The Role of Complement in the Mechanism of Action of Therapeutic Anti-Cancer mAbs



Abstract
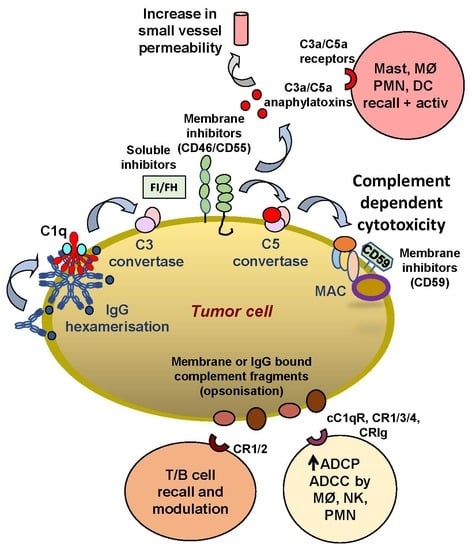
1. Introduction
2. Complement Activation by Human IgG1 mAbs
3. The Interaction of Complement Components with Immune Cells
4. Main Factors Affecting Complement Activation by IgG1 Anti-Tumor Antibodies
4.1. Antigen Density and Hexamerization
4.2. Membrane and Soluble Complement Inhibitors
5. The Role of Complement in the Therapeutic Activity of Anti-Tumor mAbs
5.1. Studies In Vitro and in Animal Models
5.2. Ex Vivo and In Vivo Human Studies
6. Conclusions and Future Perspective
Funding
Conflicts of Interest
References
- Melis, J.P.; Strumane, K.; Ruuls, S.R.; Beurskens, F.J.; Schuurman, J.; Parren, P.W. Complement in therapy and disease: Regulating the complement system with antibody-based therapeutics. Mol. Immunol. 2015, 67, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.P.; Lindorfer, M.A. Cytotoxic mechanisms of immunotherapy: Harnessing complement in the action of anti-tumor monoclonal antibodies. Semin. Immunol. 2016, 28, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Weiner, G.J. Building better monoclonal antibody-based therapeutics. Nat. Rev. Cancer 2015, 15, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Golay, J. Direct targeting of cancer cells with antibodies: What can we learn from the successes and failure of unconjugated antibodies for lymphoid neoplasias? J. Autoimmun. 2017, 85, 6–19. [Google Scholar] [CrossRef] [PubMed]
- Lindorfer, M.A.; Koehl, J.; Taylor, R.P. Interactions between the complement system and Fcgamma receptors. In IgG Fc: Linking Adaptive and Innate Immunity; Nimmerhahn, F., Ackerman, M.E., Eds.; Elsevier Press: Amsterdam, The Netherlands, 2013; pp. 49–74. [Google Scholar]
- Pierpont, T.M.; Limper, C.B.; Richards, K.L. Past, Present, and Future of Rituximab-The World’s First Oncology Monoclonal Antibody Therapy. Front. Oncol. 2018, 8, 163. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.P.; Lindorfer, M.A. Immunotherapeutic mechanisms of anti-CD20 monoclonal antibodies. Curr. Opin. Immunol. 2008, 20, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Carter, P.J.; Lazar, G.A. Next generation antibody drugs: Pursuit of the ‘high-hanging fruit’. Nat. Rev. Drug Discov. 2018, 17, 197–223. [Google Scholar] [CrossRef]
- Garcia-Foncillas, J.; Sunakawa, Y.; Aderka, D.; Wainberg, Z.; Ronga, P.; Witzler, P.; Stintzing, S. Distinguishing Features of Cetuximab and Panitumumab in Colorectal Cancer and Other Solid Tumors. Front. Oncol. 2019, 9, 849. [Google Scholar] [CrossRef]
- Hudis, C.A. Trastuzumab—Mechanism of action and use in clinical practice. NEJM 2007, 357, 39–51. [Google Scholar] [CrossRef]
- Moessner, E.; Bruenker, P.; Moser, S.; Puentener, U.; Schmidt, C.; Herter, S.; Grau, R.; Gerdes, C.; Nopora, A.; van Puijenbroek, E.; et al. Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 with enhanced direct and immune effector cell-mediated B-cell cytotoxicity. Blood 2010, 115, 4393–4402. [Google Scholar] [CrossRef]
- Jiang, H.; Acharya, C.; An, G.; Zhong, M.; Feng, X.; Wang, L.; Dasilva, N.; Song, Z.; Yang, G.; Adrian, F.; et al. SAR650984 directly induces multiple myeloma cell death via lysosomal-associated and apoptotic pathways, which is further enhanced by pomalidomide. Leukemia 2016, 30, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Lapalombella, R.; Yeh, Y.Y.; Wang, L.; Ramanunni, A.; Rafiq, S.; Jha, S.; Staubli, J.; Lucas, D.M.; Mani, R.; Herman, S.E.; et al. Tetraspanin CD37 directly mediates transduction of survival and apoptotic signals. Cancer Cell 2012, 21, 694–708. [Google Scholar] [CrossRef]
- Plesner, T.; Krejcik, J. Daratumumab for the Treatment of Multiple Myeloma. Front. Immunol. 2018, 9, 1228. [Google Scholar] [CrossRef]
- Musolino, A.; Boggiani, D.; Pellegrino, B.; Zanoni, D.; Sikokis, A.; Missale, G.; Silini, E.M.; Maglietta, G.; Frassoldati, A.; Michiara, M. Role of innate and adaptive immunity in the efficacy of anti-HER2 monoclonal antibodies for HER2-positive breast cancer. Crit. Rev. Oncol./Hematol. 2020, 149, 102927. [Google Scholar] [CrossRef]
- Costa, D.; Vene, R.; Benelli, R.; Romairone, E.; Scabini, S.; Catellani, S.; Rebesco, B.; Mastracci, L.; Grillo, F.; Minghelli, S.; et al. Targeting the Epidermal Growth Factor Receptor Can Counteract the Inhibition of Natural Killer Cell Function Exerted by Colorectal Tumor-Associated Fibroblasts. Front. Immunol. 2018, 9, 1150. [Google Scholar] [CrossRef]
- Ferris, R.L.; Jaffee, E.M.; Ferrone, S. Tumor antigen-targeted, monoclonal antibody-based immunotherapy: Clinical response, cellular immunity, and immunoescape. J. Clin. Oncol. 2010, 28, 4390–4399. [Google Scholar] [CrossRef] [PubMed]
- Zent, C.S.; Elliott, M.R. Maxed out macs: Physiologic cell clearance as a function of macrophage phagocytic capacity. FEBS J. 2017, 284, 1021–1039. [Google Scholar] [CrossRef] [PubMed]
- VanDerMeid, K.R.; Elliott, M.R.; Baran, A.M.; Barr, P.M.; Chu, C.C.; Zent, C.S. Cellular Cytotoxicity of Next-Generation CD20 Monoclonal Antibodies. Cancer Immunol. Res. 2018, 6, 1150–1160. [Google Scholar] [CrossRef]
- Campbell, K.S.; Cohen, A.D.; Pazina, T. Mechanisms of NK Cell Activation and Clinical Activity of the Therapeutic SLAMF7 Antibody, Elotuzumab in Multiple Myeloma. Front. Immunol. 2018, 9, 2551. [Google Scholar] [CrossRef]
- de Weers, M.; Tai, Y.T.; van der Veer, M.S.; Bakker, J.M.; Vink, T.; Jacobs, D.C.; Oomen, L.A.; Peipp, M.; Valerius, T.; Slootstra, J.W.; et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J. Immunol. 2011, 186, 1840–1848. [Google Scholar] [CrossRef]
- Zent, C.S.; Chen, J.B.; Kurten, R.C.; Kaushal, G.P.; Marie Lacy, H.; Schichman, S.A. Alemtuzumab (CAMPATH 1H) does not kill chronic lymphocytic leukemia cells in serum free medium. Leuk. Res. 2004, 28, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Golay, J.; Manganini, M.; Rambaldi, A.; Introna, M. Effect of alemtuzumab on neoplastic B cells. Haematologica 2004, 89, 1476–1483. [Google Scholar] [PubMed]
- Diebolder, C.A.; Beurskens, F.J.; de Jong, R.N.; Koning, R.I.; Strumane, K.; Lindorfer, M.A.; Voorhorst, M.; Ugurlar, D.; Rosati, S.; Heck, A.J.; et al. Complement is activated by IgG hexamers assembled at the cell surface. Science 2014, 343, 1260–1263. [Google Scholar] [CrossRef]
