Design and Production of Bispecific Antibodies

 , , and
, , and
Abstract
:1. Introduction
2. Strategies to Improve Bispecific Antibody Production and Quality
2.1. Single-Chain Variable Fragment (scFv) Antibodies
2.1.1. Antibody Fragment Types
2.1.2. Linker Engineering
2.1.3. Stability Engineering of scFv Antibodies
2.1.4. Bispecific scFv Antibody Expression and Production
2.2. Full-Size IgG-like Asymmetric Bispecific Antibody
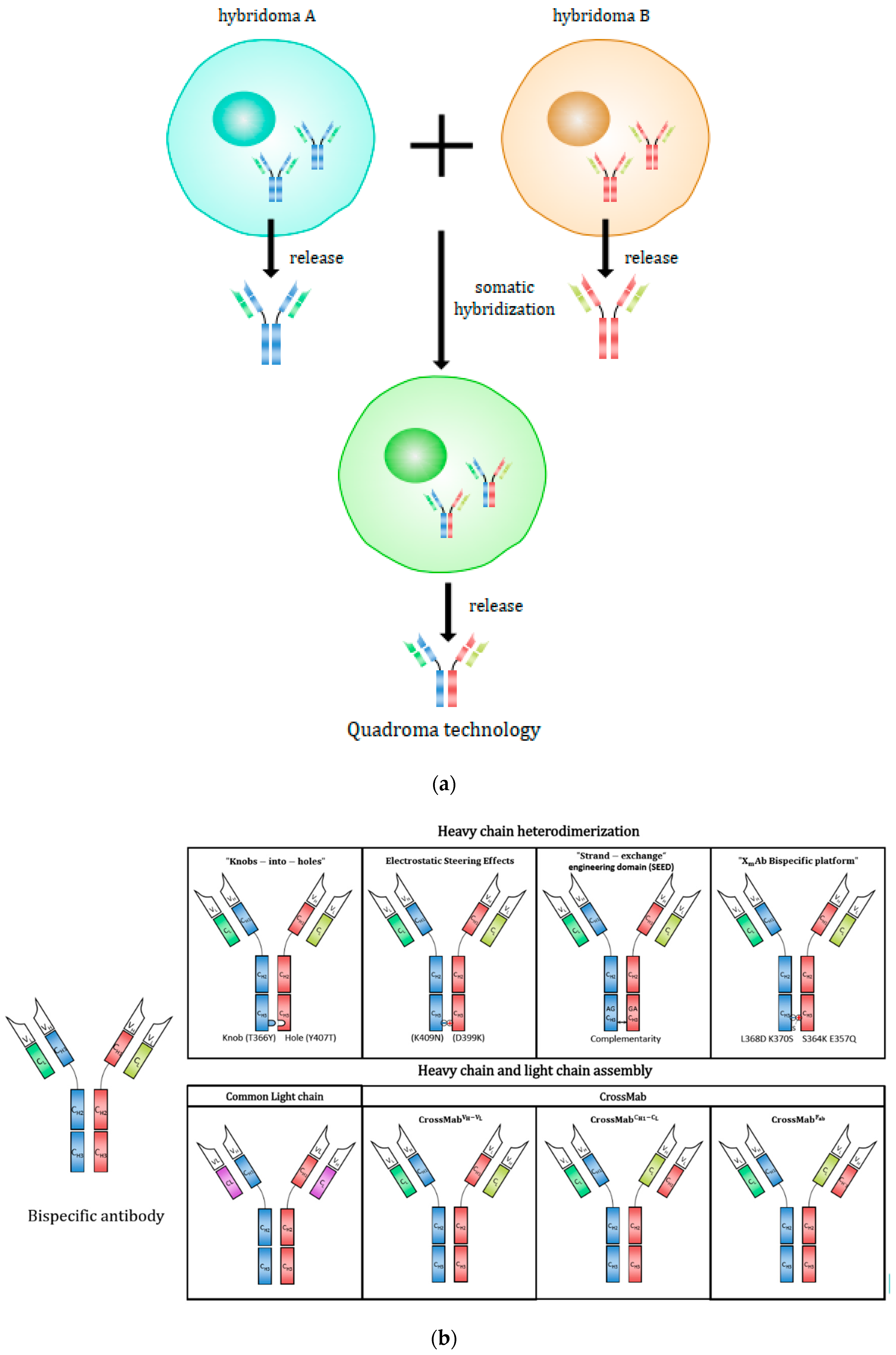
2.2.1. Quadroma (or Hybrid-Hybridoma) Technology
2.2.2. Heavy-Chain Assembly
2.2.3. Heavy Chain and Light-Chain Assembly
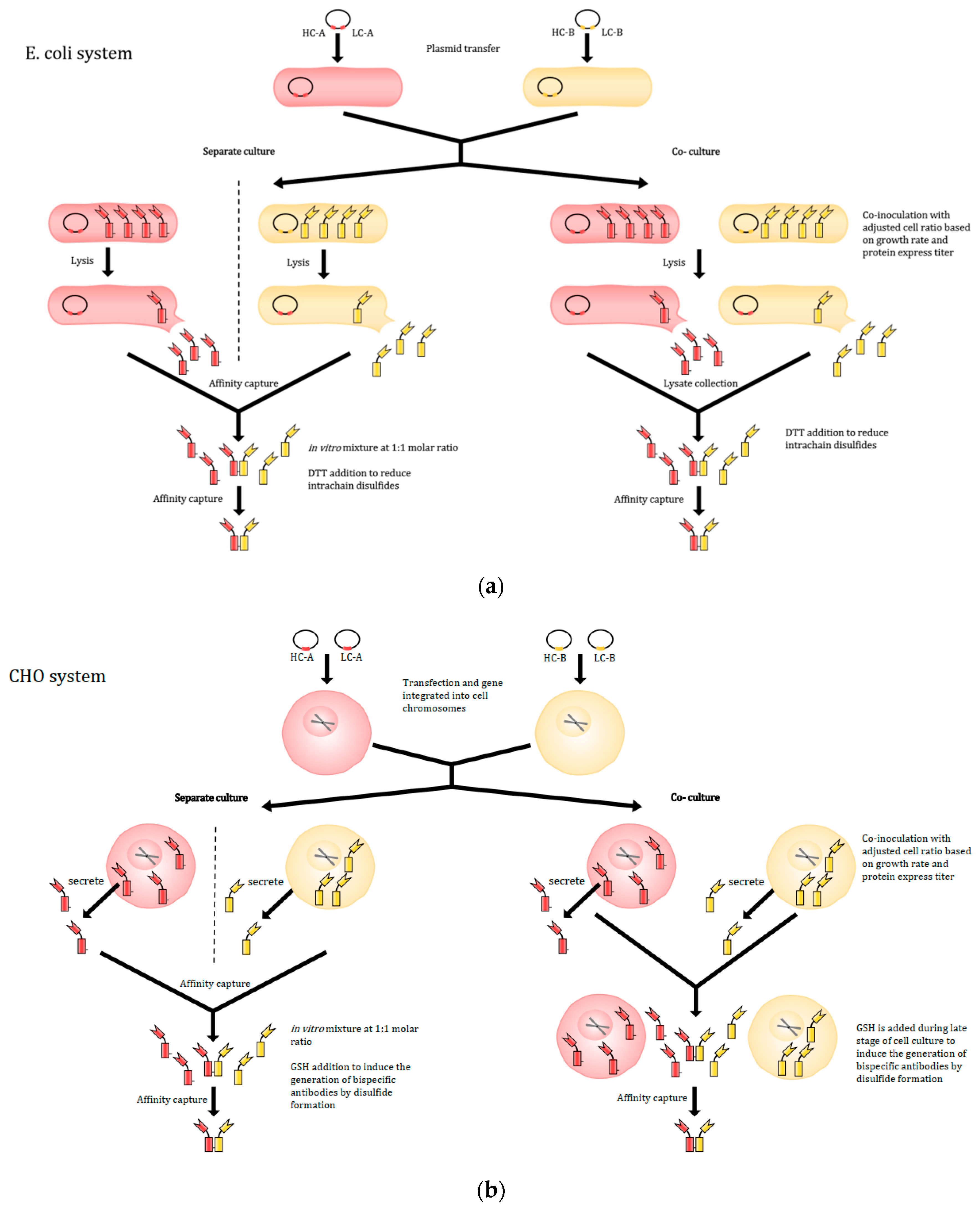
2.2.4. Co-Culture Method
2.2.5. Expression and Production of IgG-like Bispecific Antibodies
3. Conclusions and Future Thoughts
Author Contributions
Funding
Conflicts of Interest
References
- Kimiz-Gebologlu, I.; Gulce-Iz, S.; Biray-Avci, C. Monoclonal antibodies in cancer immunotherapy. Mol. Biol. Rep. 2018, 45, 2935–2940. [Google Scholar] [CrossRef] [PubMed]
- Howard, G.C.; Bethell, D.R. Basic Methods in Antibody Production and Characterization; CRC Press: Boca Raton, FL, USA, 2000. [Google Scholar]
- Wang, Q.; Chung, C.Y.; Chough, S.; Betenbaugh, M.J. Antibody glycoengineering strategies in mammalian cells. Biotechnol. Bioeng. 2018, 115, 1378–1393. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Saxena, A.; Sidhu, S.S.; Wu, D. Fc Engineering for Developing Therapeutic Bispecific Antibodies and Novel Scaffolds. Front. Immunol. 2017, 8, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedykh, S.E.; Prinz, V.V.; Buneva, V.N.; Nevinsky, G.A. Bispecific antibodies: Design, therapy, perspectives. Drug Des. Dev. Ther. 2018, 12, 195–208. [Google Scholar] [CrossRef]
- Sela-Culang, I.; Kunik, V.; Ofran, Y. The structural basis of antibody-antigen recognition. Front. Immunol. 2013, 4, 302. [Google Scholar] [CrossRef]
- Kiyoshi, M.; Tsumoto, K.; Ishii-Watabe, A.; Caaveiro, J.M. Glycosylation of IgG-Fc: A molecular perspective. Int. Immunol. 2017, 29, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Erbe, A.K.; Hank, J.A.; Morris, Z.S.; Sondel, P.M. NK Cell-Mediated Antibody-Dependent Cellular Cytotoxicity in Cancer Immunotherapy. Front. Immunol. 2015, 6, 368. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Hu, W.; Qin, X. The Role of Complement in the Mechanism of Action of Rituximab for B-Cell Lymphoma: Implications for Therapy. Oncologist 2008, 13, 954–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, A.; Wurch, T.; Bailly, C.; Corvaia, N. Strategies and challenges for the next generation of therapeutic antibodies. Nat. Rev. Immunol. 2010, 10, 345–352. [Google Scholar] [CrossRef]
- Jefferis, R. Glycosylation as a strategy to improve antibody-based therapeutics. Nat. Rev. Drug Discov. 2009, 8, 226–234. [Google Scholar] [CrossRef]
- Kamen, L.A.; Kho, E.; Ordonia, B.; Langsdorf, C.; Chung, S. A method for determining antibody-dependent cellular phagocytosis. J. Immunol. 2017, 198, 157.17. [Google Scholar] [CrossRef]
- Kontermann, R.E. Dual targeting strategies with bispecific antibodies. MAbs 2012, 4, 182–197. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Witsch, E.; Sela, M.; Yarden, Y. Roles for growth factors in cancer progression. Physiology (Bethesda) 2010, 25, 85–101. [Google Scholar] [CrossRef]
- Tai, W.; Mahato, R.; Cheng, K. The role of HER2 in cancer therapy and targeted drug delivery. J. Control. Release 2010, 146, 264–275. [Google Scholar] [CrossRef] [Green Version]
- Pabla, B.; Bissonnette, M.; Konda, V.J. Colon cancer and the epidermal growth factor receptor: Current treatment paradigms, the importance of diet, and the role of chemoprevention. World J. Clin. Oncol. 2015, 6, 133. [Google Scholar] [CrossRef]
- Ponz-Sarvise, M.; Rodriguez, J.; Viudez, A.; Chopitea, A.; Calvo, A.; Garcia-Foncillas, J.; Gil-Bazo, I. Epidermal growth factor receptor inhibitors in colorectal cancer treatment: What’s new? World J. Gastroenterol. 2007, 13, 5877. [Google Scholar] [CrossRef]
- Beckman, R.A.; Weiner, L.M.; Davis, H.M. Antibody constructs in cancer therapy. Cancer 2007, 109, 170–179. [Google Scholar] [CrossRef]
- Speirs, C.K.; Hwang, M.; Kim, S.; Li, W.; Chang, S.; Varki, V.; Mitchell, L.; Schleicher, S.; Lu, B. Harnessing the cell death pathway for targeted cancer treatment. Am. J. Cancer Res. 2011, 1, 43–61. [Google Scholar]
- Yang, F.; Wen, W.; Qin, W. Bispecific Antibodies as a Development Platform for New Concepts and Treatment Strategies. Int. J. Mol. Sci. 2016, 18. [Google Scholar] [CrossRef]
- Brinkmann, U.; Kontermann, R.E. The making of bispecific antibodies. MAbs 2017, 9, 182–212. [Google Scholar] [CrossRef]
- Fraser, L. Engineering Bispecific Antibodies is Challenging, Creating Unwanted Side Products. CrossMAb Technology, Designed as a Simple Platform for Complex Molecules, Solves those Problems. Available online: https://www.roche.com/research_and_development/what_we_are_working_on/research_technologies/bispecific-antibodies.htm (accessed on 17 May 2019).
