tiraby, G.; et al. IgG1 Allotypes Influence the Pharmacokinetics of Therapeutic Monoclonal Antibodies through FcRn Binding. J. Immunol. 2016, 196, 607–613. [Google Scholar] [CrossRef]
- Brusco, A.; Saviozzi, S.; Cinque, F.; DeMarchi, M.; Boccazzi, C.; de Lange, G.; van Leeuwen, A.M.; Carbonara, A.O. Molecular characterization of immunoglobulin G4 gene isoallotypes. Eur. J. Immunogenet. 1998, 25, 349–355. [Google Scholar] [CrossRef]
- Howie, H.L.; Delaney, M.; Wang, X.; Er, L.S.; Kapp, L.; Lebedev, J.N.; Zimring, J.C. Errors in data interpretation from genetic variation of human analytes. JCI Insight 2017, 2, 1–9. [Google Scholar] [CrossRef]
- Howie, H.L.; Delaney, M.; Wang, X.; Er, L.S.; Vidarsson, G.; Stegmann, T.C.; Kapp, L.; Lebedev, J.N.; Wu, Y.; AuBuchon, J.P.; et al. Serological blind spots for variants of human IgG3 and IgG4 by a commonly used anti-immunoglobulin reagent. Transfusion 2016, 56, 2953–2962. [Google Scholar] [CrossRef] [Green Version]
- Subedi, G.P.; Barb, A.W. The Structural Role of Antibody N-Glycosylation in Receptor Interactions. Structure 2015, 23, 1573–1583. [Google Scholar] [CrossRef] [PubMed]
- Niwa, R.; Natsume, A.; Uehara, A.; Wakitani, M.; Iida, S.; Uchida, K.; Satoh, M.; Shitara, K. IgG subclass-independent improvement of antibody-dependent cellular cytotoxicity by fucose removal from Asn297-linked oligosaccharides. J. Immunol. Methods 2005, 306, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Shinkawa, T.; Nakamura, K.; Yamane, N.; Shoji-Hosaka, E.; Kanda, Y.; Sakurada, M.; Uchida, K.; Anazawa, H.; Satoh, M.; Yamasaki, M.; et al. The Absence of Fucose but Not the Presence of Galactose or Bisecting N -Acetylglucosamine of Human IgG1 Complex-type Oligosaccharides Shows the Critical Role of Enhancing Antibody-dependent Cellular Cytotoxicity. J. Biol. Chem. 2003, 278, 3466–3473. [Google Scholar] [CrossRef] [PubMed]
- Pereira, N.A.; Chan, K.F.; Lin, P.C.; Song, Z. The “less-is-more” in therapeutic antibodies: Afucosylated anti-cancer antibodies with enhanced antibody-dependent cellular cytotoxicity. MAbs 2018, 10, 693–711. [Google Scholar] [CrossRef]
- Dekkers, G.; Bentlage, A.E.H.; Plomp, R.; Visser, R.; Koeleman, C.A.M.; Beentjes, A.; Mok, J.Y.; van Esch, W.J.E.; Wuhrer, M.; Rispens, T.; et al. Conserved FcγR- glycan discriminates between fucosylated and afucosylated IgG in humans and mice. Mol. Immunol. 2018, 94, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.R.; Roberts, J.T.; Subedi, G.P.; Barb, A.W. Restricted processing of CD16a/Fc receptor IIIa N-glycans from primary human NK cells impacts structure and function. J. Biol. Chem. 2018, 293, 3477–3489. [Google Scholar] [CrossRef]
- Falconer, D.J.; Subedi, G.P.; Marcella, A.M.; Barb, A.W. Antibody fucosylation lowers the FcγRIIIa/CD16a affinity by limiting the conformations sampled by the N162-glycan. ACS Chem. Biol. 2018, 13, 2179–2189. [Google Scholar] [CrossRef]
