1. Introduction
Typical monoclonal antibodies (mAbs) with specificity towards a target antigen are composed of heavy (HC) and light (LC) chains containing conserved and variable regions. Previously, heavy- chain only antibody (HCAb) formation was reported to occur in various species with significant human therapeutic potential [
1]. Camelids are long known to express functional HC-only antibodies that are composed of a homodimeric V
HH domain [
2,
3]. Further, sharks produce functional heavy-chain only antibodies, that like camelid antibodies, are smaller in nature, and formed the basis of nanobody technology [
4,
5]. Like camelid V
HH domains and shark nanobodies, both lacking C
H1 and LC domains, HCAbs have been reported to be secreted in LC-deficient mice lacking the C
H1 domain [
6]. Separately, hybrid llama/human antibody HCAbs, lacking the C
H1 domain and having swapped the llama V
HH regions with human V
H, have been reported [
7]. In addition, HC-only transcripts, lacking the C
H1 domain and in the absence of LC, can be expressed on the cell surface of mammalian pro-B cells [
8]. What is noteworthy here with these examples of HC-only antibodies found in camelids, sharks, LC-deficient mice, and mammalian pro-B cells is that the presence of these molecules does not contradict the longstanding views on antibody mAb or Fab assembly, where LC assembly to the HC, or in particular to the C
H1 domain, is required for C
H1 domain folding and dissociation from the molecular chaperone BiP [
9,
10,
11]. Interestingly, it has been reported that full- length HC-only antibody dimers are formed from a stable Drosophila cell line via a BiP mediated pathway [
12]. This observation challenges the long-held hypothesis that the unfolded C
H1 domain in complex to the molecular chaperone BiP requires association with LC to fold and release BiP chaperone, enabling export and secretion. Nonetheless, the formation of full-length HC-only antibodies is uncommon, and aside from the normal requirement of the LC to bind chaperoned C
H1 and release BiP, additional mechanisms may be required to neutralize their potential toxicity in the absence of LC as previously reported in plasma cells [
13].
Human HCAbs have only recently been reported by Stoyle and coworkers to occur from transient Chinese Hamster Ovary (CHO) expression [
14]. Like antibody producing B cells, CHO cells have a similar quality control system and mechanism of antibody assembly, utilizing BiP, prolyl isomerases, and disulfide reductases [
15]. Therein, HCAbs containing the constant C
H1 and V
H regions humanized from rodent sources were found to form homodimers and be secreted even in the absence of light chain. These HC dimers were found to form from both HC/LC cotransfected cells and HC-only transfection, and both full-length HC dimers and HC dimers lacking the Fc domain (V
H + C
H1 only) were able to form. The LC-independent secretion of HC dimers was inferred to be variable region dependent since only certain HCs were able to form and be secreted as folded molecules. One characteristic noted for some of the molecules being able to form HC2s was the increased number of positively charged amino acids in the HC-CDR3 [
14].
Although Stoyle and coworkers clearly showed that both full-length heavy-chain dimers (HC2s) and HCs lacking the Fc domain (analogous to the Fab domain, herein described as “Fd2”) are able to be formed and secreted in the absence of LC, it is not clear what their mechanism of assembly is, nor is it clear what are the sequence and structural determinants that drive their formation. Also lacking are biophysical and structural characterizations of these novel molecules. The formation of human HC2s is unique and offers extraordinary insights into antibody assembly, as well as potentially enabling different, novel antibody formats with unique advantages and properties. One potential advantage is that common LC mispairing in multi-specific formats can be avoided. However, without further understanding their biophysical properties and how structural and sequence elements impact their formation, engineering and modulating HC dimers into a novel potential modality cannot be achieved. Aside from assumed differences in the HC variable sequence that impact HC2 formation, the role of the LC is also not well understood and how this competing pathway to normal HC-LC assembly could impact HC-HC (HC2) assembly.
Herein, we report robust expression and formation of human HC2s for two different humanized antibodies with unrelated variable sequences against different biological targets. Robust formation of HC2 impurities originally constituted a severe, negative mAb developability attribute. In HC/LC cotransfected transient CHO cells, HC2 formation was robust and prevented further developability of these clones as acceptable purity/heterogeneity of the desired mAb species was unattainable under representative process conditions. To characterize further these novel and unique “impurities”, we performed a series of HC-only transfections and characterized the secreted and purified HC dimers. For both full-length and Fd2 versions, we found these molecules can achieve high purity, and to have similar or better expression titer, thermal stability, and accelerated storage stability than their mAb and Fab counterparts, respectively. Additionally, we solved the first ever crystal structure at 2.9 Angstroms (Å) of a human HC homodimer (herein named Fd2-A) that reveals exposed CDR regions and a symmetrical dimerization complex analogous to HC-LC association in Fabs, where one opposing HC (CH1 + VH or Fd) replaces the LC. Disulfide formation is overall conserved between this Fd2 complex in comparison to the Fd domain in Fabs. Mutagenesis of key, variable CDR residues in the dimerization interface indicated by the novel Fd2-A crystal structure reduced full-length HC dimer formation. Similar mutations were made for a second recombinant antibody based on a sequence alignment to Fd2-A and similar HC2 dimer reduction was achieved, suggesting that this heavy-chain only dimer structure is conserved for two antibody sequences with different germlines against two different biological targets. These results indicate that we have solved a novel crystal structure representing the human HC dimer (Fd2) structure, provided new insights into their formation, structural and sequence determinants, and demonstrated that their application as a novel and functional antibody format is realistic.
2. Materials and Methods
2.1. Transient Protein Expression
For the small-scale protein production, transient transfections were done in TubeSpin® bioreactors (TPP Techno Plastic Products AG, Trasadingen, Switzerland) using the ExpiCHO Expression System (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocol. Briefly, the cells were grown and maintained in ExpiCHO Expression Medium (Thermo Fisher Scientific, Waltham, MA, USA) and seeded in 10 mL of media at 6 × 106 cells/mL on the day of transfection. Complexes were formed with 8 ug of DNA and 32 μL of Expifectamine in OptiPRO™ SFM and incubated for 1 min followed by addition to the cells. The transfected cultures were grown at 37 °C, 5% CO2, 80% humidity, and 300 rpm rotation in a Multitron incubator (Infors HT, Basel, Switzerland) and then shifted to 32 °C 24 h post-transfection and were fed with feed and enhancer on days 1 and 5. Expression variables include no enhancer treatment (Thermo), pulling culture on Day 4, using 20% of coding DNA by weight, transfecting with 5X LC by molar mass, and using a 10 mL shake flask instead of tubespin cultures. The cultures were harvested on day 7, the cells were pelleted by centrifugation, and the supernatant was passed through a 0.2 micron filter. The protein titers were determined using a ForteBio Octet (Molecular Devices, LLC. San Jose, CA, USA) with Protein A sensors and a purified mAb to generate the standard curve.
For large-scale protein production, transient transfections were performed in 1 L shake flasks using the ExpiCHO Expression System (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocol. Transfections and expression conditions were the same as in small-scale format. The cultures were harvested on day 7, the cells were pelleted by centrifugation, and the supernatant was passed through a 0.2 micron filter. The protein titers were determined using a ForteBio Octet (Molecular Devices, LLC. San Jose, CA, USA) with Protein A sensors and a purified mAb to generate the standard curve.