- Rader, C. DARTs take aim at BiTEs. Blood 2011, 117, 4403–4404. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, Y.; Fan, D.; Xiong, D. The development of bispecific antibodies and their applications in tumor immune escape. Exp. Hematol. Oncol. 2017, 6, 12. [Google Scholar] [CrossRef]
- Ridgway, J.B.; Presta, L.G.; Carter, P. ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. 1996, 9, 617–621. [Google Scholar] [CrossRef]
- De Nardis, C.; Hendriks, L.J.A.; Poirier, E.; Arvinte, T.; Gros, P.; Bakker, A.B.H.; de Kruif, J. A new approach for generating bispecific antibodies based on a common light chain format and the stable architecture of human immunoglobulin G1. J. Biol. Chem. 2017. [Google Scholar] [CrossRef]
- Klein, C.; Schaefer, W.; Regula, J.T.; Dumontet, C.; Brinkmann, U.; Bacac, M.; Umaña, P. Engineering therapeutic bispecific antibodies using CrossMab technology. Methods 2019, 154, 21–31. [Google Scholar] [CrossRef]
- Carter, P. Bispecific human IgG by design. J. Immunol. Methods 2001, 248, 7–15. [Google Scholar] [CrossRef]
- Huehls, A.M.; Coupet, T.A.; Sentman, C.L. Bispecific T-cell engagers for cancer immunotherapy. Immunol. Cell Biol. 2015, 93, 290–296. [Google Scholar] [CrossRef]
- Huston, J.S.; Mudgett-Hunter, M.; Tai, M.S.; McCartney, J.; Warren, F.; Haber, E.; Oppermann, H. Protein engineering of single-chain Fv analogs and fusion proteins. Methods Enzymol. 1991, 203, 46–88. [Google Scholar]
- Ahmad, Z.A.; Yeap, S.K.; Ali, A.M.; Ho, W.Y.; Alitheen, N.B.; Hamid, M. scFv antibody: Principles and clinical application. Clin. Dev. Immunol. 2012, 2012, 980250. [Google Scholar] [CrossRef]
- Slaney, C.Y.; Wang, P.; Darcy, P.K.; Kershaw, M.H. CARs versus BiTEs: A Comparison between T Cell–Redirection Strategies for Cancer Treatment. Cancer Discov. 2018, 8, 924–934. [Google Scholar] [CrossRef]
- Strohl, W.R.; Naso, M. Bispecific T-Cell Redirection versus Chimeric Antigen Receptor (CAR)-T Cells as Approaches to Kill Cancer Cells. Antibodies 2019, 8, 41. [Google Scholar] [CrossRef]
- Mallender, W.D.; Voss, E.W., Jr. Construction, expression, and activity of a bivalent bispecific single-chain antibody. J. Biol. Chem. 1994, 269, 199–206. [Google Scholar]
- Wolf, E.; Hofmeister, R.; Kufer, P.; Schlereth, B.; Baeuerle, P.A. BiTEs: Bispecific antibody constructs with unique anti-tumor activity. Drug Discov. Today 2005, 10, 1237–1244. [Google Scholar] [CrossRef]
- Portell, C.A.; Wenzell, C.M.; Advani, A.S. Clinical and pharmacologic aspects of blinatumomab in the treatment of B-cell acute lymphoblastic leukemia. Clin. Pharmacol. Adv. Appl. 2013, 5, 5–11. [Google Scholar] [CrossRef] [Green Version]
- Klinger, M.; Brandl, C.; Zugmaier, G.; Hijazi, Y.; Bargou, R.C.; Topp, M.S.; Gokbuget, N.; Neumann, S.; Goebeler, M.; Viardot, A.; et al. Immunopharmacologic response of patients with B-lineage acute lymphoblastic leukemia to continuous infusion of T cell-engaging CD19/CD3-bispecific BiTE antibody blinatumomab. Blood 2012, 119, 6226–6233. [Google Scholar] [CrossRef]
- Ghaderi, D.; Zhang, M.; Hurtado-Ziola, N.; Varki, A. Production platforms for biotherapeutic glycoproteins. Occurrence, impact, and challenges of non-human sialylation. Biotechnol. Genet. Eng. Rev. 2012, 28, 147–175. [Google Scholar] [CrossRef]
- Moore, P.A.; Zhang, W.; Rainey, G.J.; Burke, S.; Li, H.; Huang, L.; Gorlatov, S.; Veri, M.C.; Aggarwal, S.; Yang, Y.; et al. Application of dual affinity retargeting molecules to achieve optimal redirected T-cell killing of B-cell lymphoma. Blood 2011, 117, 4542–4551. [Google Scholar] [CrossRef] [Green Version]
- Holliger, P.; Prospero, T.; Winter, G. “Diabodies”: Small bivalent and bispecific antibody fragments. Proc. Natl. Acad. Sci. USA 1993, 90, 6444–6448. [Google Scholar] [CrossRef]
- Walseng, E.; Nelson, C.G.; Qi, J.; Nanna, A.R.; Roush, W.R.; Goswami, R.K.; Sinha, S.C.; Burke, T.R., Jr.; Rader, C. Chemically Programmed Bispecific Antibodies in Diabody Format. J. Biol. Chem. 2016, 291, 19661–19673. [Google Scholar] [CrossRef] [Green Version]
- Arvedson, T.L.; Balazs, M.; Bogner, P.; Black, K.; Graham, K.; Henn, A.; Friedrich, M.; Hoffmann, P.; Kischel, R.; Kufer, P.; et al. Abstract 55: Generation of half-life extended anti-CD33 BiTE® antibody constructs compatible with once-weekly dosing. Cancer Res. 2017, 77, 55. [Google Scholar] [CrossRef]
- Goyos, A.; Li, C.M.; Deegen, P.; Bogner, P.; Thomas, O.; Matthias, K.; Wahl, J.; Goldstein, R.; Coxon, A.; Balazs, M. Generation of half-life extended anti-BCMA Bite® antibody construct compatible with once-weekly dosing for treatment of multiple myeloma (MM). Am. Soc. Hematol. 2017, 130, 5389. [Google Scholar]
- Lorenczewski, G.; Friedrich, M.; Kischel, R.; Dahlhoff, C.; Anlahr, J.; Balazs, M.; Rock, D.; Boyle, M.C.; Goldstein, R.; Coxon, A.; et al. Generation of a Half-Life Extended Anti-CD19 BiTE® Antibody Construct Compatible with Once-Weekly Dosing for Treatment of CD19-Positive Malignancies. Blood 2017, 130, 2815. [Google Scholar]
- Sloan, D.D.; Lam, C.Y.; Irrinki, A.; Liu, L.; Tsai, A.; Pace, C.S.; Kaur, J.; Murry, J.P.; Balakrishnan, M.; Moore, P.A.; et al. Targeting HIV Reservoir in Infected CD4 T Cells by Dual-Affinity Re-targeting Molecules (DARTs) that Bind HIV Envelope and Recruit Cytotoxic T Cells. PLoS Pathog. 2015, 11, e1005233. [Google Scholar] [CrossRef]
- McDonagh, C.F.; Huhalov, A.; Harms, B.D.; Adams, S.; Paragas, V.; Oyama, S.; Zhang, B.; Luus, L.; Overland, R.; Nguyen, S.; et al. Antitumor activity of a novel bispecific antibody that targets the ErbB2/ErbB3 oncogenic unit and inhibits heregulin-induced activation of ErbB3. Mol. Cancer Ther. 2012, 11, 582–593. [Google Scholar] [CrossRef]
- Reusch, U.; Harrington, K.H.; Gudgeon, C.J.; Fucek, I.; Ellwanger, K.; Weichel, M.; Knackmuss, S.H.; Zhukovsky, E.A.; Fox, J.A.; Kunkel, L.A.; et al. Characterization of CD33/CD3 Tetravalent Bispecific Tandem Diabodies (TandAbs) for the Treatment of Acute Myeloid Leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 5829–5838. [Google Scholar] [CrossRef] [Green Version]
- Reusch, U.; Burkhardt, C.; Fucek, I.; Le Gall, F.; Le Gall, M.; Hoffmann, K.; Knackmuss, S.H.; Kiprijanov, S.; Little, M.; Zhukovsky, E.A. A novel tetravalent bispecific TandAb (CD30/CD16A) efficiently recruits NK cells for the lysis of CD30+ tumor cells. MAbs 2014, 6, 728–739. [Google Scholar] [CrossRef]
- Compte, M.; Alvarez-Cienfuegos, A.; Nunez-Prado, N.; Sainz-Pastor, N.; Blanco-Toribio, A.; Pescador, N.; Sanz, L.; Alvarez-Vallina, L. Functional comparison of single-chain and two-chain anti-CD3-based bispecific antibodies in gene immunotherapy applications. Oncoimmunology 2014, 3, e28810. [Google Scholar] [CrossRef] [Green Version]
- Kontermann, R.E.; Brinkmann, U. Bispecific antibodies. Drug Discov. Today 2015, 20, 838–847. [Google Scholar] [CrossRef] [Green Version]
- Pantoliano, M.W.; Bird, R.E.; Johnson, S.; Asel, E.D.; Dodd, S.W.; Wood, J.F.; Hardman, K.D. Conformational stability, folding, and ligand-binding affinity of single-chain Fv immunoglobulin fragments expressed in Escherichia coli. Biochemistry 1991, 30, 10117–10125. [Google Scholar] [CrossRef]
- Dorai, H.; McCartney, J.E.; Hudziak, R.M.; Tai, M.-S.; Laminet, A.A.; Houston, L.L.; Huston, J.S.; Oppermann, H. Mammalian Cell Expression of Single–Chain Fv (sFv) Antibody Proteins and Their C–terminal Fusions with Interleukin–2 and Other Effector Domains. Biol. Technol. 1994, 12, 890–897. [Google Scholar] [CrossRef]
- Desplancq, D.; King, D.J.; Lawson, A.D.; Mountain, A. Multimerization behaviour of single chain Fv variants for the tumour-binding antibody B72.3. Protein Eng. 1994, 7, 1027–1033. [Google Scholar] [CrossRef]
- Long, N.E.; Sullivan, B.J.; Ding, H.; Doll, S.; Ryan, M.A.; Hitchcock, C.L.; Martin, E.W.; Kumar, K.; Tweedle, M.F.; Magliery, T.J. Linker engineering in anti-TAG-72 antibody fragments optimizes biophysical properties, serum half-life, and high-specificity tumor imaging. J. Biol. Chem. 2018, 293, 9030–9040. [Google Scholar] [CrossRef] [Green Version]