- Wang, G.; de Jong, R.N.; van den Bremer, E.T.; Beurskens, F.J.; Labrijn, A.F.; Ugurlar, D.; Gros, P.; Schuurman, J.; Parren, P.W.; Heck, A.J. Molecular Basis of Assembly and Activation of Complement Component C1 in Complex with Immunoglobulin G1 and Antigen. Mol. Cell 2016, 63, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part I—Molecular Mechanisms of Activation and Regulation. Front. Immunol. 2015, 6, 262. [Google Scholar] [CrossRef]
- Goldberg, B.S.; Ackerman, M.E. Antibody-mediated complement activation in pathology and protection. Immunol. Cell Biol. 2020, 98, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Bordron, A.; Bagacean, C.; Tempescul, A.; Berthou, C.; Bettacchioli, E.; Hillion, S.; Renaudineau, Y. Complement System: A Neglected Pathway in Immunotherapy. Clin. Rev. Allergy Immunol. 2020, 58, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Lindorfer, M.A.; Cook, E.M.; Tupitza, J.C.; Zent, C.S.; Burack, R.; de Jong, R.N.; Beurskens, F.J.; Schuurman, J.; Parren, P.W.; Taylor, R.P. Real-time analysis of the detailed sequence of cellular events in mAb-mediated complement-dependent cytotoxicity of B-cell lines and of chronic lymphocytic leukemia B-cells. Mol. Immunol. 2016, 70, 13–23. [Google Scholar] [CrossRef]
- Morgan, B.P.; Walters, D.; Serna, M.; Bubeck, D. Terminal complexes of the complement system: New structural insights and their relevance to function. Immunol. Rev. 2016, 274, 141–151. [Google Scholar] [CrossRef]
- Reis, E.S.; Mastellos, D.C.; Ricklin, D.; Mantovani, A.; Lambris, J.D. Complement in cancer: Untangling an intricate relationship. Nat. Rev. Immunol. 2018, 18, 5–18. [Google Scholar] [CrossRef]
- Merle, N.S.; Noe, R.; Halbwachs-Mecarelli, L.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part II: Role in Immunity. Front. Immunol. 2015, 6, 257. [Google Scholar] [CrossRef] [PubMed]
- Geller, A.; Yan, J. The Role of Membrane Bound Complement Regulatory Proteins in Tumor Development and Cancer Immunotherapy. Front. Immunol. 2019, 10, 1074. [Google Scholar] [CrossRef]
- Laumonnier, Y.; Karsten, C.M.; Kohl, J. Novel insights into the expression pattern of anaphylatoxin receptors in mice and men. Mol. Immunol. 2017, 89, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Karsten, C.M.; Pandey, M.K.; Figge, J.; Kilchenstein, R.; Taylor, P.R.; Rosas, M.; McDonald, J.U.; Orr, S.J.; Berger, M.; Petzold, D.; et al. Anti-inflammatory activity of IgG1 mediated by Fc galactosylation and association of FcgammaRIIB and dectin-1. Nat. Med. 2012, 18, 1401–1406. [Google Scholar] [CrossRef]
- Lukacsi, S.; Macsik-Valent, B.; Nagy-Balo, Z.; Kovacs, K.G.; Kliment, K.; Bajtay, Z.; Erdei, A. Utilization of complement receptors in immune cell-microbe interaction. FEBS Lett. 2020, 594, 2695–2713. [Google Scholar] [CrossRef]
- Fries, L.F.; Siwik, S.A.; Malbran, A.; Frank, M.M. Phagocytosis of target particles bearing C3b-IgG covalent complexes by human monocytes and polymorphonuclear leucocytes. Immunology 1987, 62, 45–51. [Google Scholar] [PubMed]
- Brown, E.J.; Joiner, K.A.; Cole, R.M.; Berger, M. Localization of complement component 3 on Streptococcus pneumoniae: Anti-capsular antibody causes complement deposition on the pneumococcal capsule. Infect. Immun. 1983, 39, 403–409. [Google Scholar] [CrossRef]
- Ehlenberger, A.G.; Nussenzweig, V. The role of membrane receptors for C3b and C3d in phagocytosis. J. Exp. Med. 1977, 145, 357–371. [Google Scholar] [CrossRef]
- Schreiber, A.D.; Frank, M.M. Role of antibody and complement in the immune clearance and destruction of erythrocytes. I. In vivo effects of IgG and IgM complement-fixing sites. J. Clin. Investig. 1972, 51, 575–582. [Google Scholar] [CrossRef]
- Atkinson, J.P.; Frank, M.M. Studies on the in vivo effects of antibody. Interaction of IgM antibody and complement in the immune clearance and destruction of erythrocytes in man. J. Clin. Investig. 1974, 54, 339–348. [Google Scholar] [CrossRef]
- Zaal, A.; van Ham, S.M.; Ten Brinke, A. Differential effects of anaphylatoxin C5a on antigen presenting cells, roles for C5aR1 and C5aR2. Immunol. Lett. 2019, 209, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, H.; He, Y.W. The Complement Receptors C3aR and C5aR Are a New Class of Immune Checkpoint Receptor in Cancer Immunotherapy. Front. Immunol. 2019, 10, 1574. [Google Scholar] [CrossRef]
- Tammen, A.; Derer, S.; Schwanbeck, R.; Rosner, T.; Kretschmer, A.; Beurskens, F.J.; Schuurman, J.; Parren, P.W.; Valerius, T. Monoclonal Antibodies against Epidermal Growth Factor Receptor Acquire an Ability To Kill Tumor Cells through Complement Activation by Mutations That Selectively Facilitate the Hexamerization of IgG on Opsonized Cells. J. Immunol. 2017, 198, 1585–1594. [Google Scholar] [CrossRef] [PubMed]
- Cook, E.M.; Lindorfer, M.A.; van der Horst, H.; Oostindie, S.; Beurskens, F.J.; Schuurman, J.; Zent, C.S.; Burack, R.; Parren, P.W.; Taylor, R.P. Antibodies That Efficiently Form Hexamers upon Antigen Binding Can Induce Complement-Dependent Cytotoxicity under Complement-Limiting Conditions. J. Immunol. 2016, 197, 1762–1775. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.P.; Lindorfer, M.A.; Cook, E.M.; Beurskens, F.J.; Schuurman, J.; Parren, P.; Zent, C.S.; VanDerMeid, K.R.; Burack, R.; Mizuno, M.; et al. Hexamerization-enhanced CD20 antibody mediates complement-dependent cytotoxicity in serum genetically deficient in C9. Clin. Immunol. 2017, 181, 24–28. [Google Scholar] [CrossRef]
- Marshall, M.J.E.; Stopforth, R.J.; Cragg, M.S. Therapeutic Antibodies: What Have We Learnt from Targeting CD20 and Where Are We Going? Front. Immunol. 2017, 8, 1245. [Google Scholar] [CrossRef] [PubMed]
- Teeling, J.L.; French, R.R.; Cragg, M.S.; van den Brakel, J.; Pluyter, M.; Huang, H.; Chan, C.; Parren, P.W.; Hack, C.E.; Dechant, M.; et al. Characterization of new human CD20 monoclonal antibodies with potent cytolytic activity against non-Hodgkin lymphomas. Blood 2004, 104, 1793–1800. [Google Scholar] [CrossRef]
- Pawluczkowycz, A.W.; Beurskens, F.J.; Beum, P.V.; Lindorfer, M.A.; van de Winkel, J.G.; Parren, P.W.; Taylor, R.P. Binding of submaximal C1q promotes complement-dependent cytotoxicity (CDC) of B cells opsonized with anti-CD20 mAbs ofatumumab (OFA) or rituximab (RTX): Considerably higher levels of CDC are induced by OFA than by RTX. J. Immunol. 2009, 183, 749–758. [Google Scholar] [CrossRef]