2.2. Small-Scale Protein Purification
The clarified cell culture supernatants were loaded onto a Tecan Freedom EVO 200 (Tecan Life Sciences, Männedorf, Switzerland) for antibody purification utilizing miniature columns manufactured by Repligen (Waltham, MA, USA) and packed with MabSelect™ SuRe™ LX (GE Healthcare Life Sciences, Pittsburgh, PA, USA). The antibodies were eluted with 20 mM sodium acetate at pH 3.5 and immediately neutralized with 0.33 M Tris, 1 M sodium acetate pH 8.0 and buffer exchanged into 20 mM sodium acetate pH 5.5 using 10K MWCO Slide-A-Lyzer dialysis cassettes (Thermo Fisher Scientific, Waltham, MA, USA).
2.3. Large-Scale mAb and Full-Length HC Dimer Purification
Cell culture supernatant was incubated with Protein A (ProA) affinity MabSelect SuRe LX resin (GE Healthcare, Pittsburgh, PA, USA) overnight (1 mL resin for 40 mg target antibody or HC dimer estimated by ForteBio Octet) for batch binding. Affinity resin with bound protein was collected by filtration and transferred to a disposable column. Bound resin was washed with 20X column volumes of 1X Gibco Phosphate buffered saline (PBS) (Thermo Fisher Scientific, Waltham, MA, USA) in batch mode. Desired protein was eluted by approximately 5 column volumes of 20 mM sodium acetate pH 3.5 buffer. Eluate was immediately buffer exchanged into 20 mM sodium acetate pH 5.5 buffer using 10K MWCO Slide-A-Lyzer dialysis cassettes. To further polish material for analytical and biophysical characterization efforts (DSC, SEC-MALS, LC-MS), desired product was purified to >98% purity (by SE-UPLC and cSDS) on a Superdex 200 Increase 10/300GL column (GE Healthcare Bio-Science AB, Uppsala, Sweden). Mobile phase was 20 mM sodium acetate, 200 mM sodium chloride pH 5.5 buffer. All purified protein was buffer exchanged overnight using 10K MWCO Slide-A-Lyzer dialysis cassettes into 1X Gibco PBS pH 7.4 and normalized to 1 mg/mL for characterization. Concentration was determined by UV absorbance at 280 nm on a Nanodrop 2000 1- position Spectrophotometer (Thermo Scientific).
2.4. Fab and Fd Dimer Purification
Cell culture supernatant was incubated with CaptureSelect IgG-CH1 affinity matrix (Thermo Scientific) overnight for batch binding (1 mL resin for 10 mg protein estimated by ForteBio Octet). Resin was collected by filtration, transferred to a disposable column, and then washed with 20 column volumes of 1X Gibco PBS in batch mode. Desired protein was eluted with 5 column volumes of 0.1 M glycine pH 3.0 buffer. Eluate was immediately buffer exchanged into 20 mM sodium acetate pH 5.5 buffer. To further polish material for biophysical characterization and crystallization efforts, desired product was purified to >98% purity (by SE-UPLC and cSDS) on a Superdex 200 increase 10/300GL (GE Healthcare Bio-Science AB, Uppsala, Sweden) column. Mobile phase was 20 mM sodium acetate, 200 mM sodium chloride pH 5.5 buffer. All purified protein was buffer exchanged overnight into 1X Gibco PBS pH 7.4 and normalized to 1 mg/mL for characterization. For crystallization efforts, Fd2 for Molecule A was reformulated into 20 mM sodium acetate pH 5.5 (low salt) and concentrated to 20 mg/mL using a 4 mL Vivaspin™ ultrafiltration spin column with 10K MWCO membrane. Purity was verified to be unchanged following concentration by SE-UPLC.
2.5. Size-Exclusion Ultra-Pressure Liquid Chromatography (SE-UPLC)
A Waters Acquity UPLC H-Class PLUS system (Waters, Milford, MA, USA) was used to separate molecules based on differences in their hydrodynamic size. Samples (10 μg) were injected into an Acquity BEH200 SEC column (Waters, Milford, MA, USA) and eluted at a 0.5 mL/min flow rate. Mobile phase contained 100 mM sodium phosphate and 200 mM sodium chloride pH 7.0. Waters BEH200 SEC protein standard mix (Waters, Milford, MA, USA) was used as molecular weight marker and for column quality control purposes. Samples were detected by UV absorbance at 280 nm. Chromatograms were integrated manually and reported as a % of integrated area for each species.
2.6. Capillary Sodium Dodecyl-Sulfate Electrophoresis (cSDS)
A LabChip GXII Clipper (Perkin Elmer, Waltham, MA, USA) was used to determine purity and approximate molecular weight under non-reduced and reduced conditions. All sample and chip preparation was done according to manufacturer’s protocol using the Protein Express Assay reagent kit (Perkin Elmer). Five μL of sample was mixed with 35 μL of reduced Protein Express Sample Buffer (35 mM Dithiothreitol or DTT added to kit sample buffer) or 35 μL non-reduced sample buffer (35 mM iodoacetamide added to kit sample buffer). An HT Protein Express Assay LabChip (Perkin Elmer) was primed using the Protein Express Assay Reagent kit (Perkin Elmer). LabChip GX Reviewer software was used for data analysis. Chromatograms were integrated manually and reported as % for each species. This method produces system or method-related peaks that appear at before 10 KDa in size and include reagents and the internal 10 KDa molecular weight standard.
2.7. Size Exclusion Chromatography Coupled to Multi-Angle Light Scattering (SEC-MALS)
A µDAWN (Wyatt Technology, Santa Barbara, CA, USA) was coupled online to a Waters UPLC H-Class system (Waters, Milford, MA, USA) to measure molecular weight (Mw) using static light scattering. Light scattering wavelength used was 650 nm. The concentration detector was an online uTrex equipped with a refractive index detector. Dn/Dc value used in Mw calculations was 0.185 mL/g. PBS was used as the mobile phase and 20 μg samples were injected. Other SE-UPLC running conditions used are described here in the SE-UPLC methods section. Bovine serum albumin (Thermo Scientific) was used as an isotropic standard for molecular weight normalization. Astra 7 software (Wyatt Technology, Santa Barbara, CA, USA) was used for data acquisition and analysis. Detector alignment, band broadening, and normalization coefficient parameters were set to achieve a BSA Mw within 5% of 66,500 Da.
2.8. Differential Scanning Calorimetry (DSC)
A MicroCal PEAQ-DSC (Malvern Panalytical, Malvern, UK) was used to measure protein melting temperatures. An amount of 500 μL of protein solution for each sample at 1 mg/mL in 20 mM sodium acetate pH 5.5 was added to a 96-well 500 μL volume plate (Wheaton). Temperature was ramped from 25 °C to 95 °C at 1 °C per min. Origin 7 software was used for data acquisition and analysis where melting transition temperatures (Tm) were fitted using a non-two state algorithm.