- Argos, P. An investigation of oligopeptides linking domains in protein tertiary structures and possible candidates for general gene fusion. J. Mol. Biol. 1990, 211, 943–958. [Google Scholar] [CrossRef]
- Ekerljung, L.; Wallberg, H.; Sohrabian, A.; Andersson, K.; Friedman, M.; Frejd, F.Y.; Stahl, S.; Gedda, L. Generation and evaluation of bispecific affibody molecules for simultaneous targeting of EGFR and HER2. Bioconjugate Chem. 2012, 23, 1802–1811. [Google Scholar] [CrossRef]
- Whitlow, M.; Bell, B.A.; Feng, S.L.; Filpula, D.; Hardman, K.D.; Hubert, S.L.; Rollence, M.L.; Wood, J.F.; Schott, M.E.; Milenic, D.E.; et al. An improved linker for single-chain Fv with reduced aggregation and enhanced proteolytic stability. Protein Eng. 1993, 6, 989–995. [Google Scholar] [CrossRef]
- Tang, Y.; Jiang, N.; Parakh, C.; Hilvert, D. Selection of Linkers for a Catalytic Single-chain Antibody Using Phage Display Technology. J. Biol. Chem. 1996, 271, 15682–15686. [Google Scholar] [CrossRef] [Green Version]
- Atwell, J.L.; Breheney, K.A.; Lawrence, L.J.; McCoy, A.J.; Kortt, A.A.; Hudson, P.J. scFv multimers of the anti-neuraminidase antibody NC10: Length of the linker between VH and VL domains dictates precisely the transition between diabodies and triabodies. Protein Eng. 1999, 12, 597–604. [Google Scholar] [CrossRef]
- Dolezal, O.; Pearce, L.A.; Lawrence, L.J.; McCoy, A.J.; Hudson, P.J.; Kortt, A.A. ScFv multimers of the anti-neuraminidase antibody NC10: Shortening of the linker in single-chain Fv fragment assembled in VL to VH orientation drives the formation of dimers, trimers, tetramers and higher molecular mass multimers. Protein Eng. Des. Sel. 2000, 13, 565–574. [Google Scholar] [CrossRef]
- Alfthan, K.; Takkinen, K.; Sizmann, D.; Söderlund, H.; Teeri, T.T. Properties of a single-chain antibody containing different linker peptides. Protein Eng. Des. Sel. 1995, 8, 725–731. [Google Scholar] [CrossRef]
- Gil, D.; Schrum, A.G. Strategies to stabilize compact folding and minimize aggregation of antibody-based fragments. Adv. Biosci. Biotechnol. (Print) 2013, 4, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Pavlinkova, G.; Beresford, G.W.; Booth, B.J.M.; Batra, S.K.; Colcher, D. Pharmacokinetics and Biodistribution of Engineered Single-Chain Antibody Constructs of MAb CC49 in Colon Carcinoma Xenografts. J. Nucl. Med. 1999, 40, 1536–1546. [Google Scholar]
- Goel, A.; Colcher, D.; Baranowska-Kortylewicz, J.; Augustine, S.; Booth, B.J.M.; Pavlinkova, G.; Batra, S.K. Genetically Engineered Tetravalent Single-Chain Fv of the Pancarcinoma Monoclonal Antibody CC49: Improved Biodistribution and Potential for Therapeutic Application. Cancer Res. 2000, 60, 6964–6971. [Google Scholar]
- Goel, A.; Baranowska-Kortylewicz, J.; Hinrichs, S.H.; Wisecarver, J.; Pavlinkova, G.; Augustine, S.; Colcher, D.; Booth, B.J.M.; Batra, S.K. 99mTc-Labeled Divalent and Tetravalent CC49 Single-Chain Fv’s: Novel Imaging Agents for Rapid In Vivo Localization of Human Colon Carcinoma. J. Nucl. Med. 2001, 42, 1519–1527. [Google Scholar]
- Gruber, M.; Schodin, B.A.; Wilson, E.R.; Kranz, D.M. Efficient tumor cell lysis mediated by a bispecific single chain antibody expressed in Escherichia coli. J. Immunol. 1994, 152, 5368–5374. [Google Scholar]
- Hao, C.H.; Han, Q.H.; Shan, Z.J.; Hu, J.T.; Zhang, N.; Zhang, X.P. Effects of different interchain linkers on biological activity of an anti-prostate cancer single-chain bispecific antibody. Theor. Biol. Med Model. 2015, 12, 14. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, T.; Tsumoto, K.; Yokota, A.; Kondo, H.; Kumagai, I. Critical contribution of VH-VL interaction to reshaping of an antibody: The case of humanization of anti-lysozyme antibody, HyHEL-10. Protein Sci. Publ. Protein Soc. 2008, 17, 261–270. [Google Scholar] [CrossRef] [Green Version]
- Nagorsen, D.; Kufer, P.; Baeuerle, P.A.; Bargou, R. Blinatumomab: A historical perspective. Pharmacol. Ther. 2012, 136, 334–342. [Google Scholar] [CrossRef]
- Reusch, U.; Duell, J.; Ellwanger, K.; Herbrecht, C.; Knackmuss, S.H.; Fucek, I.; Eser, M.; McAleese, F.; Molkenthin, V.; Gall, F.L.; et al. A tetravalent bispecific TandAb (CD19/CD3), AFM11, efficiently recruits T cells for the potent lysis of CD19(+) tumor cells. MAbs 2015, 7, 584–604. [Google Scholar] [CrossRef]
- Miller, B.R.; Demarest, S.J.; Lugovskoy, A.; Huang, F.; Wu, X.; Snyder, W.B.; Croner, L.J.; Wang, N.; Amatucci, A.; Michaelson, J.S.; et al. Stability engineering of scFvs for the development of bispecific and multivalent antibodies. Protein Eng. Des. Sel. 2010, 23, 549–557. [Google Scholar] [CrossRef]
- Wörn, A.; Auf der Maur, A.; Escher, D.; Honegger, A.; Barberis, A.; Plückthun, A. Correlation between in Vitro Stability and in Vivo Performance of Anti-GCN4 Intrabodies as Cytoplasmic Inhibitors. J. Biol. Chem. 2000, 275, 2795–2803. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.; Honegger, A.; Plückthun, A. Selection for improved protein stability by phage display. J. Mol. Biol. 1999, 294, 163–180. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.; Plückthun, A. Improving in vivo folding and stability of a single-chain Fv antibody fragment by loop grafting. Protein Eng. Des. Sel. 1997, 10, 959–966. [Google Scholar] [CrossRef] [Green Version]
- Willuda, J.; Honegger, A.; Waibel, R.; Schubiger, P.A.; Stahel, R.; Zangemeister-Wittke, U.; Plückthun, A. High Thermal Stability Is Essential for Tumor Targeting of Antibody Fragments. Eng. Humaniz. Anti Epithel. Glycoprotein 2 (Epithel. Cell Adhes. Mol.) Single Chain Fv Fragm. 1999, 59, 5758–5767. [Google Scholar]
- Ewert, S.; Honegger, A.; Plückthun, A. Stability improvement of antibodies for extracellular and intracellular applications: CDR grafting to stable frameworks and structure-based framework engineering. Methods 2004, 34, 184–199. [Google Scholar] [CrossRef]
- Jones, P.T.; Dear, P.H.; Foote, J.; Neuberger, M.S.; Winter, G. Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature 1986, 321, 522–525. [Google Scholar] [CrossRef]
- Borras, L.; Gunde, T.; Tietz, J.; Bauer, U.; Hulmann-Cottier, V.; Grimshaw, J.P.A.; Urech, D.M. Generic approach for the generation of stable humanized single-chain Fv fragments from rabbit monoclonal antibodies. J. Biol. Chem. 2010, 285, 9054–9066. [Google Scholar] [CrossRef]
- Wörn, A.; Plückthun, A. Mutual Stabilization of VL and VH in Single-Chain Antibody Fragments, Investigated with Mutants Engineered for Stability. Biochemistry 1998, 37, 13120–13127. [Google Scholar] [CrossRef]
- Steipe, B. Consensus-based engineering of protein stability: From intrabodies to thermostable enzymes. Methods Enzymol. 2004, 388, 176–186. [Google Scholar] [CrossRef]
- Steipe, B.; Schiller, B.; Plückthun, A.; Steinbacher, S. Sequence Statistics Reliably Predict Stabilizing Mutations in a Protein Domain. J. Mol. Biol. 1994, 240, 188–192. [Google Scholar] [CrossRef]
- Ewert, S.; Honegger, A.; Plückthun, A. Structure-Based Improvement of the Biophysical Properties of Immunoglobulin VH Domains with a Generalizable Approach. Biochemistry 2003, 42, 1517–1528. [Google Scholar] [CrossRef]
- Monsellier, E.; Bedouelle, H. Improving the Stability of an Antibody Variable Fragment by a Combination of Knowledge-based Approaches: Validation and Mechanisms. J. Mol. Biol. 2006, 362, 580–593. [Google Scholar] [CrossRef]
- Jermutus, L.; Honegger, A.; Schwesinger, F.; Hanes, J.; Plückthun, A. Tailoring in vitro evolution for protein affinity or stability. Proc. Natl. Acad. Sci. USA 2001, 98, 75–80. [Google Scholar] [CrossRef]
- Proba, K.; Wörn, A.; Honegger, A.; Plückthun, A. Antibody scFv fragments without disulfide bonds, made by molecular evolution11Edited by I. A. Wilson. J. Mol. Biol. 1998, 275, 245–253. [Google Scholar] [CrossRef]