- Bologna, L.; Gotti, E.; Manganini, M.; Rambaldi, A.; Intermesoli, T.; Introna, M.; Golay, J. Mechanism of action of type II, glycoengineered, anti-CD20 monoclonal antibody GA101 in B-chronic lymphocytic leukemia whole blood assays in comparison with rituximab and alemtuzumab. J. Immunol. 2011, 186, 3762–3769. [Google Scholar] [CrossRef]
- Cragg, M.S.; Morgan, S.M.; Chan, H.T.; Morgan, B.P.; Filatov, A.V.; Johnson, P.W.; French, R.R.; Glennie, M.J. Complement-mediated lysis by anti-CD20 mAb correlates with segregation into lipid rafts. Blood 2003, 101, 1045–1052. [Google Scholar] [CrossRef]
- Rouge, L.; Chiang, N.; Steffek, M.; Kugel, C.; Croll, T.I.; Tam, C.; Estevez, A.; Arthur, C.P.; Koth, C.M.; Ciferri, C.; et al. Structure of CD20 in complex with the therapeutic monoclonal antibody rituximab. Science 2020, 367, 1224–1230. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Planchais, C.; Fronzes, R.; Mouquet, H.; Reyes, N. Binding mechanisms of therapeutic antibodies to human CD20. Science 2020, 369, 793–799. [Google Scholar] [CrossRef]
- Niederfellner, G.; Lammens, A.; Mundigl, O.; Georges, G.J.; Schaefer, W.; Schwaiger, M.; Franke, A.; Wiechmann, K.; Jenewein, S.; Slootstra, J.W.; et al. Epitope characterization and crystal structure of GA101 provide insights into the molecular basis for type I/II distinction of CD20 antibodies. Blood 2010, 118, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Cleary, K.L.S.; Chan, H.T.C.; James, S.; Glennie, M.J.; Cragg, M.S. Antibody Distance from the Cell Membrane Regulates Antibody Effector Mechanisms. J. Immunol. 2017, 198, 3999–4011. [Google Scholar] [CrossRef] [PubMed]
- Beum, P.V.; Lindorfer, M.A.; Peek, E.M.; Stukenberg, P.T.; de Weers, M.; Beurskens, F.J.; Parren, P.W.; van de Winkel, J.G.; Taylor, R.P. Penetration of antibody-opsonized cells by the membrane attack complex of complement promotes Ca(2+) influx and induces streamers. Eur. J. Immunol. 2011, 41, 2436–2446. [Google Scholar] [CrossRef]
- de Jong, R.N.; Beurskens, F.J.; Verploegen, S.; Strumane, K.; van Kampen, M.D.; Voorhorst, M.; Horstman, W.; Engelberts, P.J.; Oostindie, S.C.; Wang, G.; et al. A Novel Platform for the Potentiation of Therapeutic Antibodies Based on Antigen-Dependent Formation of IgG Hexamers at the Cell Surface. PLoS Biol. 2016, 14, e1002344. [Google Scholar] [CrossRef]
- Schutze, K.; Petry, K.; Hambach, J.; Schuster, N.; Fumey, W.; Schriewer, L.; Rockendorf, J.; Menzel, S.; Albrecht, B.; Haag, F.; et al. CD38-Specific Biparatopic Heavy Chain Antibodies Display Potent Complement-Dependent Cytotoxicity Against Multiple Myeloma Cells. Front. Immunol. 2018, 9, 2553. [Google Scholar] [CrossRef]
- Oostindie, S.C.; van der Horst, H.J.; Kil, L.P.; Strumane, K.; Overdijk, M.B.; van den Brink, E.N.; van den Brakel, J.H.N.; Rademaker, H.J.; van Kessel, B.; van den Noort, J.; et al. DuoHexaBody-CD37((R)), a novel biparatopic CD37 antibody with enhanced Fc-mediated hexamerization as a potential therapy for B-cell malignancies. Blood Cancer J. 2020, 10, 30. [Google Scholar] [CrossRef]
- Gulati, S.; Beurskens, F.J.; de Kreuk, B.J.; Roza, M.; Zheng, B.; DeOliveira, R.B.; Shaughnessy, J.; Nowak, N.A.; Taylor, R.P.; Botto, M.; et al. Complement alone drives efficacy of a chimeric antigonococcal monoclonal antibody. PLoS Biol. 2019, 17, e3000323. [Google Scholar] [CrossRef]
- van Meerten, T.; van Rijn, R.S.; Hol, S.; Hagenbeek, A.; Ebeling, S.B. Complement-induced cell death by rituximab depends on CD20 expression level and acts complementary to antibody-dependent cellular cytotoxicity. Clin. Cancer Res. 2006, 12, 4027–4035. [Google Scholar] [CrossRef]
- van Meerten, T.; Rozemuller, H.; Hol, S.; Moerer, P.; Zwart, M.; Hagenbeek, A.; Mackus, W.J.; Parren, P.W.; van de Winkel, J.G.; Ebeling, S.B.; et al. HuMab-7D8, a monoclonal antibody directed against the membrane-proximal small loop epitope of CD20 can effectively eliminate CD20 low expressing tumor cells that resist rituximab-mediated lysis. Haematologica 2010, 95, 2063–2071. [Google Scholar] [CrossRef]
- Golay, J.; Lazzari, M.; Facchinetti, V.; Bernasconi, S.; Borleri, G.; Barbui, T.; Rambaldi, A.; Introna, M. CD20 levels determine the in vitro susceptibility to rituximab and complement of B-cell chronic lymphocytic leukemia: Further regulation by CD55 and CD59. Blood 2001, 98, 3383–3389. [Google Scholar] [CrossRef] [PubMed]
- Bologna, L.; Gotti, E.; Da Roit, F.; Intermesoli, T.; Rambaldi, A.; Introna, M.; Golay, J. Ofatumumab is more efficient than rituximab in lysing B chronic lymphocytic leukemia cells in whole blood and in combination with chemotherapy. J. Immunol. 2013, 190, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Golay, J.; Zaffaroni, L.; Vaccari, T.; Lazzari, M.; Borleri, G.M.; Bernasconi, S.; Tedesco, F.; Rambaldi, A.; Introna, M. Biologic response of B lymphoma cells to anti-CD20 monoclonal antibody rituximab in vitro: CD55 and CD59 regulate complement-mediated cell lysis. Blood 2000, 95, 3900–3908. [Google Scholar] [CrossRef]
- Sebejova, L.; Borsky, M.; Jaskova, Z.; Potesil, D.; Navrkalova, V.; Malcikova, J.; Sramek, M.; Doubek, M.; Loja, T.; Pospisilova, S.; et al. Distinct in vitro sensitivity of p53-mutated and ATM-mutated chronic lymphocytic leukemia cells to ofatumumab and rituximab. Exp. Hematol. 2014, 42, 867–874.e861. [Google Scholar] [CrossRef] [PubMed]
- Terui, Y.; Sakurai, T.; Mishima, Y.; Mishima, Y.; Sugimura, N.; Sasaoka, C.; Kojima, K.; Yokoyama, M.; Mizunuma, N.; Takahashi, S.; et al. Blockade of bulky lymphoma-associated CD55 expression by RNA interference overcomes resistance to complement-dependent cytotoxicity with rituximab. Cancer Sci. 2006, 97, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Ge, X.; You, T.; Xu, T.; Zhang, J.; Wu, G.; Peng, Z.; Chorev, M.; Aktas, B.H.; Halperin, J.A.; et al. Human CD59 inhibitor sensitizes rituximab-resistant lymphoma cells to complement-mediated cytolysis. Cancer Res. 2011, 71, 2298–2307. [Google Scholar] [CrossRef]
- Ge, X.; Wu, L.; Hu, W.; Fernandes, S.; Wang, C.; Li, X.; Brown, J.R.; Qin, X. rILYd4, a human CD59 inhibitor, enhances complement-dependent cytotoxicity of ofatumumab against rituximab-resistant B-cell lymphoma cells and chronic lymphocytic leukemia. Clin. Cancer Res. 2011, 17, 6702–6711. [Google Scholar] [CrossRef]
- Barth, M.J.; Hernandez-Ilizaliturri, F.J.; Mavis, C.; Tsai, P.C.; Gibbs, J.F.; Deeb, G.; Czuczman, M.S. Ofatumumab demonstrates activity against rituximab-sensitive and -resistant cell lines, lymphoma xenografts and primary tumour cells from patients with B-cell lymphoma. Br. J. Haematol. 2011, 156, 490–498. [Google Scholar] [CrossRef]