2.9. Nanoscale-Differential Scanning Fluorimetry (Nano-DSF)
All nano-DSF studies were performed using the Nanotemper Prometheus NT.48 instrument and data analysis software. Samples were evaluated at 1 mg/mL in 20 mM sodium acetate pH 5.5. Samples were introduced by capillary action into glass capillaries (Prometheus) prior to placing into the instrument capillary holder. Temperature was ramped from 20 °C to 95 °C at 1 °C/minute. Thermal melting transitions (Tm) were measured by identifying changes in inflection of intrinsic fluorescence intensity ratios (F350 nm/F330 nm) at specific temperatures.
2.10. Surface Plasmon Resonance (SPR) Affinity Measurements by BIAcore
Binding kinetics of the mAbs to the target was determined by SPR on a BIAcore T200 (GE Healthcare, Chicago, IL, USA). The running buffer, 10 mM HEPES, 150 mM NaCl, 0.05% (v/v) Surfactant P20, 3 mM EDTA, pH 7.4 (HBS-EP+, GE Healthcare), was used for immobilization and reagent dilutions. All binding kinetics were measured at 25 °C. For each injection cycle, mAbs were first captured in different flow cells with an anti-human Fc antibody (Human Antibody Capture Kit, GE Healthcare) immobilized to the sensor chip (Series S CM5, GE Healthcare). Reference flow cell with no captured mAb was also used. Serial dilutions (1:2) of the target protein, ranging in concentration from 1 μM to 32 μM, and buffer blanks were injected in multiple cycles over the captured mAbs and reference surfaces for a 1-min association followed by a 3-min dissociation. The surfaces were regenerated with a 30 s injection of 3 M MgCl2 after each cycle. Double-referenced titration data was globally fit to a 1:1 Langmuir binding model to determine the association rate constant, ka (M−1 s−1), and the dissociation rate constant, kd (s−1), using the BIAcore T200 Evaluation Software version 2.0 (GE Healthcare). The equilibrium dissociation constant was calculated as KD (M) = kd/ka.
2.11. Liquid Chromatography Mass Spectrometry (LC-MS or Intact Mass)
Sample was diluted to 0.2 mg/mL with 50 mM ammonium bicarbonate and 4 μL was injected to a POROS R2/10 2.1 × 30 mm Column (Life Technologies, Carlsbad, CA, USA, 1-1112-12). A gradient from 30 to 58% Buffer B (acetonitrile, 0.1% formic acid) in Buffer A (water, 0.1% formic acid) was applied to the chromatographic column with flow rate of 100 μL/min. Data was acquired on a Waters Synapt G2-S Mass Spectrometer and deconvoluted to monoisotopic and singly-charged using the Waters MassEnt 1 software.
2.12. Storage Stability Study
All samples were formulated in 20 mM sodium acetate pH 5.5 at 5 mg/mL. Solutions were filtered through a Millex-GV 0.2 μM PVDF 33 mm syringe filter (Millipore, Burlington, MA, USA) and aliquoted into 2 mL screw-top microcentrifuge tubes (Fisher). Samples subjected to accelerated storage were placed into a temperature-controlled stability chamber (Thermo Scientific) at 40 °C. Samples were pulled at specific timepoints and then analyzed for purity by SE-UPLC and cSDS.
2.13. Crystallization and Data Collection
Crystallization plates were set up in 3 sub-well plates (Intelli, Art Robbins) by vapor diffusion using Mosquito (TTP Labtech, Boston, MA, USA) at 4, 18, and 30 °C, and images were acquired using RockImager 1000 (Formulatrix Bedford, MA, USA). Crystals appeared in well of H7 of JCSG Plus Screen (Molecular Dimensions, Maumee, OH, USA) within a few hours and were fully grown after 3 days. Crystals (length, 80–200 μm) were present in condition H7 (0.2 M ammonium sulfate, 0.1 M Bis-Tris pH 5.5, 25%
w/v PEG 3350) in 1:1, 2:1 and 1:2 protein to precipitant ratio in 200 nL drops. Further optimization of condition resulted in optimal crystal in 2:1 protein to precipitant ratio at 30 °C of 200 nL drops. Crystals were cryo-protected in reservoir solution supplemented with 5% glycerol and flash-cooled in liquid nitrogen. We noticed that crystals harvested after 2–3 days resulted in optimal diffraction. Data collection was performed at the Industrial Macromolecular Crystallography Association (IMCA) beam line, sector 17 of the Advanced Photon Source (APS) at the Argonne National Laboratory (ANL, Lemont, IL, USA). Data were collected at a wavelength of 1.0 Å using a Pilatus 6M detector (Dectris A G, Baden Dättwil, Switzerland). The data were processed using the autoPROC [
16,
17] automated processing software. AutoPROC utilizes XDS for indexing and integration and, AIMLESS for scaling, POINT LESS for data analysis, and STARANISO for applying anisotropic diffraction limits.
2.14. Structure Determination and Model Building
The structure was solved by Molecular Replacement using MOLREP [
18] and Phaser [
19]. The partial model was further extended by AutoBuild [
20]. The structure was then refined using autoBUSTER [
21] and phenix.refine [
22]. The initial maps had poor density for several regions, including some of the CDR-like loops, which were removed from the model and gradually rebuilt during refinement. The electron density map was consistent with most sequence substitutions and insertions or deletions between the starting molecular replacement model and the final structure. The sequence was manually corrected using COOT [
23]. The resulting structure was refined using Phenix and rebuilt several times leading to final values of R
free and R
work. The final model contained 3 dimers in the asymmetric unit.
2.15. Constructs Used, Sequence Alignment, and Numbering
All mAbs, Fabs, HC2s, and Fd2 molecules used in this study were prepared by gene synthesis. Full length mAb versions contained all residues within the VH and CH domains. mAbs A and B were humanized from mouse and rat immunization campaigns, respectively. For full-length HC2 molecules, the entire HC was used without alterations or truncations. Mutated HC2 molecules were prepared by site-directed mutagenesis. For design of Fabs and Fd2s, engineered IgG1 HC constructs were terminated just prior to the hinge region (or residues 1-224 for Fab-A and Fd2-A and residues 1-235 for Fab-B and Fd2-B) ending in the conserved sequence THT. For the LC, the full-length LC sequence was used (residues 1-219 for Fab-A and residues 1-213 for Fab-B). Both LC sequences terminated in the conserved cysteine involved in the HC:LC intermolecular disulfide. Sequences were aligned and CDRs annotated using proprietary AbacusTM Antibody & Engineering Analysis software using standard pre-sets and reference antibody sets. Default numbering referenced throughout the text is based on sequential numbering, with the exception of the sequence alignments generated by AbacusTM, where default sequential numbering was used throughout the alignment by the software.
4. Discussion
Herein, we structurally and biophysically characterized unexpectedly occurring human heavy- chain only antibodies arising directly from transient CHO expression. We found these non- artefactual HC-only antibodies to be homodimeric in nature, with an extended interface joining both CH1 and VH domains, and to be linked by a conserved intermolecular disulfide bond similar to how Fabs are linked. Overall, these HC dimers are efficiently expressed with or without the LC and conserved Fc domain. Further, these HC-only molecules have similar or better accelerated storage stability characteristics and thermal stability compared to their mAb or Fab counterparts. Further, the HC dimer is structurally analogous to the Fab, and this is an interesting result that has implications concerning its mechanism of assembly and its potential application as a novel antibody format. The solved crystal structure and structure-guided, rational mutagenesis to the dimer interface reveals for the first time structural and sequence determinants to human heavy-chain only antibody formation.