- Ferrara, C.; Grau, S.; Jäger, C.; Sondermann, P.; Brünker, P.; Waldhauer, I.; Hennig, M.; Ruf, A.; Rufer, A.C.; Stihle, M.; et al. Unique carbohydrate-carbohydrate interactions are required for high affinity binding between FcgammaRIII and antibodies lacking core fucose. Proc. Natl. Acad. Sci. USA 2011, 108, 12669–12674. [Google Scholar] [CrossRef]
- Thomann, M.; Reckermann, K.; Reusch, D.; Prasser, J.; Tejada, M.L. Fc-galactosylation modulates antibody-dependent cellular cytotoxicity of therapeutic antibodies. Mol. Immunol. 2016, 73, 69–75. [Google Scholar] [CrossRef]
- Beers, S.A.; Chan, C.H.T.; French, R.R.; Cragg, M.S.; Glennie, M.J. CD20 as a Target for Therapeutic Type I and II Monoclonal Antibodies. Semin. Hematol. 2010, 47, 107–114. [Google Scholar] [CrossRef]
- Dekkers, G.; Rispens, T.; Vidarsson, G. Novel Concepts of Altered Immunoglobulin G Galactosylation in Autoimmune Diseases. Front. Immunol. 2018, 9, 553. [Google Scholar] [CrossRef]
- Pincetic, A.; Bournazos, S.; DiLillo, D.J.; Maamary, J.; Wang, T.T.; Dahan, R.; Fiebiger, B.-M.; Ravetch, J. V Type I and type II Fc receptors regulate innate and adaptive immunity. Nat. Immunol. 2014, 15, 707–716. [Google Scholar] [CrossRef]
- van de Bovenkamp, F.S.; Derksen, N.I.L.; Ooijevaar-de Heer, P.; van Schie, K.A.; Kruithof, S.; Berkowska, M.A.; van der Schoot, C.E.; IJspeert, H.; van der Burg, M.; Gils, A.; et al. Adaptive antibody diversification through N -linked glycosylation of the immunoglobulin variable region. Proc. Natl. Acad. Sci. USA 2018, 115, 1901–1906. [Google Scholar] [CrossRef]
- van de Bovenkamp, F.S.; Derksen, N.I.L.; van Breemen, M.J.; de Taeye, S.W.; Ooijevaar-de Heer, P.; Sanders, R.W.; Rispens, T. Variable Domain N-Linked Glycans Acquired During Antigen-Specific Immune Responses Can Contribute to Immunoglobulin G Antibody Stability. Front. Immunol. 2018, 9, 740. [Google Scholar] [CrossRef]
- Hamza, N.; Hershberg, U.; Kallenberg, C.G.M.; Vissink, A.; Spijkervet, F.K.L.; Bootsma, H.; Kroese, F.G.M.; Bos, N.A. Ig Gene Analysis Reveals Altered Selective Pressures on Ig-Producing Cells in Parotid Glands of Primary Sjögren’s Syndrome Patients. J. Immunol. 2015, 194, 514–521. [Google Scholar] [CrossRef]
- Youings, A.; Chang, S.C.; Dwek, R.A.; Scragg, I.G. Site-specific glycosylation of human immunoglobulin G is altered in four rheumatoid arthritis patients. Biochem. J. 1996, 314, 621–630. [Google Scholar] [CrossRef] [Green Version]
- Koers, J.; Derksen, N.I.L.; Ooijevaar-de Heer, P.; Nota, B.; van de Bovenkamp, F.S.; Vidarsson, G.; Rispens, T. Biased N-Glycosylation Site Distribution and Acquisition across the Antibody V Region during B Cell Maturation. J. Immunol. 2019, 202, 2220–2228. [Google Scholar] [CrossRef]
- Willemze, A.; Trouw, L.A.; Toes, R.E.M.; Huizinga, T.W.J. The influence of ACPA status and characteristics on the course of RA. Nat. Rev. Rheumatol. 2012, 8, 144–152. [Google Scholar] [CrossRef]