- Demarest, S.J.; Chen, G.; Kimmel, B.E.; Gustafson, D.; Wu, J.; Salbato, J.; Poland, J.; Elia, M.; Tan, X.; Wong, K.; et al. Engineering stability into Escherichia coli secreted Fabs leads to increased functional expression. Protein Eng. Des. Sel. 2006, 19, 325–336. [Google Scholar] [CrossRef] [Green Version]
- Jespers, L.; Schon, O.; Famm, K.; Winter, G. Aggregation-resistant domain antibodies selected on phage by heat denaturation. Nat. Biotechnol. 2004, 22, 1161. [Google Scholar] [CrossRef]
- Martineau, P.; Jones, P.; Winter, G. Expression of an antibody fragment at high levels in the bacterial cytoplasm11Edited by J. Karn. J. Mol. Biol. 1998, 280, 117–127. [Google Scholar] [CrossRef]
- Graff, C.P.; Chester, K.; Begent, R.; Wittrup, K.D. Directed evolution of an anti-carcinoembryonic antigen scFv with a 4-day monovalent dissociation half-time at 37 °C. Protein Eng. Des. Sel. 2004, 17, 293–304. [Google Scholar] [CrossRef]
- Brockmann, E.-C.; Cooper, M.; Strömsten, N.; Vehniäinen, M.; Saviranta, P. Selecting for antibody scFv fragments with improved stability using phage display with denaturation under reducing conditions. J. Immunol. Methods 2005, 296, 159–170. [Google Scholar] [CrossRef]
- Glockshuber, R.; Malia, M.; Pfitzinger, I.; Plueckthun, A. A comparison of strategies to stabilize immunoglobulin Fv-fragments. Biochemistry 1990, 29, 1362–1367. [Google Scholar] [CrossRef]
- Reiter, Y.; Brinkmann, U.; Lee, B.; Pastan, I. Engineering antibody Fv fragments for cancer detection and therapy: Disulfide-stabilized Fv fragments. Nat. Biotechnol. 1996, 14, 1239–1245. [Google Scholar] [CrossRef]
- Zhao, J.X.; Yang, L.; Gu, Z.N.; Chen, H.Q.; Tian, F.W.; Chen, Y.Q.; Zhang, H.; Chen, W. Stabilization of the single-chain fragment variable by an interdomain disulfide bond and its effect on antibody affinity. Int. J. Mol. Sci. 2010, 12, 1–11. [Google Scholar] [CrossRef]
- Pokkuluri, P.R.; Gu, M.; Cai, X.; Raffen, R.; Stevens, F.J.; Schiffer, M. Factors contributing to decreased protein stability when aspartic acid residues are in β-sheet regions. Protein Sci. 2002, 11, 1687–1694. [Google Scholar] [CrossRef]
- Kaufmann, M.; Lindner, P.; Honegger, A.; Blank, K.; Tschopp, M.; Capitani, G.; Plückthun, A.; Grütter, M.G. Crystal Structure of the Anti-His Tag Antibody 3D5 Single-chain Fragment Complexed to its Antigen. J. Mol. Biol. 2002, 318, 135–147. [Google Scholar] [CrossRef]
- Vaks, L.; Benhar, I. Production of stabilized scFv antibody fragments in the E. coli bacterial cytoplasm. Methods Mol. Biol. 2014, 1060, 171–184. [Google Scholar] [CrossRef]
- Miller, K.D.; Weaver-Feldhaus, J.; Gray, S.A.; Siegel, R.W.; Feldhaus, M.J. Production, purification, and characterization of human scFv antibodies expressed in Saccharomyces cerevisiae, Pichia pastoris, and Escherichia coli. Protein Expr. Purif. 2005, 42, 255–267. [Google Scholar] [CrossRef]
- Verma, R.; Boleti, E.; George, A.J. Antibody engineering: Comparison of bacterial, yeast, insect and mammalian expression systems. J. Immunol. Methods 1998, 216, 165–181. [Google Scholar] [CrossRef]
- Galeffi, P.; Lombardi, A.; Pietraforte, I.; Novelli, F.; Di Donato, M.; Sperandei, M.; Tornambé, A.; Fraioli, R.; Martayan, A.; Natali, P.G.; et al. Functional expression of a single-chain antibody to ErbB-2 in plants and cell-free systems. J. Transl. Med. 2006, 4, 39. [Google Scholar] [CrossRef]
- McCall, A.M.; Adams, G.P.; Amoroso, A.R.; Nielsen, U.B.; Zhang, L.; Horak, E.; Simmons, H.; Schier, R.; Marks, J.D.; Weiner, L.M. Isolation and characterization of an anti-CD16 single-chain Fv fragment and construction of an anti-HER2/neu/anti-CD16 bispecific scFv that triggers CD16-dependent tumor cytolysis. Mol. Immunol. 1999, 36, 433–445. [Google Scholar] [CrossRef]
- Offner, S.; Hofmeister, R.; Romaniuk, A.; Kufer, P.; Baeuerle, P.A. Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol. Immunol. 2006, 43, 763–771. [Google Scholar] [CrossRef]
- Mack, M.; Riethmüller, G.; Kufer, P. A small bispecific antibody construct expressed as a functional single-chain molecule with high tumor cell cytotoxicity. Proc. Natl. Acad. Sci. USA 1995, 92, 7021–7025. [Google Scholar] [CrossRef]
- Mack, M.; Gruber, R.; Schmidt, S.; Riethmüller, G.; Kufer, P. Biologic properties of a bispecific single-chain antibody directed against 17-1A (EpCAM) and CD3: Tumor cell-dependent T cell stimulation and cytotoxic activity. J. Immunol. 1997, 158, 3965–3970. [Google Scholar]
- Lin, L.; Li, L.; Zhou, C.; Li, J.; Liu, J.; Shu, R.; Dong, B.; Li, Q.; Wang, Z. A HER2 bispecific antibody can be efficiently expressed in Escherichia coli with potent cytotoxicity. Oncol. Lett. 2018, 16, 1259–1266. [Google Scholar] [CrossRef]
- Kuo, S.R.; Wong, L.; Liu, J.S. Engineering a CD123xCD3 bispecific scFv immunofusion for the treatment of leukemia and elimination of leukemia stem cells. Protein Eng. Des. Sel. PEDS 2012, 25, 561–569. [Google Scholar] [CrossRef]
- Root, R.A.; Cao, W.; Li, B.; LaPan, P.; Meade, C.; Sanford, J.; Jin, M.; O’Sullivan, C.; Cummins, E.; Lambert, M.; et al. Development of PF-06671008, a Highly Potent Anti-P-cadherin/Anti-CD3 Bispecific DART Molecule with Extended Half-Life for the Treatment of Cancer. Antibodies 2016, 5, 6. [Google Scholar] [CrossRef]
- Brischwein, K.; Schlereth, B.; Guller, B.; Steiger, C.; Wolf, A.; Lutterbuese, R.; Offner, S.; Locher, M.; Urbig, T.; Raum, T.; et al. MT110: A novel bispecific single-chain antibody construct with high efficacy in eradicating established tumors. Mol. Immunol. 2006, 43, 1129–1143. [Google Scholar] [CrossRef]
- Stadler, C.R.; Bahr-Mahmud, H.; Plum, L.M.; Schmoldt, K.; Kolsch, A.C.; Tureci, O.; Sahin, U. Characterization of the first-in-class T-cell-engaging bispecific single-chain antibody for targeted immunotherapy of solid tumors expressing the oncofetal protein claudin 6. Oncoimmunology 2016, 5, e1091555. [Google Scholar] [CrossRef]
- Cheal, S.M.; Xu, H.; Guo, H.F.; Zanzonico, P.B.; Larson, S.M.; Cheung, N.K. Preclinical evaluation of multistep targeting of diasialoganglioside GD2 using an IgG-scFv bispecific antibody with high affinity for GD2 and DOTA metal complex. Mol. Cancer Ther. 2014, 13, 1803–1812. [Google Scholar] [CrossRef]
- Rosano, G.L.; Ceccarelli, E.A. Recombinant protein expression in Escherichia coli: Advances and challenges. Front. Microbiol. 2014, 5. [Google Scholar] [CrossRef]
- Arbabi-Ghahroudi, M.; Tanha, J.; MacKenzie, R. Prokaryotic expression of antibodies. Cancer Metastasis Rev. 2005, 24, 501–519. [Google Scholar] [CrossRef]
- Power, B.E.; Hudson, P.J. Synthesis of high avidity antibody fragments (scFv multimers) for cancer imaging. J. Immunol. Methods 2000, 242, 193–204. [Google Scholar] [CrossRef]
- Vendel, M.C.; Favis, M.; Snyder, W.B.; Huang, F.; Capili, A.D.; Dong, J.; Glaser, S.M.; Miller, B.R.; Demarest, S.J. Secretion from bacterial versus mammalian cells yields a recombinant scFv with variable folding properties. Arch. Biochem. Biophys. 2012, 526, 188–193. [Google Scholar] [CrossRef]
- Joosten, V.; Lokman, C.; van den Hondel, C.A.; Punt, P.J. The production of antibody fragments and antibody fusion proteins by yeasts and filamentous fungi. Microb. Cell Fact. 2003, 2, 1. [Google Scholar] [CrossRef]
- Geng, S.; Chang, H.; Qin, W.; Li, Y.; Feng, J.; Shen, B. Overexpression, effective renaturation, and bioactivity of novel single-chain antibodies against TNF-alpha. Prep. Biochem. Biotechnol. 2008, 38, 74–86. [Google Scholar] [CrossRef]
- Liu, M.; Wang, X.; Yin, C.; Zhang, Z.; Lin, Q.; Zhen, Y.; Huang, H. A novel bivalent single-chain variable fragment (scFV) inhibits the action of tumour necrosis factor alpha. Biotechnol. Appl. Biochem. 2008, 50, 173–179. [Google Scholar] [CrossRef]
- Carrio, M.M.; Cubarsi, R.; Villaverde, A. Fine architecture of bacterial inclusion bodies. FEBS Lett. 2000, 471, 7–11. [Google Scholar] [CrossRef] [Green Version]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. The Immunoglobulin Fold Consists of a Beta-Sandwich Framework with Hypervariable Loops. 2002. Available online: https://www.ncbi.nlm.nih.gov/books/NBK22461/ (accessed on 14 March 2019).