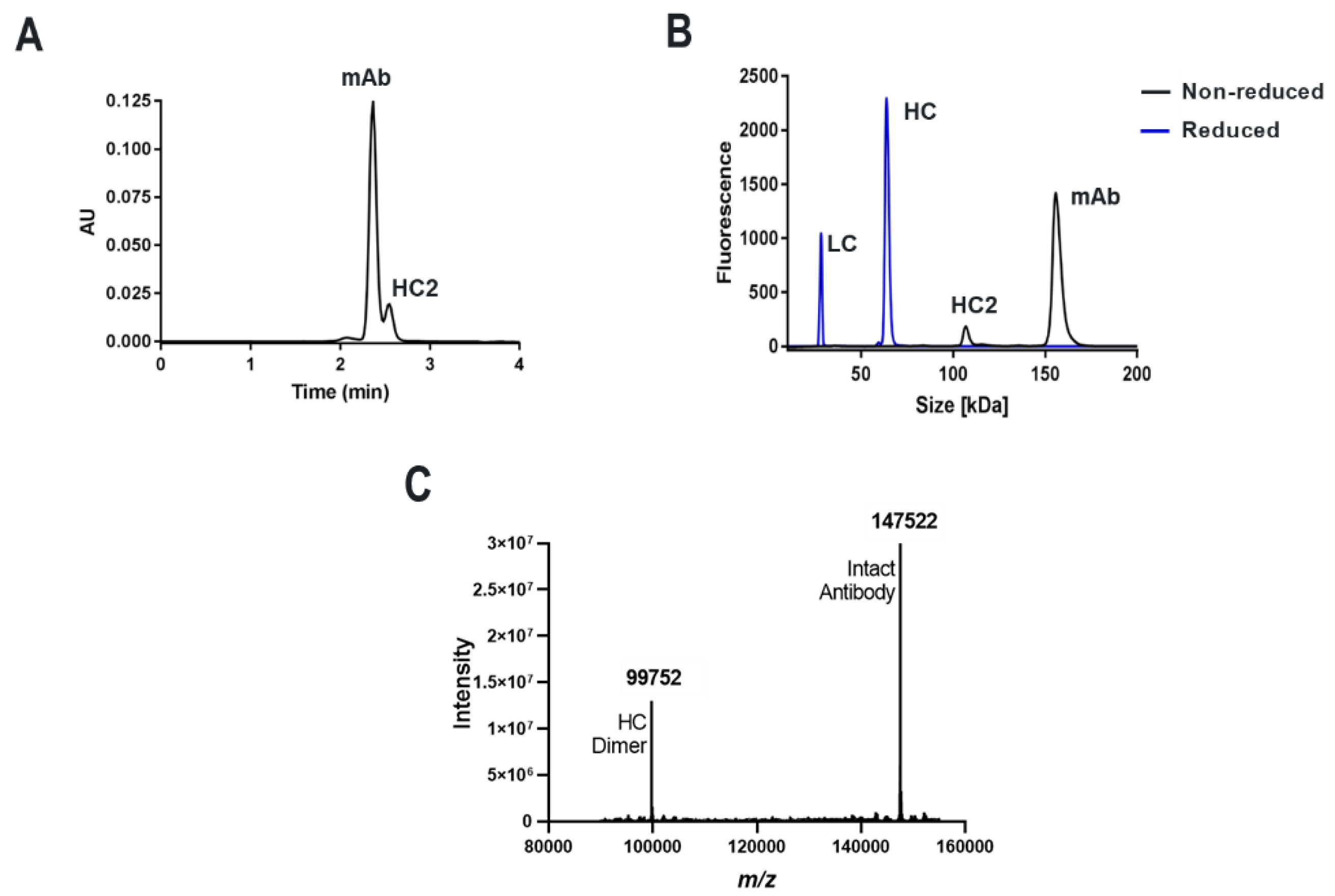
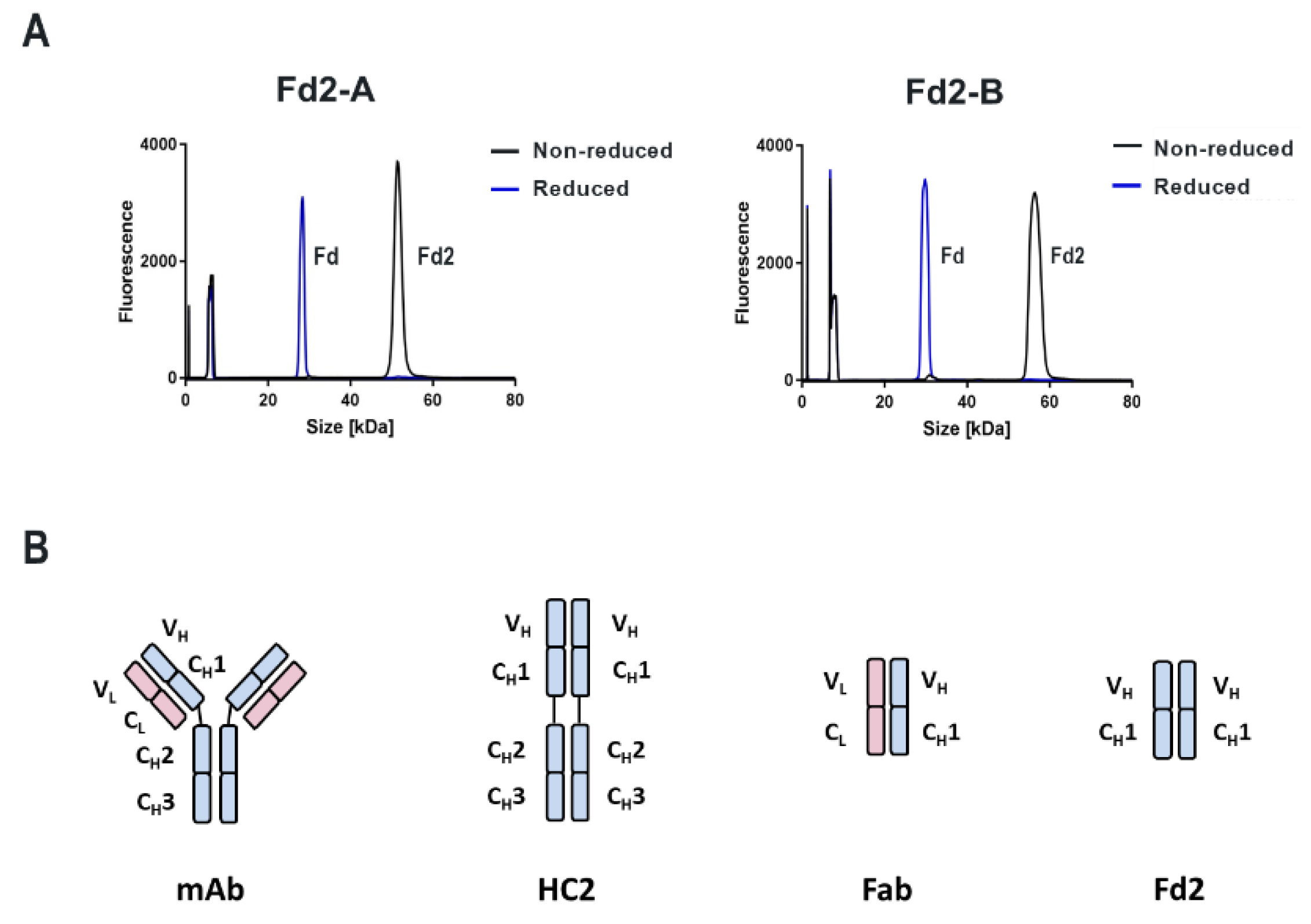
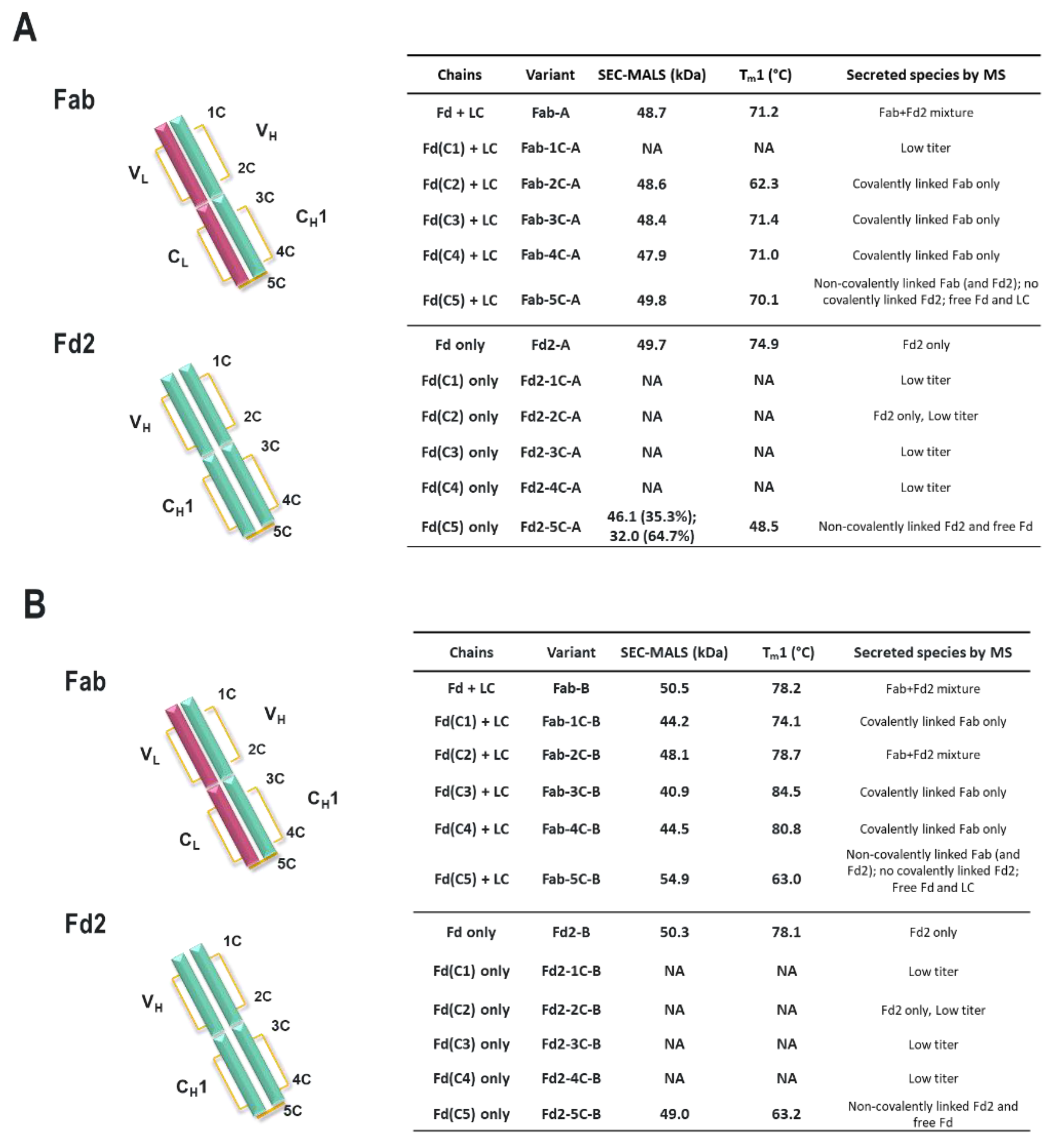
For both mAbs A and B presented in this study, which are two humanized and functional recombinant antibodies with different germlines (HV1/KV2D and HV4/KV1 respectively) against different and unrelated biological targets, HC-only expression yields stable HC dimers (HC2s). These HC2s are reducible into monomeric components as demonstrated by denaturing R-cSDS of Fd2 forms and NR-cSDS of C219S-Fd2 forms (lacking the intermolecular disulfide, see
Figure 2 and
Figure 4). In
Table 1, we see that expression variables and isotype do not eliminate or significantly impact HC dimer formation for molecule A (HC2-A), and HC dimer remains robust, although varied, across conditions. The only exception to this is with the addition of 5X transfected LC, which can effectively eliminate HC2-A formation but was not considered to be a viable downstream process solution.