- Rombouts, Y.; Willemze, A.; van Beers, J.J.B.C.; Shi, J.; Kerkman, P.F.; van Toorn, L.; Janssen, G.M.C.; Zaldumbide, A.; Hoeben, R.C.; Pruijn, G.J.M.; et al. Extensive glycosylation of ACPA-IgG variable domains modulates binding to citrullinated antigens in rheumatoid arthritis. Ann. Rheum. Dis. 2016, 75, 578–585. [Google Scholar] [CrossRef]
- Visser, A.; Doorenspleet, M.E.; de Vries, N.; Spijkervet, F.K.L.; Vissink, A.; Bende, R.J.; Bootsma, H.; Kroese, F.G.M.; Bos, N.A. Acquisition of N-Glycosylation Sites in Immunoglobulin Heavy Chain Genes During Local Expansion in Parotid Salivary Glands of Primary Sjögren Patients. Front. Immunol. 2018, 9, 491. [Google Scholar] [CrossRef]
- van de Bovenkamp, F.S.; Hafkenscheid, L.; Rispens, T.; Rombouts, Y. The Emerging Importance of IgG Fab Glycosylation in Immunity. J. Immunol. 2016, 196. [Google Scholar] [CrossRef]
- Stavenhagen, J.B.; Gorlatov, S.; Tuaillon, N.; Rankin, C.T.; Li, H.; Burke, S.; Huang, L.; Vijh, S.; Johnson, S.; Bonvini, E.; et al. Fc optimization of therapeutic antibodies enhances their ability to kill tumor cells in vitro and controls tumor expansion in vivo via low-affinity activating Fcgamma receptors. Cancer Res. 2007, 67, 8882–8890. [Google Scholar] [CrossRef]
- Nordstrom, J.L.; Gorlatov, S.; Zhang, W.; Yang, Y.; Huang, L.; Burke, S.; Li, H.; Ciccarone, V.; Zhang, T.; Stavenhagen, J.; et al. Anti-tumor activity and toxicokinetics analysis of MGAH22, an anti-HER2 monoclonal antibody with enhanced Fcγ receptor binding properties. Breast Cancer Res. 2011, 13, R123. [Google Scholar] [CrossRef]
- Romain, G.; Senyukov, V.; Rey-Villamizar, N.; Merouane, A.; Kelton, W.; Liadi, I.; Mahendra, A.; Charab, W.; Georgiou, G.; Roysam, B.; et al. Antibody Fc engineering improves frequency and promotes kinetic boosting of serial killing mediated by NK cells. Blood 2014, 124, 3241–3249. [Google Scholar] [CrossRef] [Green Version]
- Bruhns, P.; Jönsson, F. Mouse and human FcR effector functions. Immunol. Rev. 2015, 268, 25–51. [Google Scholar] [CrossRef]
- Vafa, O.; Gilliland, G.L.; Brezski, R.J.; Strake, B.; Wilkinson, T.; Lacy, E.R.; Scallon, B.; Teplyakov, A.; Malia, T.J.; Strohl, W.R. An engineered Fc variant of an IgG eliminates all immune effector functions via structural perturbations. Methods 2014, 65, 114–126. [Google Scholar] [CrossRef]
- An, Z.; Forrest, G.; Moore, R.; Cukan, M.; Haytko, P.; Huang, L.; Vitelli, S.; Zhao, J.Z.; Lu, P.; Hua, J.; et al. IgG2m4, an engineered antibody isotype with reduced Fc function. MAbs 2009, 1, 572–579. [Google Scholar] [CrossRef] [Green Version]
- Stapleton, N.M.; Armstrong-Fisher, S.S.; Andersen, J.T.; van der Schoot, C.E.; Porter, C.; Page, K.R.; Falconer, D.; de Haas, M.; Williamson, L.M.; Clark, M.R.; et al. Human IgG lacking effector functions demonstrate lower FcRn-binding and reduced transplacental transport. Mol. Immunol. 2018, 95, 1–9. [Google Scholar] [CrossRef]