- Guglielmi, L.; Martineau, P. Expression of single-chain Fv fragments in E. coli cytoplasm. Methods Mol. Biol. 2009, 562, 215–224. [Google Scholar] [CrossRef]
- Singh, A.; Upadhyay, V.; Upadhyay, A.K.; Singh, S.M.; Panda, A.K. Protein recovery from inclusion bodies of Escherichia coli using mild solubilization process. Microb. Cell Fact. 2015, 14, 41. [Google Scholar] [CrossRef]
- Kipriyanov, S.M. High-level periplasmic expression and purification of scFvs. Methods Mol. Biol. 2009, 562, 205–214. [Google Scholar] [CrossRef]
- Chi, W.-J.; Kim, H.; Yoo, H.; Kim, Y.P.; Hong, S.-K. Periplasmic expression, purification, and characterization of an anti-epidermal growth factor receptor antibody fragment in Escherichia coli. Biotechnol. Bioprocess Eng. 2016, 21, 321–330. [Google Scholar] [CrossRef]
- Dewi, K.S.; Retnoningrum, D.S.; Riani, C.; Fuad, A.M. Construction and Periplasmic Expression of the Anti-EGFRvIII ScFv Antibody Gene in Escherichia coli. Sci. Pharm. 2016, 84, 141–152. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Zhong, Y.; Wang, J.; Zhang, Q.; Li, X.; Ling, S.; Wang, S.; Wang, R. Screening of a ScFv Antibody With High Affinity for Application in Human IFN-gamma Immunoassay. Front. Microbiol. 2018, 9, 261. [Google Scholar] [CrossRef] [PubMed]
- Nogi, T.; Sangawa, T.; Tabata, S.; Nagae, M.; Tamura-Kawakami, K.; Beppu, A.; Hattori, M.; Yasui, N.; Takagi, J. Novel affinity tag system using structurally defined antibody-tag interaction: Application to single-step protein purification. Protein Sci. 2008, 17, 2120–2126. [Google Scholar] [CrossRef]
- Wang, R.; Xiang, S.; Feng, Y.; Srinivas, S.; Srinivas, S.; Lin, M.; Wang, S. Engineering production of functional scFv antibody in E. coli by co-expressing the molecule chaperone Skp. Front. Cell. Infect. Microbiol. 2013, 3, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Marco, A.; De Marco, V. Bacteria co-transformed with recombinant proteins and chaperones cloned in independent plasmids are suitable for expression tuning. J. Biotechnol. 2004, 109, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, H.; Kumada, Y.; Katsuda, T.; Yamaji, H. Functional expression of single-chain Fv antibody in the cytoplasm of Escherichia coli by thioredoxin fusion and co-expression of molecular chaperones. Protein Expr. Purif. 2010, 70, 248–253. [Google Scholar] [CrossRef]
- Sonoda, H.; Kumada, Y.; Katsuda, T.; Yamaji, H. Effects of cytoplasmic and periplasmic chaperones on secretory production of single-chain Fv antibody in Escherichia coli. J. Biosci. Bioeng. 2011, 111, 465–470. [Google Scholar] [CrossRef]
- Jäger, V.; Büssow, K.; Wagner, A.; Weber, S.; Hust, M.; Frenzel, A.; Schirrmann, T. High level transient production of recombinant antibodies and antibody fusion proteins in HEK293 cells. BMC Biotechnol. 2013, 13, 52. [Google Scholar] [CrossRef]
- Jain, S.; Aresu, L.; Comazzi, S.; Shi, J.; Worrall, E.; Clayton, J.; Humphries, W.; Hemmington, S.; Davis, P.; Murray, E.; et al. The Development of a Recombinant scFv Monoclonal Antibody Targeting Canine CD20 for Use in Comparative Medicine. PloS ONE 2016, 11, e0148366. [Google Scholar] [CrossRef]
- Chambers, A.C.; Aksular, M.; Graves, L.P.; Irons, S.L.; Possee, R.D.; King, L.A. Overview of the Baculovirus Expression System. Curr. Protoc. Protein Sci. 2018, 91, 5.4.1–5.4.6. [Google Scholar] [CrossRef]
- Ma, J.K.; Drake, P.M.; Christou, P. The production of recombinant pharmaceutical proteins in plants. Nat. Rev. Genet. 2003, 4, 794–805. [Google Scholar] [CrossRef]
- Rech, E.; Vianna, G.; Murad, A.; Cunha, N.; Lacorte, C.; Araujo, A.; Brigido, M.; Michael, W.; Fontes, A.; Barry, O.; et al. Recombinant proteins in plants. BMC Proc. 2014, 8, 1. [Google Scholar] [CrossRef]
- Makvandi-Nejad, S.; McLean, M.D.; Hirama, T.; Almquist, K.C.; Mackenzie, C.R.; Hall, J.C. Transgenic tobacco plants expressing a dimeric single-chain variable fragment (scfv) antibody against Salmonella enterica serotype Paratyphi B. Transgenic Res. 2005, 14, 785–792. [Google Scholar] [CrossRef]
- Stech, M.; Hust, M.; Schulze, C.; Dübel, S.; Kubick, S. Cell-free eukaryotic systems for the production, engineering, and modification of scFv antibody fragments. Eng. Life Sci. 2014, 14, 387–398. [Google Scholar] [CrossRef]
- Carlson, E.D.; Gan, R.; Hodgman, C.E.; Jewett, M.C. Cell-free protein synthesis: Applications come of age. Biotechnol. Adv. 2012, 30, 1185–1194. [Google Scholar] [CrossRef]
- Spasevska, I. An Outlook on Bispecific Antibodies: Methods of Production and Therapeutic Benefits; BioSciences Master Reviews: Lyon, France, 2013. [Google Scholar]
- Krah, S.; Kolmar, H.; Becker, S.; Zielonka, S. Engineering IgG-Like Bispecific Antibodies—An Overview. Antibodies 2018, 7, 28. [Google Scholar] [CrossRef]
- Efficient Protein A Chromatography for Bispecific Antibodies. Available online: https://www.gelifesciences.com/en/us/solutions/bioprocessing/knowledge-center/purifying-bispecific-antibodies-in-a-single-step (accessed on 8 July 2019).
- Schaefer, W.; Regula, J.T.; Bähner, M.; Schanzer, J.; Croasdale, R.; Dürr, H.; Gassner, C.; Georges, G.; Kettenberger, H.; Imhof-Jung, S.; et al. Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc. Natl. Acad. Sci. USA 2011, 108, 11187. [Google Scholar] [CrossRef]