- Barth, M.J.; Mavis, C.; Czuczman, M.S.; Hernandez-Ilizaliturri, F.J. Ofatumumab Exhibits Enhanced In Vitro and In Vivo Activity Compared to Rituximab in Preclinical Models of Mantle Cell Lymphoma. Clin. Cancer Res. 2015, 21, 4391–4397. [Google Scholar] [CrossRef]
- Beum, P.V.; Mack, D.A.; Pawluczkowycz, A.W.; Lindorfer, M.A.; Taylor, R.P. Binding of rituximab, trastuzumab, cetuximab, or mAb T101 to cancer cells promotes trogocytosis mediated by THP-1 cells and monocytes. J. Immunol. 2008, 181, 8120–8132. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Ma, Z.W.; Li, H.; Xu, G.L.; Zheng, P.; Zhu, B.; Wu, Y.Z.; Zou, Q. Mapping of binding epitopes of a human decay-accelerating factor monoclonal antibody capable of enhancing rituximab-mediated complement-dependent cytotoxicity. Clin. Immunol. 2008, 128, 155–163. [Google Scholar] [CrossRef]
- Morgan, B.P.; Berg, C.W.; Harris, C.L. ”Homologous restriction” in complement lysis: Roles of membrane complement regulators. Xenotransplantation 2005, 12, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Harjunpaa, A.; Junnikkala, S.; Meri, S. Rituximab (anti-CD20) therapy of B-cell lymphomas: Direct complement killing is superior to cellular effector mechanisms. Scand. J. Immunol. 2000, 51, 634–641. [Google Scholar] [CrossRef]
- Bellone, S.; Roque, D.; Cocco, E.; Gasparrini, S.; Bortolomai, I.; Buza, N.; Abu-Khalaf, M.; Silasi, D.A.; Ratner, E.; Azodi, M.; et al. Downregulation of membrane complement inhibitors CD55 and CD59 by siRNA sensitises uterine serous carcinoma overexpressing Her2/neu to complement and antibody-dependent cell cytotoxicity in vitro: Implications for trastuzumab-based immunotherapy. Br. J. Cancer 2012, 106, 1543–1550. [Google Scholar] [CrossRef]
- Zhao, W.P.; Zhu, B.; Duan, Y.Z.; Chen, Z.T. Neutralization of complement regulatory proteins CD55 and CD59 augments therapeutic effect of herceptin against lung carcinoma cells. Oncol. Rep. 2009, 21, 1405–1411. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, Y.J.; Wang, Z.; Liao, J.; Liu, M.; Zhong, X.R.; Zheng, H.; Wang, Y.P. CD55 and CD59 expression protects HER2-overexpressing breast cancer cells from trastuzumab-induced complement-dependent cytotoxicity. Oncol. Lett. 2017, 14, 2961–2969. [Google Scholar] [CrossRef] [PubMed]
- Loeff, F.C.; van Egmond, H.M.E.; Nijmeijer, B.A.; Falkenburg, J.H.F.; Halkes, C.J.; Jedema, I. Complement-dependent cytotoxicity induced by therapeutic antibodies in B-cell acute lymphoblastic leukemia is dictated by target antigen expression levels and augmented by loss of membrane-bound complement inhibitors. Leuk. Lymphoma 2017, 58, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Nijhof, I.S.; Casneuf, T.; van Velzen, J.; van Kessel, B.; Axel, A.E.; Syed, K.; Groen, R.W.; van Duin, M.; Sonneveld, P.; Minnema, M.C.; et al. CD38 expression and complement inhibitors affect response and resistance to daratumumab therapy in myeloma. Blood 2016, 128, 959–970. [Google Scholar] [CrossRef] [PubMed]
- You, T.; Hu, W.; Ge, X.; Shen, J.; Qin, X. Application of a novel inhibitor of human CD59 for the enhancement of complement-dependent cytolysis on cancer cells. Cell. Mol. Immunol. 2011, 8, 157–163. [Google Scholar] [CrossRef]
- Takei, K.; Yamazaki, T.; Sawada, U.; Ishizuka, H.; Aizawa, S. Analysis of changes in CD20, CD55, and CD59 expression on established rituximab-resistant B-lymphoma cell lines. Leuk. Res. 2006, 30, 625–631. [Google Scholar] [CrossRef]
- Macor, P.; Tripodo, C.; Zorzet, S.; Piovan, E.; Bossi, F.; Marzari, R.; Amadori, A.; Tedesco, F. In vivo targeting of human neutralizing antibodies against CD55 and CD59 to lymphoma cells increases the antitumor activity of rituximab. Cancer Res. 2007, 67, 10556–10563. [Google Scholar] [CrossRef]
- Macor, P.; Secco, E.; Mezzaroba, N.; Zorzet, S.; Durigutto, P.; Gaiotto, T.; De Maso, L.; Biffi, S.; Garrovo, C.; Capolla, S.; et al. Bispecific antibodies targeting tumor-associated antigens and neutralizing complement regulators increase the efficacy of antibody-based immunotherapy in mice. Leukemia 2015, 29, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.E.; Densmore, J.J.; Pawluczkowycz, A.W.; Beum, P.V.; Kennedy, A.D.; Lindorfer, M.A.; Hamil, S.H.; Eggleton, J.C.; Taylor, R.P. Thrice-weekly low-dose rituximab decreases CD20 loss via shaving and promotes enhanced targeting in chronic lymphocytic leukemia. J. Immunol. 2006, 177, 7435–7443. [Google Scholar] [CrossRef] [PubMed]
- Mamidi, S.; Hone, S.; Teufel, C.; Sellner, L.; Zenz, T.; Kirschfink, M. Neutralization of membrane complement regulators improves complement-dependent effector functions of therapeutic anticancer antibodies targeting leukemic cells. Oncoimmunology 2015, 4, e979688. [Google Scholar] [CrossRef]
- Beyer, I.; Cao, H.; Persson, J.; Wang, H.; Liu, Y.; Yumul, R.; Li, Z.; Woodle, D.; Manger, R.; Gough, M.; et al. Transient removal of CD46 is safe and increases B-cell depletion by rituximab in CD46 transgenic mice and macaques. Mol. Ther. 2013, 21, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Carter, D.; Lieber, A. Protein engineering to target complement evasion in cancer. FEBS Lett. 2014, 588, 334–340. [Google Scholar] [CrossRef]
- Horl, S.; Banki, Z.; Huber, G.; Ejaz, A.; Windisch, D.; Muellauer, B.; Willenbacher, E.; Steurer, M.; Stoiber, H. Reduction of complement factor H binding to CLL cells improves the induction of rituximab-mediated complement-dependent cytotoxicity. Leukemia 2013, 27, 2200–2208. [Google Scholar] [CrossRef]
- Horl, S.; Banki, Z.; Huber, G.; Ejaz, A.; Mullauer, B.; Willenbacher, E.; Steurer, M.; Stoiber, H. Complement factor H-derived short consensus repeat 18–20 enhanced complement-dependent cytotoxicity of ofatumumab on chronic lymphocytic leukemia cells. Haematologica 2013, 98, 1939–1947. [Google Scholar] [CrossRef]
- Winkler, M.T.; Bushey, R.T.; Gottlin, E.B.; Campa, M.J.; Guadalupe, E.S.; Volkheimer, A.D.; Weinberg, J.B.; Patz, E.F., Jr. Enhanced CDC of B cell chronic lymphocytic leukemia cells mediated by rituximab combined with a novel anti-complement factor H antibody. PLoS ONE 2017, 12, e0179841. [Google Scholar] [CrossRef]