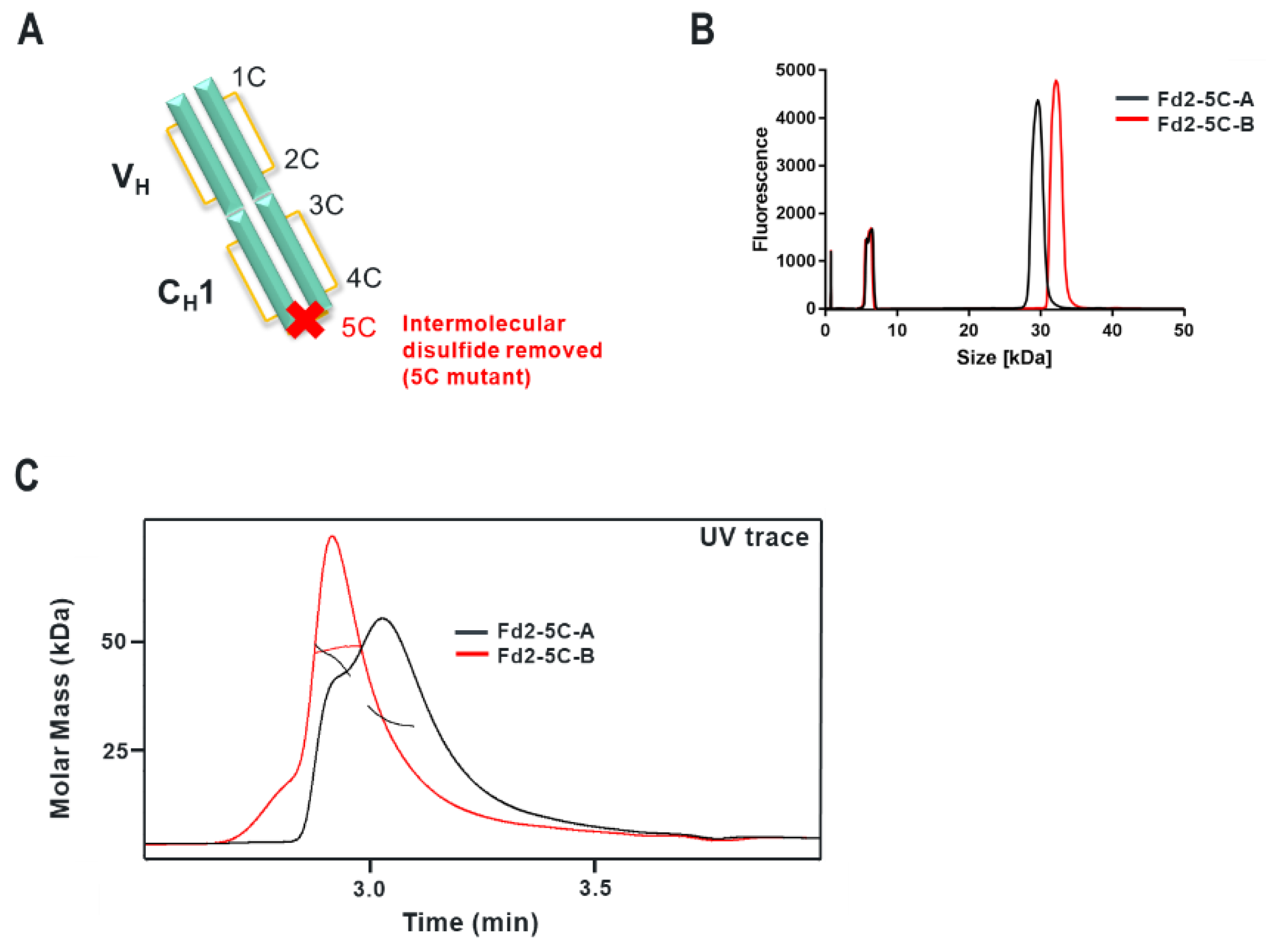
Specifically, the HC cysteine involved in the intermolecular disulfide bond connecting each Fd domain in the Fd2 molecule is the same HC cysteine that disulfide links the Fd and LC together in Fabs. Interestingly, when the Fd-Fd (no Fc and hinge disulfides present) intermolecular disulfide bond is broken through C219S/C230S mutagenesis for Fd-A and FD-B respectively, these HC (Fd) dimers are still able to be expressed and yield molecules that are partially or predominantly non- covalently dimerized in solution at low μM concentrations. This was demonstrated by native SEC-MALS, suggesting there is a real dimerization interface with low μM affinity. This result also justified crystallographic studies in an attempt to structurally reveal the nature of this dimerization interface. This same experiment was conducted for Fab expression (HC or Fd +LC), however, since a mixture of non-covalent Fab and Fd2 was formed, it is difficult to parse out comparisons between non-covalent Fd2 and Fab properties.
Interestingly, while both IgG1 mAbs A and B efficiently yield HC dimer both in the full-length and Fd2 (without Fc) forms from HC-only CHO transient transfection, in HC/LC cotransfected CHO transient expression, HC2-B clearly is more efficiently expressed than HC2-A (46.2% vs. 4.9% in control tubespin cultures). While we deemed both of these mAb molecules to have huge developability risks and concerns due to robust HC dimer formation, mAb-B HC/LC transfection almost equally expressed HC2-B versus mAb-B. By SEC-MALS analysis, C230S-Fd2-B was also predominantly a non-covalently intact Fd2 dimer, whereas C219S-Fd2-A was predominately monomeric with some non-covalent dimer present. Considering the amount injected (20 μg) on the SE-UPLC, the measured UV signal, and the volume over which the C219/230S-Fd2-A/B analytes eluted (0.25–0.5 mL), these dimeric Fd2 species were detected by SEC-MALS in the 10–20 μM range. This same C230S-Fd2-B species had a higher Tm1 (63 °C) than did C219S-Fd2-A (49 °C), although this difference may have been due to the instability of the predominant monomer form for C219S-Fd2-A at low μM concentrations. Empirically, it is evident that mAb-B had significantly higher levels of expressed HC2 in the presence of LC than did mAb-A and it also qualitatively has a stronger dimer interface in the solution state as suggested by SEC-MALS data. The stronger dimer interface present in Fd2-B versus Fd2-A may explain why Fd2-B forms more robustly in the presence of LC, where apparently HC2 or Fd2 formation is seemingly in direct competition with mAb and Fab formation and is more favored if the Fd2 interface is strengthened. Conversely, when mutations were made to the dimer interface elucidated by the novel Fd2 crystal structure that were predicted to weaken the dimer interface for both HC2-A and HC2-B, HC dimer levels decreased in the presence of LC expression. Therefore, these dimer interface mutants selectively decreased HC dimer formation versus competing HC-LC formation as originally hypothesized.
Conversely, if HC-LC pairing were strengthened or weakened, this should lower or increase HC dimer formation, respectively. When we express mAb-A in the presence of 5X LC, mAb expression dominates and we do not obtain any measurable amounts of HC2-A. This is a clear indication that mAb assembly outcompetes potential HC2 assembly when LC is expressed in vast excess. Moreover, when we evaluate HC/LC germline pairing frequencies, we see that uncommon germlines may increase the likelihood of HC2 formation (
Table 2). In a publication by Jayaram and coworkers, 358 HC/kappa-LC human antibody pairings were evaluated, and specific germline frequencies were tabulated [
25]. Their findings suggest that germline pairings are not random and that common germline pairings may be attributable to increased stability of the V
H/V
L interface. In mAb-A, the HC and LC germlines are the result of a more uncommon germline pairing, HV1/KV2D, where only four instances out of 358 occurred. However, this uncommon pairing may not be statistically significant when considering that KV2D itself is infrequently encountered. Likewise, mAb-B also has an uncommon germline pairing, HV4/KV1 (nine out of 358 instances). When 11 additional human/humanized recombinant IgG1 and IgG4 mAbs against different targets were evaluated, nine had more common pairings (>20 instances each) and none formed HC dimers appreciably. Although our panel of molecules is limited and anecdotal, our findings may indicate germline pairing preferences play a role in enabling HC dimer formation and also explain their uncommon occurrence. However, clearly there are other intrinsic factors to HC dimer formation which are presumably sequence/structurally dependent. The heavy-chain domain still needs to be formed, folded, and result in a reasonably stable product independent of the LC, a characteristic unique to a certain subset of human HC sequences.
Another example or factor in HC2 formation may include the length of the HC-CDR3, where HC2-B has an unusually long HC-CDR3 of 19 residues (See
Figure 8). HC-CDR3 length and composition can have a huge impact on the developability properties of a mAb [
29]. Previously, HC-CDR3 length has correlated with negative developability attributes such as aggregation and high viscosity, where 137 clinical-stage antibody therapeutics had a median HC-CDR3 length of 12 residues [
30]. For HC/LC transfection of mAb-B, HC2 formation (46.2%) was nearly equal to normal mAb assembly (See
Table 2). Stoyle and coworkers reported the number of basic residues to be a determinant of HC2 expression, where an increase in basicity of the HC-CDR3 increased HC2 propensity [
14]. However, no basic residues are observed in the HC-CDR3 for HC2-B. In the case of HC2-A, a relatively short HC-CDR3 of seven residues exists, with only one basic residue present. Clearly, HC2 formation may depend on other complex and unknown antibody sequence and structural factors.