- Bloemendaal, F.M.; Levin, A.D.; Wildenberg, M.E.; Koelink, P.J.; McRae, B.L.; Salfeld, J.; Lum, J.; van der Neut Kolfschoten, M.; Claassens, J.W.; Visser, R.; et al. Anti–Tumor Necrosis Factor With a Glyco-Engineered Fc-Region Has Increased Efficacy in Mice With Colitis. Gastroenterology 2017, 153, 1351–1362.e4. [Google Scholar] [CrossRef]
- Zalevsky, J.; Chamberlain, A.K.; Horton, H.M.; Karki, S.; Leung, I.W.L.; Sproule, T.J.; Lazar, G.A.; Roopenian, D.C.; Desjarlais, J.R. Enhanced antibody half-life improves in vivo activity. Nat. Biotechnol. 2010, 28, 157–159. [Google Scholar] [CrossRef] [Green Version]
- Monnet, C.; Jorieux, S.; Souyris, N.; Zaki, O.; Jacquet, A.; Fournier, N.; Crozet, F.; de Romeuf, C.; Bouayadi, K.; Urbain, R.; et al. Combined glyco- and protein-Fc engineering simultaneously enhance cytotoxicity and half-life of a therapeutic antibody. MAbs 2014, 6, 422–436. [Google Scholar] [CrossRef] [Green Version]
- Dall’Acqua, W.F.; Kiener, P.A.; Wu, H. Properties of Human IgG1s Engineered for Enhanced Binding to the Neonatal Fc Receptor (FcRn). J. Biol. Chem. 2006, 281, 23514–23524. [Google Scholar] [CrossRef] [Green Version]
- Patel, D.A.; Puig-Canto, A.; Challa, D.K.; Montoyo, H.P.; Ober, R.J.; Ward, E.S. Neonatal Fc Receptor Blockade by Fc Engineering Ameliorates Arthritis in a Murine Model. J. Immunol. 2011, 187, 1015–1022. [Google Scholar] [CrossRef]
- Ling, L.E.; Hillson, J.L.; Tiessen, R.G.; Bosje, T.; Iersel, M.P.; Nix, D.J.; Markowitz, L.; Cilfone, N.A.; Duffner, J.; Streisand, J.B.; et al. M281, an Anti-FcRn Antibody: Pharmacodynamics, Pharmacokinetics, and Safety Across the Full Range of IgG Reduction in a First-in-Human Study. Clin. Pharmacol. Ther. 2019, 105, 1031–1039. [Google Scholar] [CrossRef]
- Kiessling, P.; Lledo-Garcia, R.; Watanabe, S.; Langdon, G.; Tran, D.; Bari, M.; Christodoulou, L.; Jones, E.; Price, G.; Smith, B.; et al. The FcRn inhibitor rozanolixizumab reduces human serum IgG concentration: A randomized phase 1 study. Sci. Transl. Med. 2017, 9, eaan1208. [Google Scholar] [CrossRef]
- Ward, E.S.; Ober, R.J. Targeting FcRn to Generate Antibody-Based Therapeutics. Trends Pharmacol. Sci. 2018, 39, 892–904. [Google Scholar] [CrossRef]
- Ulrichts, P.; Guglietta, A.; Dreier, T.; van Bragt, T.; Hanssens, V.; Hofman, E.; Vankerckhoven, B.; Verheesen, P.; Ongenae, N.; Lykhopiy, V.; et al. Neonatal Fc receptor antagonist efgartigimod safely and sustainably reduces IgGs in humans. J. Clin. Investig. 2018, 128, 4372–4386. [Google Scholar] [CrossRef]
- Cleary, K.L.S.; Chan, H.T.C.; James, S.; Glennie, M.J.; Cragg, M.S. Antibody distance from the cell membrane regulates antibody effector mechanisms. J. Immunol. 2017, 198, 3999–4011. [Google Scholar] [CrossRef] [Green Version]