- Meri, S.; Pangburn, M.K. Discrimination between activators and nonactivators of the alternative pathway of complement: Regulation via a sialic acid/polyanion binding site on factor H. Proc. Natl. Acad. Sci. USA 1990, 87, 3982–3986. [Google Scholar] [CrossRef] [PubMed]
- Bordron, A.; Bagacean, C.; Mohr, A.; Tempescul, A.; Bendaoud, B.; Deshayes, S.; Dalbies, F.; Buors, C.; Saad, H.; Berthou, C.; et al. Resistance to complement activation, cell membrane hypersialylation and relapses in chronic lymphocytic leukemia patients treated with rituximab and chemotherapy. Oncotarget 2018, 9, 31590–31605. [Google Scholar] [CrossRef] [PubMed]
- Cserhalmi, M.; Papp, A.; Brandus, B.; Uzonyi, B.; Jozsi, M. Regulation of regulators: Role of the complement factor H-related proteins. Semin. Immunol. 2019, 45, 101341. [Google Scholar] [CrossRef]
- Lindorfer, M.A.; Beum, P.V.; Taylor, R.P. CD20 mAb-Mediated Complement Dependent Cytotoxicity of Tumor Cells is Enhanced by Blocking the Action of Factor I. Antibodies 2013, 2, 598–616. [Google Scholar] [CrossRef]
- Felberg, A.; Urban, A.; Borowska, A.; Stasilojc, G.; Taszner, M.; Hellmann, A.; Blom, A.M.; Okroj, M. Mutations resulting in the formation of hyperactive complement convertases support cytocidal effect of anti-CD20 immunotherapeutics. Cancer Immunol. Immunother. 2019, 68, 587–598. [Google Scholar] [CrossRef]
- Kennedy, A.D.; Solga, M.D.; Schuman, T.A.; Chi, A.W.; Lindorfer, M.A.; Sutherland, W.M.; Foley, P.L.; Taylor, R.P. An anti-C3b(i) mAb enhances complement activation, C3b(i) deposition, and killing of CD20+ cells by rituximab. Blood 2003, 101, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Rogers, L.M.; Veeramani, S.; Weiner, G.J. Complement in monoclonal antibody therapy of cancer. Immunol. Res. 2014, 59, 203–210. [Google Scholar] [CrossRef]
- Hsu, Y.F.; Ajona, D.; Corrales, L.; Lopez-Picazo, J.M.; Gurpide, A.; Montuenga, L.M.; Pio, R. Complement activation mediates cetuximab inhibition of non-small cell lung cancer tumor growth in vivo. Mol. Cancer 2010, 9, 139. [Google Scholar] [CrossRef]
- Franssen, L.E.; Stege, C.A.M.; Zweegman, S.; van de Donk, N.; Nijhof, I.S. Resistance Mechanisms Towards CD38-Directed Antibody Therapy in Multiple Myeloma. J. Clin. Med. 2020, 9, 1195. [Google Scholar] [CrossRef]
- Plesner, T.; van de Donk, N.; Richardson, P.G. Controversy in the Use of CD38 Antibody for Treatment of Myeloma: Is High CD38 Expression Good or Bad? Cells 2020, 9, 378. [Google Scholar] [CrossRef]
- Mamidi, S.; Cinci, M.; Hasmann, M.; Fehring, V.; Kirschfink, M. Lipoplex mediated silencing of membrane regulators (CD46, CD55 and CD59) enhances complement-dependent anti-tumor activity of trastuzumab and pertuzumab. Mol. Oncol. 2013, 7, 580–594. [Google Scholar] [CrossRef] [PubMed]
- Clynes, R.A.; Towers, T.L.; Presta, L.G.; Ravetch, J.V. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nat. Med. 2000, 6, 443–446. [Google Scholar] [CrossRef]
- Beers, S.A.; Chan, C.H.; James, S.; French, R.R.; Attfield, K.E.; Brennan, C.M.; Ahuja, A.; Shlomchik, M.J.; Cragg, M.S.; Glennie, M.J. Type II (tositumomab) anti-CD20 monoclonal antibody out performs type I (rituximab-like) reagents in B-cell depletion regardless of complement activation. Blood 2008, 112, 4170–4177. [Google Scholar] [CrossRef] [PubMed]
- Uchida, J.; Hamaguchi, Y.; Oliver, J.A.; Ravetch, J.V.; Poe, J.C.; Haas, K.M.; Tedder, T.F. The innate mononuclear phagocyte network depletes B lymphocytes through Fc receptor-dependent mechanisms during anti-CD20 antibody immunotherapy. J. Exp. Med. 2004, 199, 1659–1669. [Google Scholar] [CrossRef]
- Di Gaetano, N.; Cittera, E.; Nota, R.; Vecchi, A.; Grieco, V.; Scanziani, E.; Botto, M.; Introna, M.; Golay, J. Complement activation determines the therapeutic activity of rituximab in vivo. J. Immunol. 2003, 171, 1581–1587. [Google Scholar] [CrossRef] [PubMed]
- Golay, J.; Cittera, E.; Di Gaetano, N.; Manganini, M.; Mosca, M.; Nebuloni, M.; Van Rooijen, N.; Vago, L.; Introna, M. Complement is required for the therapeutic activity of rituximab in a murine B lymphoma model homing in lymph nodes. Haematologica 2006, 91, 176–183. [Google Scholar] [PubMed]
- Minard-Colin, V.; Xiu, Y.; Poe, J.C.; Horikawa, M.; Magro, C.M.; Hamaguchi, Y.; Haas, K.M.; Tedder, T.F. Lymphoma depletion during CD20 immunotherapy in mice is mediated by macrophage FcγRI, FcγRIII, and FcγRIV. Blood 2008, 112, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, Y.; Uchida, J.; Cain, D.W.; Venturi, G.M.; Poe, J.C.; Haas, K.M.; Tedder, T.F. The peritoneal cavity provides a protective niche for B1 and conventional B lymphocytes during anti-CD20 immunotherapy in mice. J. Immunol. 2005, 174, 4389–4399. [Google Scholar] [CrossRef]
- Overdijk, M.B.; Verploegen, S.; Bogels, M.; van Egmond, M.; Lammerts van Bueren, J.J.; Mutis, T.; Groen, R.W.; Breij, E.; Martens, A.C.; Bleeker, W.K.; et al. Antibody-mediated phagocytosis contributes to the anti-tumor activity of the therapeutic antibody daratumumab in lymphoma and multiple myeloma. mAbs 2015, 7, 311–321. [Google Scholar] [CrossRef]
- Grandjean, C.L.; Montalvao, F.; Celli, S.; Michonneau, D.; Breart, B.; Garcia, Z.; Perro, M.; Freytag, O.; Gerdes, C.A.; Bousso, P. Intravital imaging reveals improved Kupffer cell-mediated phagocytosis as a mode of action of glycoengineered anti-CD20 antibodies. Sci. Rep. 2016, 6, 34382. [Google Scholar] [CrossRef]
- Montalvao, F.; Garcia, Z.; Celli, S.; Breart, B.; Deguine, J.; Van Rooijen, N.; Bousso, P. The mechanism of anti-CD20-mediated B cell depletion revealed by intravital imaging. J. Clin. Investig. 2013, 123, 5098–5103. [Google Scholar] [CrossRef]
- Gul, N.; Babes, L.; Siegmund, K.; Korthouwer, R.; Bogels, M.; Braster, R.; Vidarsson, G.; ten Hagen, T.L.; Kubes, P.; van Egmond, M. Macrophages eliminate circulating tumor cells after monoclonal antibody therapy. J. Clin. Investig. 2014, 124, 812–823. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, G.T. Three major uncertainties in the antibody therapy of cancer. Haematologica 2014, 99, 1538–1546. [Google Scholar] [CrossRef] [PubMed]
- Boross, P.; Jansen, J.H.; de Haij, S.; Beurskens, F.J.; van der Poel, C.E.; Bevaart, L.; Nederend, M.; Golay, J.; van de Winkel, J.G.; Parren, P.W.; et al. The in vivo mechanism of action of CD20 monoclonal antibodies depends on local tumor burden. Haematologica 2011, 96, 1822–1830. [Google Scholar] [CrossRef]