It is well known that the molecular chaperone protein, BiP, binds non-covalently to the HC, but not to the HC associated with LC [
31]. During mAb assembly, HC-LC pairing and folding in the Fab arm occurs following BiP release from unfolded HC (C
H1 + V
H or Fd) as a result of LC assembly [
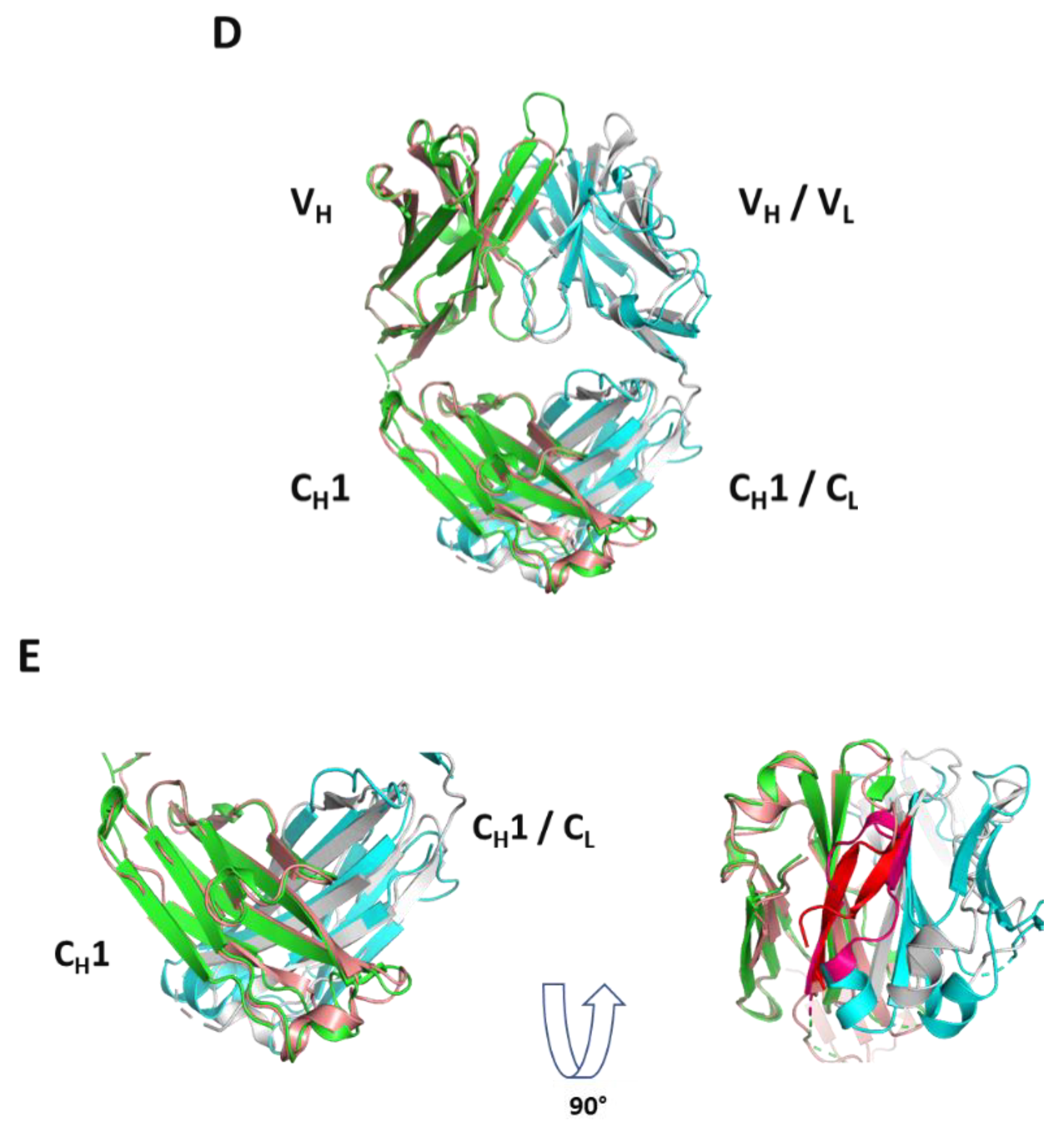
32]. Here in the case of molecules A and B, HC dimer formation occurs with or without the presence of LC, implying that perhaps one Fd domain mimics the LC, and can promote folding of another Fd domain presumably associated to BiP. Remarkably, as depicted in
Figure 6D, the novel HC2 or Fd2 complex reported herein overlays nicely with a representative Fab structure with a low overall Cα RMSD of 1.10 Å, revealing that the opposing HC, or Fd, likely mimics the LC structurally during assembly and promotes complete folding and assembly of HC2s/Fd2s. While it is known that HC dimers are common degradant products of formulated mAb (HC2-LC2) drug products [
33], resulting from the loss of two LC domains, these HC2s described herein form during expression without LC, and are secreted and captured as fully folded and stable products. Indeed, we show that these novel HC-only antibodies are highly stable with T
ms similar or better than their mAb or Fab versions (see
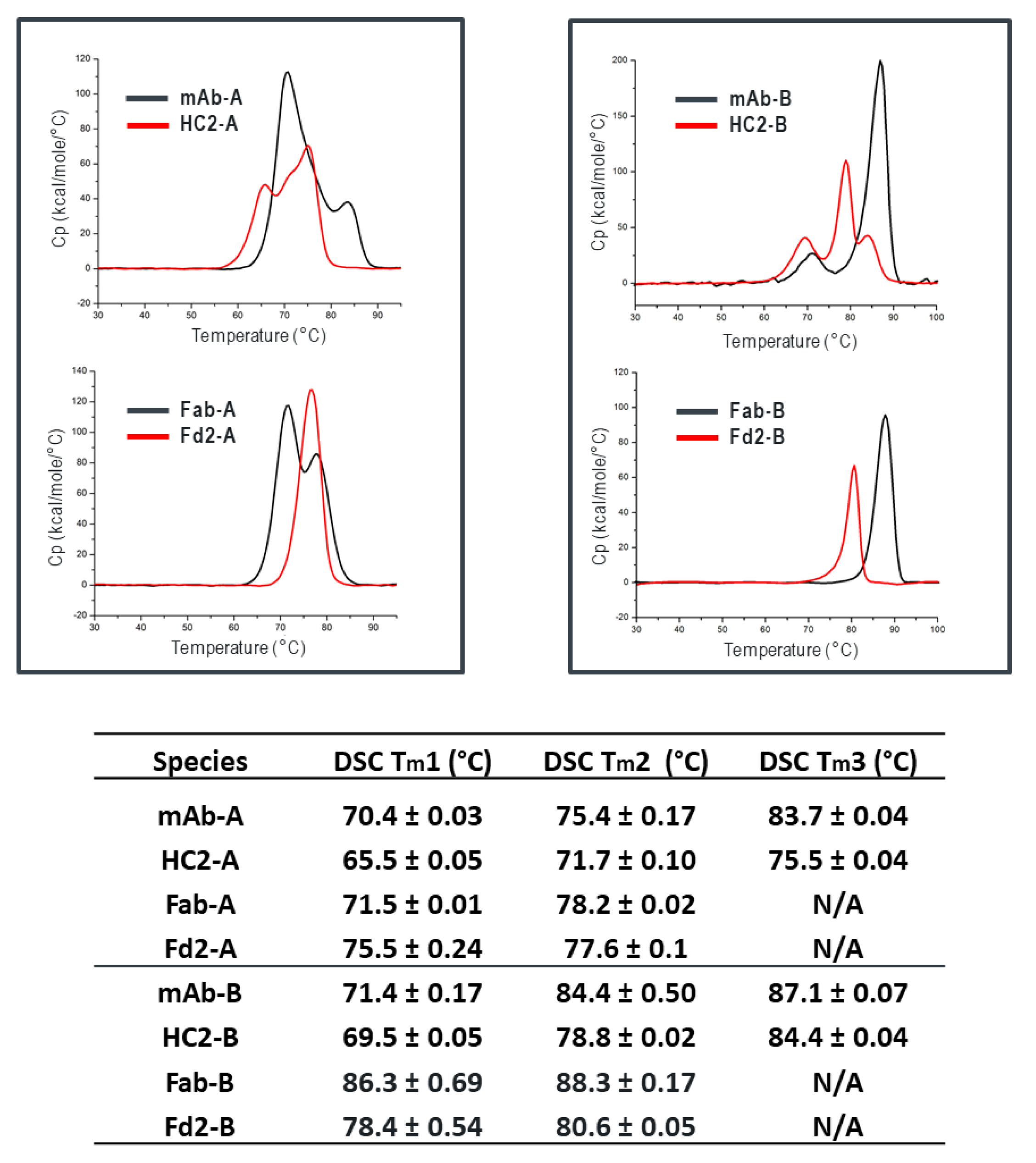
Figure 3), where specifically Fd2-A had a measured T
m1 of 75.5 °C compared to 71.5 °C for Fab-A. Additionally, we found that full-length HC2-A had excellent accelerated storage stability attributes that were superior to mAb-A, where after 1-month storage at 40 °C, purity attributes by SE-UPLC and NR-cSDS were both >98% (See
Table 4).