- Xu, Y.; Lee, J.; Tran, C.; Heibeck, T.H.; Wang, W.D.; Yang, J.; Stafford, R.L.; Steiner, A.R.; Sato, A.K.; Hallam, T.J.; et al. Production of bispecific antibodies in “knobs-into-holes” using a cell-free expression system. MAbs 2015, 7, 231–242. [Google Scholar] [CrossRef]
- Gunasekaran, K.; Pentony, M.; Shen, M.; Garrett, L.; Forte, C.; Woodward, A.; Ng, S.B.; Born, T.; Retter, M.; Manchulenko, K.; et al. Enhancing antibody Fc heterodimer formation through electrostatic steering effects: applications to bispecific molecules and monovalent IgG. J. Biol. Chem. 2010, 285, 19637–19646. [Google Scholar] [CrossRef]
- Huang, Y.; Yu, J.; Lanzi, A.; Yao, X.; Andrews, C.D.; Tsai, L.; Gajjar, M.R.; Sun, M.; Seaman, M.S.; Padte, N.N.; et al. Engineered Bispecific Antibodies with Exquisite HIV-1-Neutralizing Activity. Cell 2016, 165, 1621–1631. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Xie, F.; Tong, X.; Li, H.; Chen, Y.; Qian, W.; Duan, S.; Zheng, J.; Zhao, Z.; Li, B.; et al. Combating non-Hodgkin lymphoma by targeting both CD20 and HLA-DR through CD20-243 CrossMab. MABs 2014, 6, 740–748. [Google Scholar] [CrossRef]
- Shatz, W.; Ng, D.; Dutina, G.; Wong, A.W.; Dunshee, D.R.; Sonoda, J.; Shen, A.; Scheer, J.M. An efficient route to bispecific antibody production using single-reactor mammalian co-culture. MAbs 2016, 8, 1487–1497. [Google Scholar] [CrossRef] [Green Version]
- Spiess, C.; Merchant, M.; Huang, A.; Zheng, Z.; Yang, N.Y.; Peng, J.; Ellerman, D.; Shatz, W.; Reilly, D.; Yansura, D.G.; et al. Bispecific antibodies with natural architecture produced by co-culture of bacteria expressing two distinct half-antibodies. Nat. Biotechnol. 2013, 31, 753–758. [Google Scholar] [CrossRef]
- Suresh, M.R.; Cuello, A.C.; Milstein, C. Bispecific monoclonal antibodies from hybrid hybridomas. Methods Enzymol. 1986, 121, 210–228. [Google Scholar]
- Kroesen, B.J.; Helfrich, W.; Molema, G.; de Leij, L. Bispecific antibodies for treatment of cancer in experimental animal models and man. Adv. Drug Deliv. Rev. 1998, 31, 105–129. [Google Scholar] [CrossRef]
- Tustian, A.D.; Endicott, C.; Adams, B.; Mattila, J.; Bak, H. Development of purification processes for fully human bispecific antibodies based upon modification of protein A binding avidity. MAbs 2016, 8, 828–838. [Google Scholar] [CrossRef]
- Krishnamurthy, A.; Jimeno, A. Bispecific antibodies for cancer therapy: A review. Pharmacol. Ther. 2018, 185, 122–134. [Google Scholar] [CrossRef]
- Lindhofer, H.; Mocikat, R.; Steipe, B.; Thierfelder, S. Preferential species-restricted heavy/light chain pairing in rat/mouse quadromas. Implications for a single-step purification of bispecific antibodies. J. Immunol. (Baltimore) 1995, 155, 219–225. [Google Scholar]
- Linke, R.; Klein, A.; Seimetz, D. Catumaxomab: Clinical development and future directions. MAbs 2010, 2, 129–136. [Google Scholar] [CrossRef]
- Dall’Acqua, W.; Simon, A.L.; Mulkerrin, M.G.; Carter, P. Contribution of domain interface residues to the stability of antibody CH3 domain homodimers. Biochemistry 1998, 37, 9266–9273. [Google Scholar] [CrossRef]
- Crick, F.H.C. The packing of α-helices: Simple coiled-coils. Acta Crystallogr. 1953, 6, 689–697. [Google Scholar] [CrossRef]
- Atwell, S.; Ridgway, J.B.; Wells, J.A.; Carter, P. Stable heterodimers from remodeling the domain interface of a homodimer using a phage display library. J. Mol. Biol. 1997, 270, 26–35. [Google Scholar] [CrossRef]
- Merchant, A.M.; Zhu, Z.; Yuan, J.Q.; Goddard, A.; Adams, C.W.; Presta, L.G.; Carter, P. An efficient route to human bispecific IgG. Nat. Biotechnol. 1998, 16, 677–681. [Google Scholar] [CrossRef]
- Moore, G.L.; Bernett, M.J.; Rashid, R.; Pong, E.W.; Nguyen, D.-H.T.; Jacinto, J.; Eivazi, A.; Nisthal, A.; Diaz, J.E.; Chu, S.Y.; et al. A robust heterodimeric Fc platform engineered for efficient development of bispecific antibodies of multiple formats. Methods 2019, 154, 38–50. [Google Scholar] [CrossRef]
- Zhang, H.M.; Li, C.; Lei, M.; Lundin, V.; Lee, H.Y.; Ninonuevo, M.; Lin, K.; Han, G.; Sandoval, W.; Lei, D.; et al. Structural and Functional Characterization of a Hole–Hole Homodimer Variant in a “Knob-Into-Hole” Bispecific Antibody. Anal. Chem. 2017, 89, 13494–13501. [Google Scholar] [CrossRef]
- Shatz, W.; Chung, S.; Li, B.; Marshall, B.; Tejada, M.; Phung, W.; Sandoval, W.; Kelley, R.F.; Scheer, J.M. Knobs-into-holes antibody production in mammalian cell lines reveals that asymmetric afucosylation is sufficient for full antibody-dependent cellular cytotoxicity. MAbs 2013, 5, 872–881. [Google Scholar] [CrossRef] [Green Version]
- Davis, J.H.; Aperlo, C.; Li, Y.; Kurosawa, E.; Lan, Y.; Lo, K.M.; Huston, J.S. SEEDbodies: Fusion proteins based on strand-exchange engineered domain (SEED) CH3 heterodimers in an Fc analogue platform for asymmetric binders or immunofusions and bispecific antibodies. Protein Eng. Des. Sel. PEDS 2010, 23, 195–202. [Google Scholar] [CrossRef]
- Muda, M.; Gross, A.W.; Dawson, J.P.; He, C.; Kurosawa, E.; Schweickhardt, R.; Dugas, M.; Soloviev, M.; Bernhardt, A.; Fischer, D.; et al. Therapeutic assessment of SEED: A new engineered antibody platform designed to generate mono- and bispecific antibodies. Protein Eng. Des. Sel. PEDS 2011, 24, 447–454. [Google Scholar] [CrossRef]