- Gong, Q.; Ou, Q.; Ye, S.; Lee, W.P.; Cornelius, J.; Diehl, L.; Lin, W.Y.; Hu, Z.; Lu, Y.; Chen, Y.; et al. Importance of cellular microenvironment and circulatory dynamics in B cell immunotherapy. J. Immunol. 2005, 174, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Lux, A.; Seeling, M.; Baerenwaldt, A.; Lehmann, B.; Schwab, I.; Repp, R.; Meidenbauer, N.; Mackensen, A.; Hartmann, A.; Heidkamp, G.; et al. A humanized mouse identifies the bone marrow as a niche with low therapeutic IgG activity. Cell Rep. 2014, 7, 236–248. [Google Scholar] [CrossRef][Green Version]
- Gordan, S.; Albert, H.; Danzer, H.; Lux, A.; Biburger, M.; Nimmerjahn, F. The Immunological Organ Environment Dictates the Molecular and Cellular Pathways of Cytotoxic Antibody Activity. Cell Rep. 2019, 29, 3033–3046.e4. [Google Scholar] [CrossRef]
- Mollnes, T.E.; Brekke, O.L.; Fung, M.; Fure, H.; Christiansen, D.; Bergseth, G.; Videm, V.; Lappegard, K.T.; Kohl, J.; Lambris, J.D. Essential role of the C5a receptor in E coli-induced oxidative burst and phagocytosis revealed by a novel lepirudin-based human whole blood model of inflammation. Blood 2002, 100, 1869–1877. [Google Scholar]
- Lee, C.H.; Romain, G.; Yan, W.; Watanabe, M.; Charab, W.; Todorova, B.; Lee, J.; Triplett, K.; Donkor, M.; Lungu, O.I.; et al. IgG Fc domains that bind C1q but not effector Fcgamma receptors delineate the importance of complement-mediated effector functions. Nat. Immunol. 2017, 18, 889–898. [Google Scholar] [CrossRef]
- Verma, M.K.; Clemens, J.; Burzenski, L.; Sampson, S.B.; Brehm, M.A.; Greiner, D.L.; Shultz, L.D. A novel hemolytic complement-sufficient NSG mouse model supports studies of complement-mediated antitumor activity in vivo. J. Immunol. Methods 2017, 446, 47–53. [Google Scholar] [CrossRef]
- Cittera, E.; Leidi, M.; Buracchi, C.; Pasqualini, F.; Sozzani, S.; Vecchi, A.; Waterfield, J.D.; Introna, M.; Golay, J. The CCL3 family of chemokines and innate immunity cooperate in vivo in the eradication of an established lymphoma xenograft by rituximab. J. Immunol. 2007, 178, 6616–6623. [Google Scholar] [CrossRef]
- Betting, D.J.; Yamada, R.E.; Kafi, K.; Said, J.; van Rooijen, N.; Timmerman, J.M. Intratumoral but not systemic delivery of CpG oligodeoxynucleotide augments the efficacy of anti-CD20 monoclonal antibody therapy against B cell lymphoma. J. Immunother. 2009, 32, 622–631. [Google Scholar] [CrossRef] [PubMed]
- Derer, S.; Cossham, M.; Rosner, T.; Kellner, C.; Beurskens, F.J.; Schwanbeck, R.; Lohse, S.; Sina, C.; Peipp, M.; Valerius, T. A Complement-Optimized EGFR Antibody Improves Cytotoxic Functions of Polymorphonuclear Cells against Tumor Cells. J. Immunol. 2015, 195, 5077–5087. [Google Scholar] [CrossRef]
- Wang, S.Y.; Veeramani, S.; Racila, E.; Cagley, J.; Fritzinger, D.; Vogel, C.W.; St John, W.; Weiner, G.J. Depletion of the C3 component of complement enhances the ability of rituximab-coated target cells to activate human NK cells and improves the efficacy of monoclonal antibody therapy in an in vivo model. Blood 2009, 114, 5322–5330. [Google Scholar] [CrossRef]
- Kern, D.J.; James, B.R.; Blackwell, S.; Gassner, C.; Klein, C.; Weiner, G.J. GA101 induces NK-cell activation and antibody-dependent cellular cytotoxicity more effectively than rituximab when complement is present. Leuk. Lymphoma 2013, 54, 2500–2505. [Google Scholar] [CrossRef][Green Version]
- Abes, R.; Gelize, E.; Fridman, W.H.; Teillaud, J.L. Long-lasting antitumor protection by anti-CD20 antibody through cellular immune response. Blood 2010, 116, 926–934. [Google Scholar] [CrossRef]
- Deligne, C.; Metidji, A.; Fridman, W.H.; Teillaud, J.L. Anti-CD20 therapy induces a memory Th1 response through the IFN-gamma/IL-12 axis and prevents protumor regulatory T-cell expansion in mice. Leukemia 2015, 29, 947–957. [Google Scholar] [CrossRef] [PubMed]
- DiLillo, D.J.; Ravetch, J.V. Differential Fc-Receptor Engagement Drives an Anti-tumor Vaccinal Effect. Cell 2015, 161, 1035–1045. [Google Scholar] [CrossRef]
- Erdei, A.; Lukacsi, S.; Macsik-Valent, B.; Nagy-Balo, Z.; Kurucz, I.; Bajtay, Z. Non-identical twins: Different faces of CR3 and CR4 in myeloid and lymphoid cells of mice and men. Semin. Cell Dev. Biol. 2019, 85, 110–121. [Google Scholar] [CrossRef]
- Nijhof, I.S.; Lammerts van Bueren, J.J.; van Kessel, B.; Andre, P.; Morel, Y.; Lokhorst, H.M.; van de Donk, N.W.; Parren, P.W.; Mutis, T. Daratumumab-mediated lysis of primary multiple myeloma cells is enhanced in combination with the human anti-KIR antibody IPH2102 and lenalidomide. Haematologica 2015, 100, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Krejcik, J.; Casneuf, T.; Nijhof, I.S.; Verbist, B.; Bald, J.; Plesner, T.; Syed, K.; Liu, K.; van de Donk, N.W.; Weiss, B.M.; et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood 2016, 128, 384–394. [Google Scholar] [CrossRef] [PubMed]
- van de Donk, N.; Usmani, S.Z. CD38 Antibodies in Multiple Myeloma: Mechanisms of Action and Modes of Resistance. Front. Immunol. 2018, 9, 2134. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.P.; Lindorfer, M.A. Fcgamma-receptor-mediated trogocytosis impacts mAb-based therapies: Historical precedence and recent developments. Blood 2015, 125, 762–766. [Google Scholar] [CrossRef]
- Hogan, K.A.; Chini, C.C.S.; Chini, E.N. The Multi-faceted Ecto-enzyme CD38: Roles in Immunomodulation, Cancer, Aging, and Metabolic Diseases. Front. Immunol. 2019, 10, 1187. [Google Scholar] [CrossRef]
- Bruhns, P. Properties of mouse and human IgG receptors and their contribution to disease models. Blood 2012, 119, 5640–5649. [Google Scholar] [CrossRef] [PubMed]
- Natsume, A.; Shimizu-Yokoyama, Y.; Satoh, M.; Shitara, K.; Niwa, R. Engineered anti-CD20 antibodies with enhanced complement-activating capacity mediate potent anti-lymphoma activity. Cancer Sci. 2009, 100, 2411–2418. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.D.; Beum, P.V.; Solga, M.D.; DiLillo, D.J.; Lindorfer, M.A.; Hess, C.E.; Densmore, J.J.; Williams, M.E.; Taylor, R.P. Rituximab infusion promotes rapid complement depletion and acute CD20 loss in chronic lymphocytic leukemia. J. Immunol. 2004, 172, 3280–3288. [Google Scholar] [CrossRef] [PubMed]
- Beurskens, F.J.; Lindorfer, M.A.; Farooqui, M.; Beum, P.V.; Engelberts, P.; Mackus, W.J.; Parren, P.W.; Wiestner, A.; Taylor, R.P. Exhaustion of cytotoxic effector systems may limit monoclonal antibody-based immunotherapy in cancer patients. J. Immunol. 2012, 188, 3532–3541. [Google Scholar] [CrossRef] [PubMed]