- Rösner, T.; Derer, S.; Kellner, C.; Dechant, M.; Lohse, S.; Vidarsson, G.; Peipp, M.; Valerius, T. An IgG3 switch variant of rituximab mediates enhanced complement-dependent cytotoxicity against tumour cells with low CD20 expression levels. Br. J. Haematol. 2013, 161, 282–286. [Google Scholar] [CrossRef] [Green Version]
- Carter, P. Bispecific human IgG by design. J. Immunol. Methods 2001, 248, 7–15. [Google Scholar] [CrossRef]
- Merchant, A.M.; Zhu, Z.; Yuan, J.Q.; Goddard, A.; Adams, C.W.; Presta, L.G.; Carter, P. An efficient route to human bispecific IgG. Nat. Biotechnol. 1998, 16, 677–681. [Google Scholar] [CrossRef]
- Labrijn, A.F.; Meesters, J.I.; de Goeij, B.E.C.G.; van den Bremer, E.T.J.; Neijssen, J.; van Kampen, M.D.; Strumane, K.; Verploegen, S.; Kundu, A.; Gramer, M.J.; et al. Efficient generation of stable bispecific IgG1 by controlled Fab-arm exchange. Proc. Natl. Acad. Sci. USA 2013, 110, 5145–5150. [Google Scholar] [CrossRef] [Green Version]
- Brinkmann, U.; Kontermann, R.E. The making of bispecific antibodies. MAbs 2017, 9, 182–212. [Google Scholar] [CrossRef] [Green Version]
- Sedykh, S.E.; Prinz, V.V.; Buneva, V.N.; Nevinsky, G.A. Bispecific antibodies: design, therapy, perspectives. Drug Des. Devel. Ther. 2018, 12, 195. [Google Scholar] [CrossRef]
- Bakalar, M.H.; Joffe, A.M.; Schmid, E.M.; Son, S.; Podolski, M.; Fletcher, D.A. Size-dependent segregation controls macrophage phagocytosis of antibody-opsonized targets. Cell 2018, 174, 131–142. [Google Scholar] [CrossRef]
- Bohn, T.; Rapp, S.; Luther, N.; Klein, M.; Bruehl, T.-J.; Kojima, N.; Aranda Lopez, P.; Hahlbrock, J.; Muth, S.; Endo, S.; et al. Tumor immunoevasion via acidosis-dependent induction of regulatory tumor-associated macrophages. Nat. Immunol. 2018, 19, 1319–1329. [Google Scholar] [CrossRef]
- Li, H.Y.; McSharry, M.; Bullock, B.; Nguyen, T.T.; Kwak, J.; Poczobutt, J.M.; Sippel, T.R.; Heasley, L.E.; Weiser-Evans, M.C.; Clambey, E.T.; et al. The Tumor Microenvironment Regulates Sensitivity of Murine Lung Tumors to PD-1/PD-L1 Antibody Blockade. Cancer Immunol. Res. 2017, 5, 767–777. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Taeye, S.W.; Rispens, T.; Vidarsson, G. The Ligands for Human IgG and Their Effector Functions. Antibodies 2019, 8, 30. https://doi.org/10.3390/antib8020030
de Taeye SW, Rispens T, Vidarsson G. The Ligands for Human IgG and Their Effector Functions. Antibodies. 2019; 8(2):30. https://doi.org/10.3390/antib8020030
Chicago/Turabian Stylede Taeye, Steven W., Theo Rispens, and Gestur Vidarsson. 2019. "The Ligands for Human IgG and Their Effector Functions" Antibodies 8, no. 2: 30. https://doi.org/10.3390/antib8020030
APA Stylede Taeye, S. W., Rispens, T., & Vidarsson, G. (2019). The Ligands for Human IgG and Their Effector Functions. Antibodies, 8(2), 30. https://doi.org/10.3390/antib8020030