- Fischer, N.; Elson, G.; Magistrelli, G.; Dheilly, E.; Fouque, N.; Laurendon, A.; Gueneau, F.; Ravn, U.; Depoisier, J.-F.; Moine, V.; et al. Exploiting light chains for the scalable generation and platform purification of native human bispecific IgG. Nat. Commun. 2015, 6, 6113. [Google Scholar] [CrossRef] [Green Version]
- Magistrelli, G.; Poitevin, Y.; Schlosser, F.; Pontini, G.; Malinge, P.; Josserand, S.; Corbier, M.; Fischer, N. Optimizing assembly and production of native bispecific antibodies by codon de-optimization. MAbs 2017, 9, 231–239. [Google Scholar] [CrossRef]
- Shiraiwa, H.; Narita, A.; Kamata-Sakurai, M.; Ishiguro, T.; Sano, Y.; Hironiwa, N.; Tsushima, T.; Segawa, H.; Tsunenari, T.; Ikeda, Y.; et al. Engineering a bispecific antibody with a common light chain: Identification and optimization of an anti-CD3 epsilon and anti-GPC3 bispecific antibody, ERY974. Methods 2019, 154, 10–20. [Google Scholar] [CrossRef]
- Dimasi, N.; Fleming, R.; Wu, H.; Gao, C. Molecular engineering strategies and methods for the expression and purification of IgG1-based bispecific bivalent antibodies. Methods 2019, 154, 77–86. [Google Scholar] [CrossRef]
- Adamkewicz, J.I.; Chen, D.C.; Paz-Priel, I. Effects and Interferences of Emicizumab, a Humanised Bispecific Antibody Mimicking Activated Factor VIII Cofactor Function, on Coagulation Assays. Thromb. Haemost. 2019, 119, 1084–1093. [Google Scholar] [CrossRef] [Green Version]
- ART-Ig® (Bispecific Antibody Manufacturing Technology). Available online: https://www.chugai-pharm.co.jp/english/ir/rd/technologies_popup3.html (accessed on 4 July 2019).
- Husain, B.; Ellerman, D. Expanding the Boundaries of Biotherapeutics with Bispecific Antibodies. BioDrugs 2018, 32, 441–464. [Google Scholar] [CrossRef] [Green Version]
- Jackman, J.; Chen, Y.; Huang, A.; Moffat, B.; Scheer, J.M.; Leong, S.R.; Lee, W.P.; Zhang, J.; Sharma, N.; Lu, Y.; et al. Development of a two-part strategy to identify a therapeutic human bispecific antibody that inhibits IgE receptor signaling. J. Biol. Chem. 2010, 285, 20850–20859. [Google Scholar] [CrossRef]
- Van Blarcom, T.; Lindquist, K.; Melton, Z.; Cheung, W.L.; Wagstrom, C.; McDonough, D.; Valle Oseguera, C.; Ding, S.; Rossi, A.; Potluri, S.; et al. Productive common light chain libraries yield diverse panels of high affinity bispecific antibodies. MAbs 2018, 10, 256–268. [Google Scholar] [CrossRef]
- Dillon, M.; Yin, Y.; Zhou, J.; McCarty, L.; Ellerman, D.; Slaga, D.; Junttila, T.T.; Han, G.; Sandoval, W.; Ovacik, M.A. Efficient production of bispecific IgG of different isotypes and species of origin in single mammalian cells. MAbs 2017, 9, 213–230. [Google Scholar] [CrossRef]
- Klein, C.; Schaefer, W.; Regula, J.T. The use of CrossMAb technology for the generation of bi- and multispecific antibodies. MAbs 2016, 8, 1010–1020. [Google Scholar] [CrossRef] [Green Version]
- Regula, J.T.; Imhof-Jung, S.; Mølhøj, M.; Benz, J.; Ehler, A.; Bujotzek, A.; Schaefer, W.; Klein, C. Variable heavy-variable light domain and Fab-arm CrossMabs with charged residue exchanges to enforce correct light chain assembly. Protein Eng. Des. Sel. PEDS 2018, 31, 289–299. [Google Scholar] [CrossRef]
- Thomas, M.; Kienast, Y.; Scheuer, W.; Bahner, M.; Kaluza, K.; Gassner, C.; Herting, F.; Brinkmann, U.; Seeber, S.; Kavlie, A.; et al. A novel angiopoietin-2 selective fully human antibody with potent anti-tumoral and anti-angiogenic efficacy and superior side effect profile compared to Pan-Angiopoietin-1/-2 inhibitors. PLoS ONE 2013, 8, e54923. [Google Scholar] [CrossRef]
- Kienast, Y.; Klein, C.; Scheuer, W.; Raemsch, R.; Lorenzon, E.; Bernicke, D.; Herting, F.; Yu, S.; The, H.H.; Martarello, L.; et al. Ang-2-VEGF-A CrossMab, a novel bispecific human IgG1 antibody blocking VEGF-A and Ang-2 functions simultaneously, mediates potent antitumor, antiangiogenic, and antimetastatic efficacy. Clin. Cancer Res. 2013, 19, 6730–6740. [Google Scholar] [CrossRef]
- Gassner, C.; Lipsmeier, F.; Metzger, P.; Beck, H.; Schnueriger, A.; Regula, J.T.; Moelleken, J. Development and validation of a novel SPR-based assay principle for bispecific molecules. J. Pharm. Biomed. Anal. 2015, 102, 144–149. [Google Scholar] [CrossRef]
- Bratt, J.; Linderholm, A.; Monroe, B.; Chamow, S.M. Therapeutic IgG-Like Bispecific Antibodies: Modular Versatility and Manufacturing Challenges; Part 2; BioProcess International: Boston, MA, USA, 2018. [Google Scholar]
- Priola, J.J.; Calzadilla, N.; Baumann, M.; Borth, N.; Tate, C.G.; Betenbaugh, M.J. High-throughput screening and selection of mammalian cells for enhanced protein production. Biotechnol. J. 2016, 11, 853–865. [Google Scholar] [CrossRef]
- Hossler, P.; Khattak, S.F.; Li, Z.J. Optimal and consistent protein glycosylation in mammalian cell culture. Glycobiology 2009, 19, 936–949. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Alvin, K.; Latif, H.; Hsu, A.; Parikh, V.; Whitmer, T.; Tellers, M.; de la Cruz Edmonds, M.C.; Ly, J.; Salmon, P.; et al. Rapid protein production using CHO stable transfection pools. Biotechnol. Prog. 2010, 26, 1431–1437. [Google Scholar] [CrossRef]
- Rajendra, Y.; Peery, R.B.; Hougland, M.D.; Barnard, G.C.; Wu, X.; Fitchett, J.R.; Bacica, M.; Demarest, S.J. Transient and stable CHO expression, purification and characterization of novel hetero-dimeric bispecific IgG antibodies. Biotechnol. Prog. 2017, 33, 469–477. [Google Scholar] [CrossRef]