- van der Kolk, L.E.; Grillo-Lopez, A.J.; Baars, J.W.; Hack, C.E.; van Oers, M.H. Complement activation plays a key role in the side-effects of rituximab treatment. Br. J. Haematol. 2001, 115, 807–811. [Google Scholar] [CrossRef]
- Baig, N.A.; Taylor, R.P.; Lindorfer, M.A.; Church, A.K.; LaPlant, B.R.; Pettinger, A.M.; Shanafelt, T.D.; Nowakowski, G.S.; Zent, C.S. Induced resistance to ofatumumab-mediated cell clearance mechanisms, including complement-dependent cytotoxicity, in chronic lymphocytic leukemia. J. Immunol. 2014, 192, 1620–1629. [Google Scholar] [CrossRef]
- Tempescul, A.; Bagacean, C.; Riou, C.; Bendaoud, B.; Hillion, S.; Debant, M.; Buors, C.; Berthou, C.; Renaudineau, Y. Ofatumumab capacity to deplete B cells from chronic lymphocytic leukaemia is affected by C4 complement exhaustion. Eur. J. Haematol. 2016, 96, 229–235. [Google Scholar] [CrossRef]
- Middleton, O.; Cosimo, E.; Dobbin, E.; McCaig, A.M.; Clarke, C.; Brant, A.M.; Leach, M.T.; Michie, A.M.; Wheadon, H. Complement deficiencies limit CD20 monoclonal antibody treatment efficacy in CLL. Leukemia 2015, 29, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Manches, O.; Lui, G.; Chaperot, L.; Gressin, R.; Molens, J.P.; Jacob, M.C.; Sotto, J.J.; Leroux, D.; Bensa, J.C.; Plumas, J. In vitro mechanisms of action of rituximab on primary non-Hodgkin lymphomas. Blood 2003, 101, 949–954. [Google Scholar] [CrossRef]
- Baig, N.A.; Taylor, R.P.; Lindorfer, M.A.; Church, A.K.; Laplant, B.R.; Pavey, E.S.; Nowakowski, G.S.; Zent, C.S. Complement dependent cytotoxicity in chronic lymphocytic leukemia: Ofatumumab enhances alemtuzumab complement dependent cytotoxicity and reveals cells resistant to activated complement. Leuk. Lymphoma 2012, 53, 2218–2227. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R. Fresh frozen plasma as a complement source. Lancet Oncol. 2007, 8, 370–371. [Google Scholar] [CrossRef]
- Klepfish, A.; Schattner, A.; Ghoti, H.; Rachmilewitz, E.A. Addition of fresh frozen plasma as a source of complement to rituximab in advanced chronic lymphocytic leukaemia. Lancet Oncol. 2007, 8, 361–362. [Google Scholar] [CrossRef]
- Xu, W.; Miao, K.R.; Zhu, D.X.; Fang, C.; Zhu, H.Y.; Dong, H.J.; Wang, D.M.; Wu, Y.J.; Qiao, C.; Li, J.Y. Enhancing the action of rituximab by adding fresh frozen plasma for the treatment of fludarabine refractory chronic lymphocytic leukemia. Int. J. Cancer 2011, 128, 2192–2201. [Google Scholar] [CrossRef]
- Tuscano, J.; Poh, C.; Rosenberg, A.; Jonas, B.; Abedi, M.; Barisone, G.; Schwab, E.; Lundeberg, K.; Kaesberg, P. Ofatumumab and Complement Replacement in Relapsed/Refractory Chronic Lymphocytic Leukemia. J. Hematol. 2020, 9, 79–83. [Google Scholar] [CrossRef]
- Beum, P.V.; Kennedy, A.D.; Williams, M.E.; Lindorfer, M.A.; Taylor, R.P. The shaving reaction: Rituximab/CD20 complexes are removed from mantle cell lymphoma and chronic lymphocytic leukemia cells by THP-1 monocytes. J. Immunol. 2006, 176, 2600–2609. [Google Scholar] [CrossRef]
- Beum, P.V.; Lindorfer, M.A.; Taylor, R.P. Within peripheral blood mononuclear cells, antibody-dependent cellular cytotoxicity of rituximab-opsonized Daudi cells is promoted by NK cells and inhibited by monocytes due to shaving. J. Immunol. 2008, 181, 2916–2924. [Google Scholar] [CrossRef]
- Beum, P.V.; Peek, E.M.; Lindorfer, M.A.; Beurskens, F.J.; Engelberts, P.J.; Parren, P.W.; van de Winkel, J.G.; Taylor, R.P. Loss of CD20 and bound CD20 antibody from opsonized B cells occurs more rapidly because of trogocytosis mediated by Fc receptor-expressing effector cells than direct internalization by the B cells. J. Immunol. 2011, 187, 3438–3447. [Google Scholar] [CrossRef]
- Valgardsdottir, R.; Cattaneo, I.; Klein, C.; Introna, M.; Figliuzzi, M.; Golay, J. Human neutrophils mediate trogocytosis rather than phagocytosis of CLL B cells opsonized with anti-CD20 antibodies. Blood 2017, 129, 2636–2644. [Google Scholar] [CrossRef] [PubMed]
- Boross, P.; Jansen, J.H.; Pastula, A.; van der Poel, C.E.; Leusen, J.H. Both activating and inhibitory Fc gamma receptors mediate rituximab-induced trogocytosis of CD20 in mice. Immunol. Lett. 2012, 143, 44–52. [Google Scholar] [CrossRef]
- Beers, S.A.; French, R.R.; Chan, C.H.; Lim, S.H.; Jarrett, T.C.; Mora Vidal, R.; Wijayaweera, S.S.; Dixon, S.V.; Kim, H.J.; Cox, K.L.; et al. Antigenic modulation limits the efficacy of anti-CD20 antibodies: Implications for antibody selection. Blood 2010, 115, 5191–5201. [Google Scholar] [CrossRef] [PubMed]
- Glennie, M.J.; French, R.R.; Cragg, M.S.; Taylor, R.P. Mechanisms of killing by anti-CD20 monoclonal antibodies. Mol. Immunol. 2007, 44, 3823–3837. [Google Scholar] [CrossRef]
- Racila, E.; Link, B.K.; Weng, W.K.; Witzig, T.E.; Ansell, S.; Maurer, M.J.; Huang, J.; Dahle, C.; Halwani, A.; Levy, R.; et al. A polymorphism in the complement component C1qA correlates with prolonged response following rituximab therapy of follicular lymphoma. Clin. Cancer Res. 2008, 14, 6697–6703. [Google Scholar] [CrossRef] [PubMed]
- Charbonneau, B.; Maurer, M.J.; Fredericksen, Z.S.; Zent, C.S.; Link, B.K.; Novak, A.J.; Ansell, S.M.; Weiner, G.J.; Wang, A.H.; Witzig, T.E.; et al. Germline variation in complement genes and event-free survival in follicular and diffuse large B-cell lymphoma. Am. J. Hematol. 2012, 87, 880–885. [Google Scholar] [CrossRef]
- Rogers, L.M.; Mott, S.L.; Smith, B.J.; Link, B.K.; Sahin, D.; Weiner, G.J. Complement-Regulatory Proteins CFHR1 and CFHR3 and Patient Response to Anti-CD20 Monoclonal Antibody Therapy. Clin. Cancer Res. 2017, 23, 954–961. [Google Scholar] [CrossRef][Green Version]
- Song, G.; Song, G.; Ni, H.; Gu, L.; Liu, H.; Chen, B.; He, B.; Pan, Y.; Wang, S.; Cho, W.C. Deregulated expression of miR-224 and its target gene: CD59 predicts outcome of diffuse large B-cell lymphoma patients treated with R-CHOP. Curr. Cancer Drug Targets 2014, 14, 659–670. [Google Scholar] [CrossRef]
- Song, G.; Cho, W.C.; Gu, L.; He, B.; Pan, Y.; Wang, S. Increased CD59 protein expression is associated with the outcome of patients with diffuse large B-cell lymphoma treated with R-CHOP. Med. Oncol. 2014, 31, 56. [Google Scholar] [CrossRef]
- Dzietczenia, J.; Wrobel, T.; Mazur, G.; Poreba, R.; Jazwiec, B.; Kuliczkowski, K. Expression of complement regulatory proteins: CD46, CD55, and CD59 and response to rituximab in patients with CD20+ non-Hodgkin’s lymphoma. Med. Oncol. 2010, 27, 743–746. [Google Scholar] [CrossRef]
- Weng, W.K.; Levy, R. Expression of complement inhibitors CD46, CD55, and CD59 on tumor cells does not predict clinical outcome after rituximab treatment in follicular non-Hodgkin lymphoma. Blood 2001, 98, 1352–1357. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Yang, Y.J.; Zheng, H.; Zhong, X.R.; Wang, Y.; Wang, Z.; Wang, Y.G.; Wang, Y.P. Membrane-bound complement regulatory proteins are prognostic factors of operable breast cancer treated with adjuvant trastuzumab: A retrospective study. Oncol. Rep. 2014, 32, 2619–2627. [Google Scholar] [CrossRef]
- Lindorfer, M.A.; Bakker, P.W.H.I.; Parren, P.W.; Taylor, R.P. Ofatumumab: A next-generation human therapeutic CD20 antibody with potent complement-dependent cytotoxicity. In Handbook of Therapeutic Antibodies; Duebel, S., Reichert, J.M., Eds.; Wiley-VCH: Weinberg, Germany, 2013; pp. 1733–1744. [Google Scholar]
- Goede, V.; Fischer, K.; Busch, R.; Engelke, A.; Eichhorst, B.; Wendtner, C.M.; Chagorova, T.; de la Serna, J.; Dilhuydy, M.S.; Illmer, T.; et al. Obinutuzumab plus Chlorambucil in Patients with CLL and Coexisting Conditions. NEJM 2014, 370, 1101–1110. [Google Scholar] [CrossRef]
- Goede, V.; Fischer, K.; Engelke, A.; Schlag, R.; Lepretre, S.; Montero, L.F.; Montillo, M.; Fegan, C.; Asikanius, E.; Humphrey, K.; et al. Obinutuzumab as frontline treatment of chronic lymphocytic leukemia: Updated results of the CLL11 study. Leukemia 2015, 29, 1602–1604. [Google Scholar] [CrossRef] [PubMed]
- van Imhoff, G.W.; McMillan, A.; Matasar, M.J.; Radford, J.; Ardeshna, K.M.; Kuliczkowski, K.; Kim, W.; Hong, X.; Goerloev, J.S.; Davies, A.; et al. Ofatumumab Versus Rituximab Salvage Chemoimmunotherapy in Relapsed or Refractory Diffuse Large B-Cell Lymphoma: The ORCHARRD Study. J. Clin. Oncol. 2017, 35, 544–551. [Google Scholar] [CrossRef]
- Freeman, C.L.; Sehn, L.H. A tale of two antibodies: Obinutuzumab versus rituximab. Br. J. Haematol. 2018, 182, 29–45. [Google Scholar] [CrossRef] [PubMed]
- Offner, F.; Robak, T.; Janssens, A.; Govind Babu, K.; Kloczko, J.; Grosicki, S.; Mayer, J.; Panagiotidis, P.; Schuh, A.; Pettitt, A.; et al. A five-year follow-up of untreated patients with chronic lymphocytic leukaemia treated with ofatumumab and chlorambucil: Final analysis of the Complement 1 phase 3 trial. Br. J. Haematol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Capuano, C.; Romanelli, M.; Pighi, C.; Cimino, G.; Rago, A.; Molfetta, R.; Paolini, R.; Santoni, A.; Galandrini, R. Anti-CD20 Therapy Acts via FcgammaRIIIA to Diminish Responsiveness of Human Natural Killer Cells. Cancer Res. 2015, 75, 4097–4108. [Google Scholar] [CrossRef] [PubMed]
- Moreno, L.; Perez, C.; Zabaleta, A.; Manrique, I.; Alignani, D.; Ajona, D.; Blanco, L.; Lasa, M.; Maiso, P.; Rodriguez, I.; et al. The Mechanism of Action of the Anti-CD38 Monoclonal Antibody Isatuximab in Multiple Myeloma. Clin. Cancer Res. 2019, 25, 3176–3187. [Google Scholar] [CrossRef]
- Cartron, G.; Watier, H.; Golay, J.; Solal-Celigny, P. From the bench to the bedside: Ways to improve rituximab efficacy. Blood 2004, 104, 2635–2642. [Google Scholar] [CrossRef] [PubMed]
- Balasa, B.; Yun, R.; Belmar, N.A.; Fox, M.; Chao, D.T.; Robbins, M.D.; Starling, G.C.; Rice, A.G. Elotuzumab enhances natural killer cell activation and myeloma cell killing through interleukin-2 and TNF-alpha pathways. Cancer Immunol. Immunother. 2015, 64, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Oostindie, S.C.; van der Horst, H.J.; Lindorfer, M.A.; Cook, E.M.; Tupitza, J.C.; Zent, C.S.; Burack, R.; VanDerMeid, K.R.; Strumane, K.; Chamuleau, M.E.D.; et al. CD20 and CD37 antibodies synergize to activate complement by Fc-mediated clustering. Haematologica 2019, 104, 1841–1852. [Google Scholar] [CrossRef] [PubMed]
- Da Roit, F.; Engelberts, P.J.; Taylor, R.P.; Breij, E.C.; Gritti, G.; Rambaldi, A.; Introna, M.; Parren, P.W.; Beurskens, F.J.; Golay, J. Ibrutinib interferes with the cell-mediated anti-tumor activities of therapeutic CD20 antibodies: Implications for combination therapy. Haematologica 2015, 100, 77–86. [Google Scholar] [CrossRef]
- Evers, M.; Jak, M.; Leusen, J.H.W. The latest developments with anti-CD20 monoclonal antibodies in chronic lymphocytic leukemia. Expert Opin. Biol. Ther. 2018, 18, 973–982. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Name | Target Antigen | Antibody Type | 1st Indication | Year of 1st Approval 1 | Major Mechanism of Action |
|---|---|---|---|---|---|
| Rituximab | CD20 | Chimeric IgG1 | B-NHL | 1997 | CDC, ADCC, ADCP |
| Ofatumumab | CD20 | Human IgG1 | CLL | 2009 | CDC, ADCC, ADCP |
| Obinutuzumab | CD20 | Humaniz. IgG1, Glycoengin. | CLL | 2013 | ADCC, ADCP, PCD |
| Daratumumab | CD38 | Human IgG1 | MM | 2015 | CDC, ADCC, ADCP, neutral. |
| Isatuximab | CD38 | Chimeric IgG1k | MM | 2020 | Neutral. ADCC, ADCP |
| Alemtuzumab | CD52 | Humanized IgG1 | CLL | 2001 | CDC, ADCC, ADCP |
| Elotuzumab | SLAMF7 | Humanized IgG1 | MM | 2015 | ADCC. NK agonist, ADCP |
| Mogamulizumab | CCR4 | Humanized IgG1, low fucose | T leuk/lymph | 2012 Japan 2018 EU | ADCC, ADCP, Treg elimin. |
| Trastuzumab | HER2 | Humanized IgG1 | Breast cancer | 1998 | ADCC, neutral. |
| Pertuzumab | HER2 | Humanized IgG1 | Breast cancer | 2012 | Neutral. (HER2/HER3 dimerization) |
| Cetuximab | EGFR | Chimeric IgG1 | CRC | 2004 | Neutral., ADCC, CDC |
| Panitumumab | EGFR | Human IgG2 | CRC | 2006 | Neutral., PMN mediated ADCC |
| Necitumumab | EGFR | Human IgG1 | NSCLC | 2015 | ADCC, neutral. |
| Dinutuximab | GD2 | Chimeric IgG1 | Neuroblastoma | 2015 | CDC, ADCC, ADCP |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Golay, J.; Taylor, R.P. The Role of Complement in the Mechanism of Action of Therapeutic Anti-Cancer mAbs. Antibodies 2020, 9, 58. https://doi.org/10.3390/antib9040058
Golay J, Taylor RP. The Role of Complement in the Mechanism of Action of Therapeutic Anti-Cancer mAbs. Antibodies. 2020; 9(4):58. https://doi.org/10.3390/antib9040058
Chicago/Turabian StyleGolay, Josée, and Ronald P. Taylor. 2020. "The Role of Complement in the Mechanism of Action of Therapeutic Anti-Cancer mAbs" Antibodies 9, no. 4: 58. https://doi.org/10.3390/antib9040058
APA StyleGolay, J., & Taylor, R. P. (2020). The Role of Complement in the Mechanism of Action of Therapeutic Anti-Cancer mAbs. Antibodies, 9(4), 58. https://doi.org/10.3390/antib9040058