- Gomez, N.; Wieczorek, A.; Lu, F.; Bruno, R.; Diaz, L.; Agrawal, N.J.; Daris, K. Culture temperature modulates half antibody and aggregate formation in a Chinese hamster ovary cell line expressing a bispecific antibody. Biotechnol. Bioeng. 2018, 115, 2930–2940. [Google Scholar] [CrossRef]
- Ha, J.-H.; Kim, J.-E.; Kim, Y.-S. Immunoglobulin Fc heterodimer platform technology: From design to applications in therapeutic antibodies and proteins. Front. Immunol. 2016, 7, 394. [Google Scholar] [CrossRef]
- Labrijn, A.F.; Meesters, J.I.; de Goeij, B.E.C.G.; van den Bremer, E.T.J.; Neijssen, J.; van Kampen, M.D.; Strumane, K.; Verploegen, S.; Kundu, A.; Gramer, M.J.; et al. Efficient generation of stable bispecific IgG1 by controlled Fab-arm exchange. Proc. Natl. Acad. Sci. USA 2013, 110, 5145. [Google Scholar] [CrossRef]
- Labrijn, A.F.; Meesters, J.I.; Priem, P.; de Jong, R.N.; van den Bremer, E.T.J.; van Kampen, M.D.; Gerritsen, A.F.; Schuurman, J.; Parren, P.W.H.I. Controlled Fab-arm exchange for the generation of stable bispecific IgG1. Nat. Protoc. 2014, 9, 2450. [Google Scholar] [CrossRef]
- Igawa, T. Next Generation Antibody Therapeutics Using Bispecific Antibody Technology. Yakugaku Zasshi J. Pharm. Soc. Jpn. 2017, 137, 831–836. [Google Scholar] [CrossRef] [Green Version]
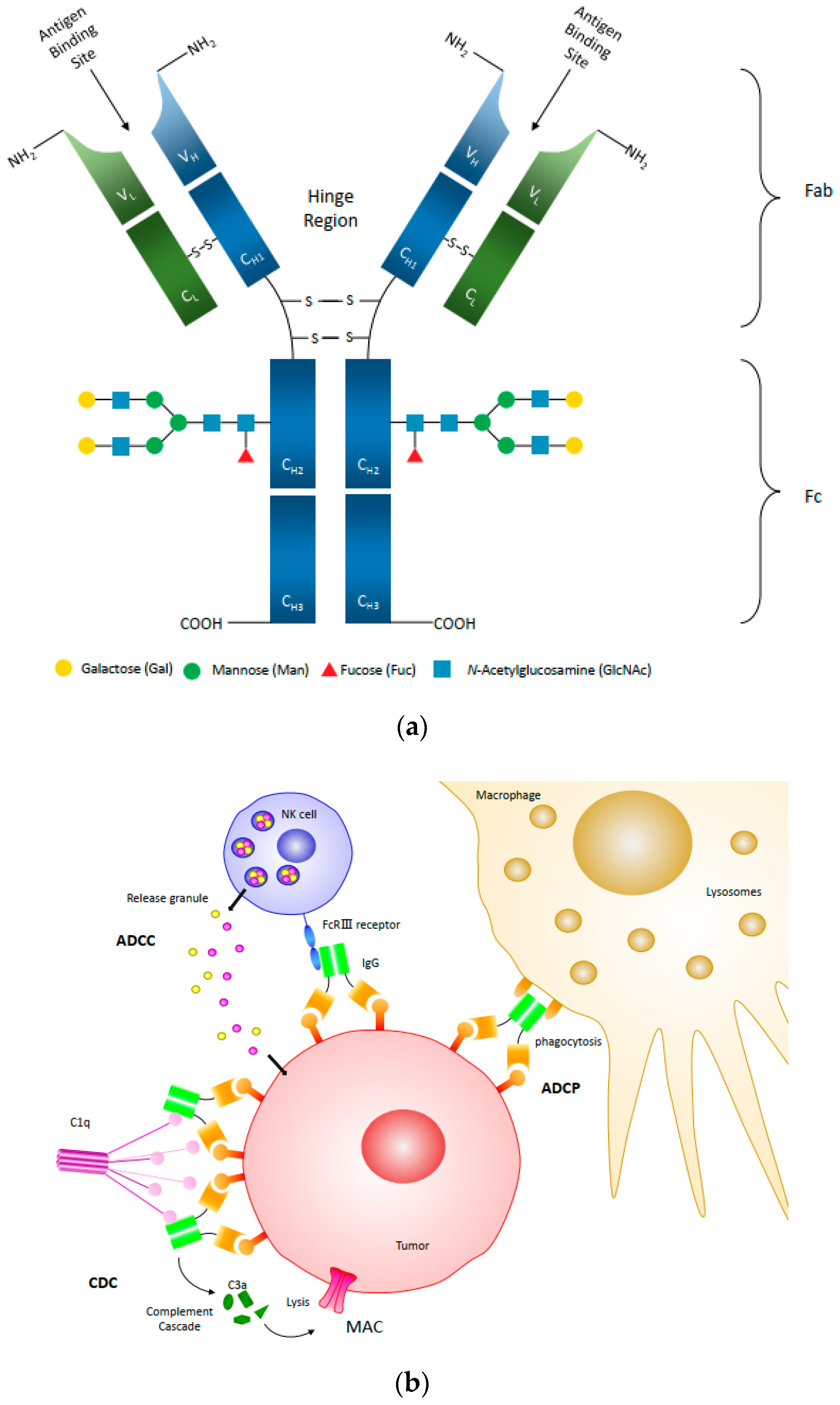





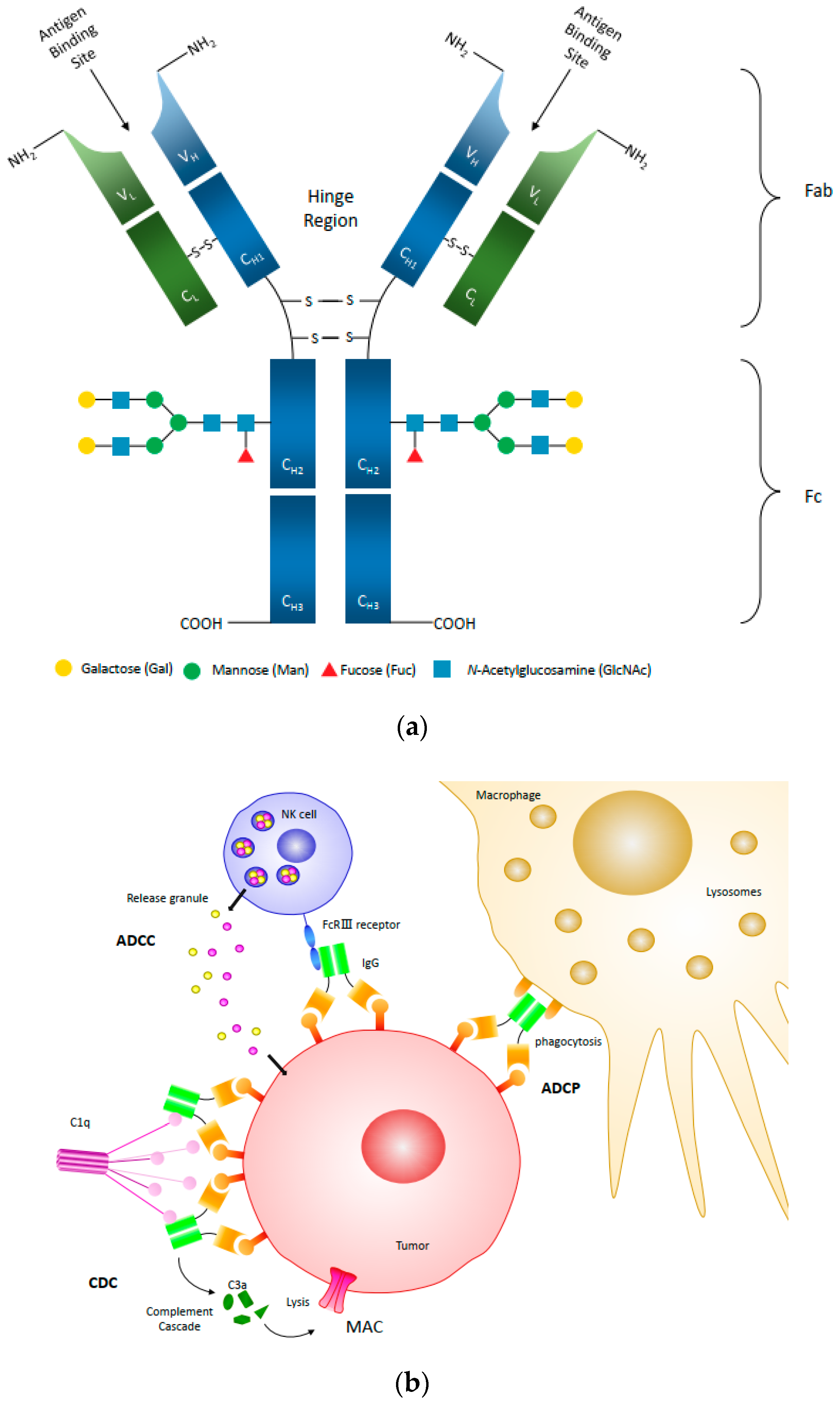{kind=link}
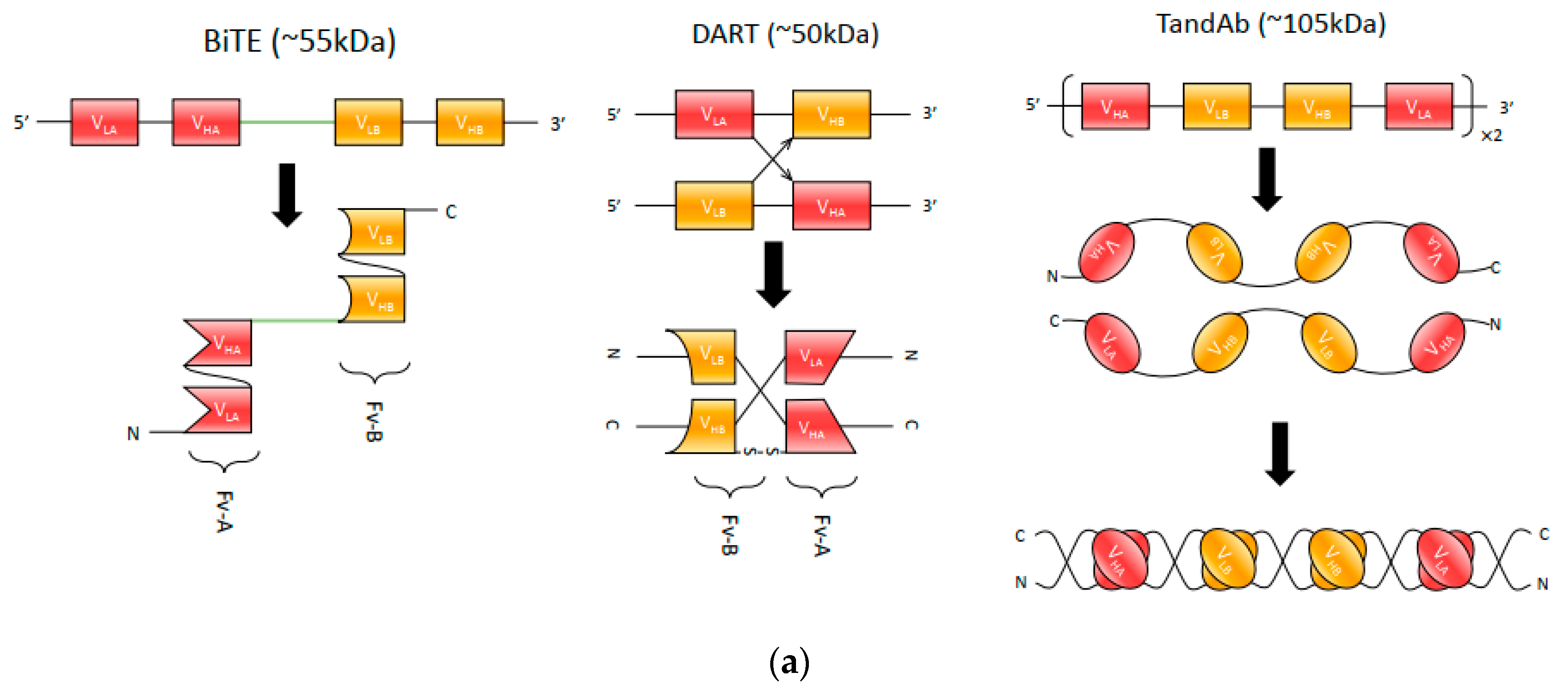{kind=link}
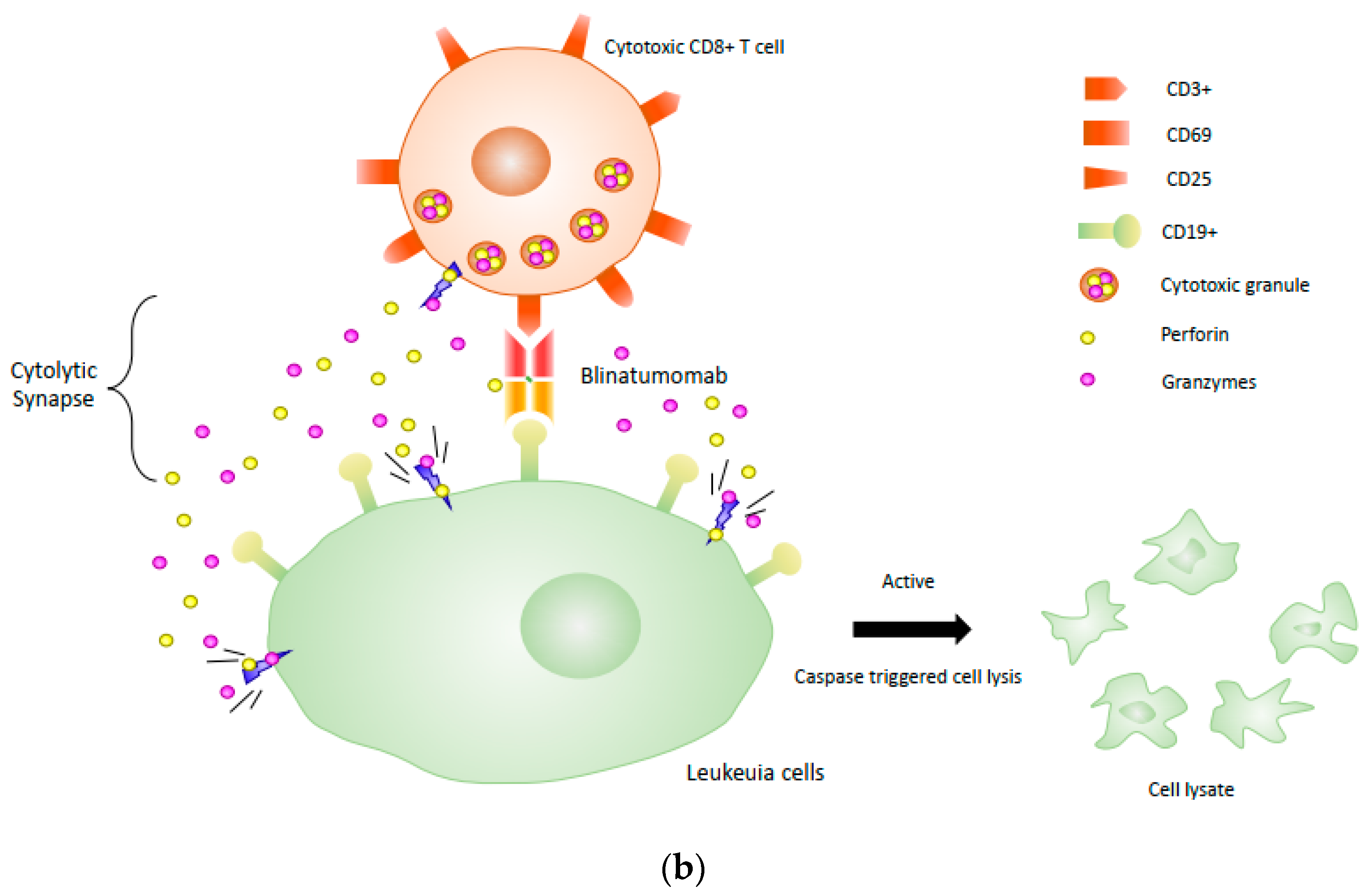{kind=link}
{kind=link}
{kind=link}
| Platform | Species | Molecule | Yield | Purification | Type | Reference |
|---|---|---|---|---|---|---|
| Bacteria | TG1 E. coli | anti-HER2/neu × anti-CD16 | around 3.7 mg/L | Ni-NTA column | BiTE | [101] |
| Bacteria | E. coli | anti-TCR × anti-fluorescein | 1 mg/L | Fluorescein affinity chromatography | Tandem bispecific scFv molecule linked by 212 and 205 c’ linkers | [67] |
| Bacteria | E. coli, periplasmic | anti-EpCAM × anti-CD3 | 12–15 mg/L | Ni-NTA column | BiTE | [102,103,104] |
| Bacteria | E. coli BL21(DE3) | anti-HER2 × anti-CD3 | 3 mg/L | Ni-NTA column | BiTE | [105] |
| Mammalian | CHO-K1 | anti-CD123 × anti-CD3 | 2–5 mg/L | Protein G chromatography | BiTE-Fc | [106] |
| Mammalian | CHO cell | anti-P-cadherin × anti-CD3 | 1300 mg/L | Protein A chromatography | DART-Fc | [107] |
| Mammalian | CHO-S | anti-CD19 × anti-CD3 | SEC | DART & BiTE | [40] | |
| Mammalian | CHO | anti-CD33 × anti-CD3 | IMAC + SEC | TandAbs | [48] | |
| Mammalian | CHO | anti-CD19 × anti-CD3 | >200 mg/L | Ni-NTA column | TandAbs | [71] |
| Mammalian | CHO cell | anti-EpCAM × ani-CD3 | IMAC + gel filtration + CEX | BiTE | [108] | |
| Mammalian | HEK 293 | anti-EpCAM × ani-CD3 | IMAC | BiTE | [109] | |
| Mammalian | CHO-S | anti-GD2/DOTA-metal complex | 5–10 mg/L | Protein A chromatography | IgG-ScFv | [110] |
| Platform | Name | Target | Heavy-Chain Engineering | Heavy/Light-Chain Engineering | Yield | Purification | Note | Reference |
|---|---|---|---|---|---|---|---|---|
| CHO-DG44 cells | MCLA-128 | Human epidermal growth factor receptors (HER2 and HER3) | knobs-into-holes | common light chain | 0.6–1.2 g/L | Protein A + IEC | stable expression | [27] |
| HEK293F suspension cells | Ang-2-VEGF-A CrossMab | angiopoietin-2 (Ang-2) and vascular endothelial growth factor A (VEGF-A) | knobs-into-holes | CrossMab (CH1-CL) | 0.03 g/L | Protein A + SEC | transient expression | [142] |
| cell-free system (E. coli extract) | ScFv-KiH, BiTE-KiH | CD3, EpCAM, HER2 | knobs-into-holes | 0.2–0.4 g/L | Protein A | in vitro | [143] | |
| HEK293 | M315-14D2 (scFv-Fc) | mouse NKG2D and mouse p55TNFR | Electrostatic Steering Effects | 0.1 g/L | protein A | transient expression | [144] | |
| Expi 293 cells * | 10E8V2.0/iMab | human CD4 and HIV-1 | knobs-into-holes | CrossMab (CH1-CL) | Protein A + SEC | transient expression | [145] | |
| HEK293F suspension cells | CD20–243 CrossMab | CD20 and HLA-DR | knobs-into-holes | CrossMab (CH1-CL) | Protein A + SEC | transient expression | [146] | |
| CHO-K1 suspension cell culture | anti-FGFR1/βKL | FGFR1/βKL | knobs-into-holes | Co-culture | 0.35 g/L | Protein A + IEC | stable expression | [147] |
| E. coli K-12 W3110 suspension cell | Anti-Her2/CD3 | Her2/CD3 | knobs-into-holes | Co-culture | 4.8 g/L | Protein A + HIC | stable expression | [148] |
| E. coli K-12 W3110 suspension cell | Anti-CD19/CD3 | CD19/CD3 | knobs-into-holes | Co-culture | 1 g/L | Protein A + HIC | stable expression | [148] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Q.; Chen, Y.; Park, J.; Liu, X.; Hu, Y.; Wang, T.; McFarland, K.; Betenbaugh, M.J. Design and Production of Bispecific Antibodies. Antibodies 2019, 8, 43. https://doi.org/10.3390/antib8030043
Wang Q, Chen Y, Park J, Liu X, Hu Y, Wang T, McFarland K, Betenbaugh MJ. Design and Production of Bispecific Antibodies. Antibodies. 2019; 8(3):43. https://doi.org/10.3390/antib8030043
Chicago/Turabian StyleWang, Qiong, Yiqun Chen, Jaeyoung Park, Xiao Liu, Yifeng Hu, Tiexin Wang, Kevin McFarland, and Michael J. Betenbaugh. 2019. "Design and Production of Bispecific Antibodies" Antibodies 8, no. 3: 43. https://doi.org/10.3390/antib8030043
APA StyleWang, Q., Chen, Y., Park, J., Liu, X., Hu, Y., Wang, T., McFarland, K., & Betenbaugh, M. J. (2019). Design and Production of Bispecific Antibodies. Antibodies, 8(3), 43. https://doi.org/10.3390/antib8030043