It is also noteworthy that intramolecular disulfide formation is necessary for HC2 or Fd2 expression. When one intramolecular disulfide bond is removed in the Fab-A and Fab-B forms, LC assembly followed by BiP removal is still able to occur, and Fab folding and secretion is achieved. The result for Fab-A and Fab-B are expressed Fab molecules with suitably high T
m values. This is not unexpected since reduced disulfides can occur naturally in antibodies [
34], although incomplete disulfide formation may result in increased aggregation propensity or decreased bioactivity [
35,
36,
37]. In HC2/Fd2 formation, one Fd domain appears to mimic or replace the role of the LC, however, when one intramolecular disulfide is removed on both Fd chains via mutagenesis, Fd2 assembly is not efficient and little to no product is yielded. This demonstrates that complete disulfide formation is necessary in one or both Fd domains to assemble to the Fd-BiP intermediate(s) and complete Fd2 folding and BiP release. While mutagenesis studies of C
H1 cysteines involved in both inter- and intramolecular disulfide formation for a mouse IgG2 enabled expression and secretion of HC-only transcripts [
38], we find complete intramolecular disulfide formation a requirement for the expression of two different humanized HC-Abs evaluated in this study.
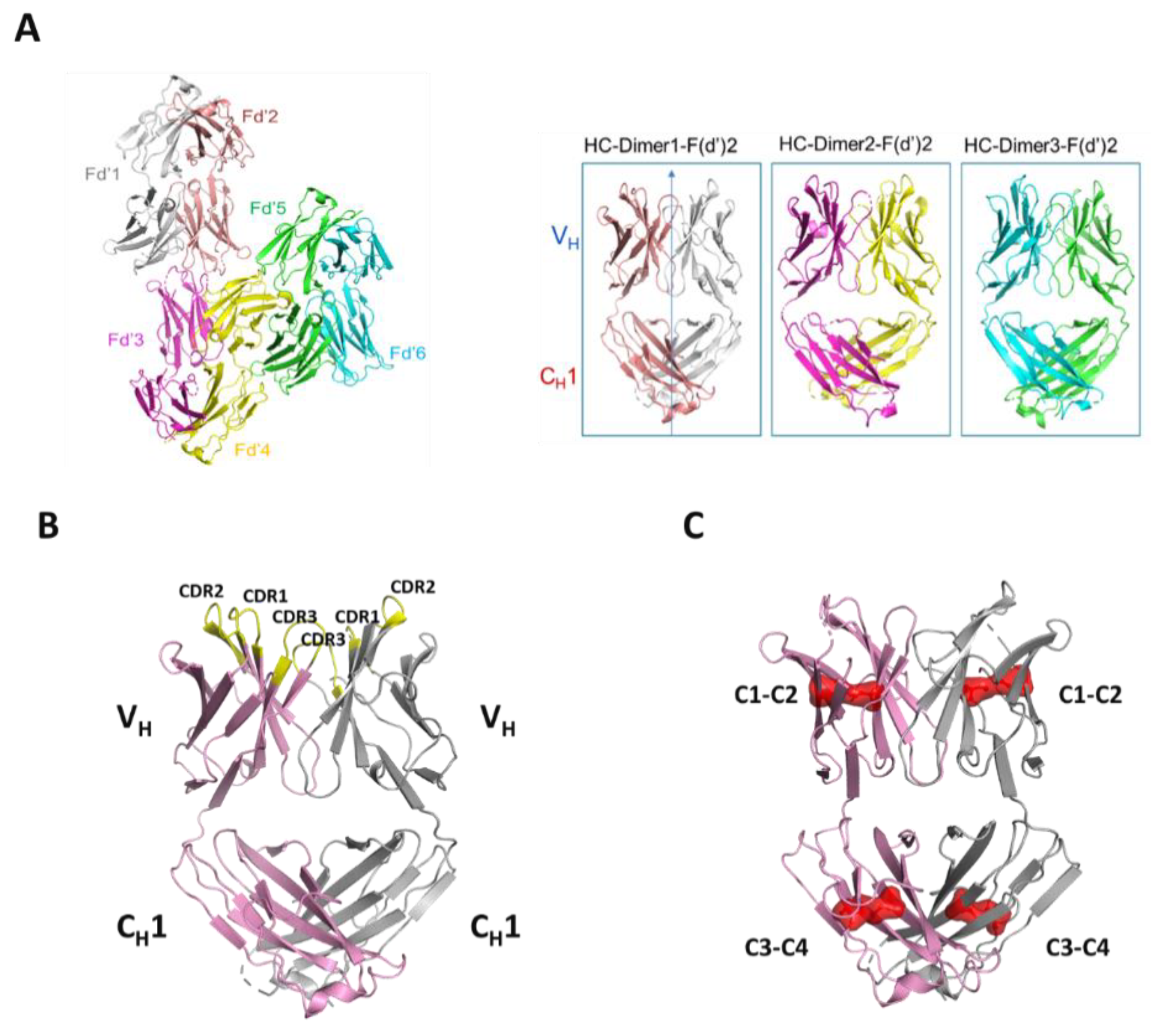
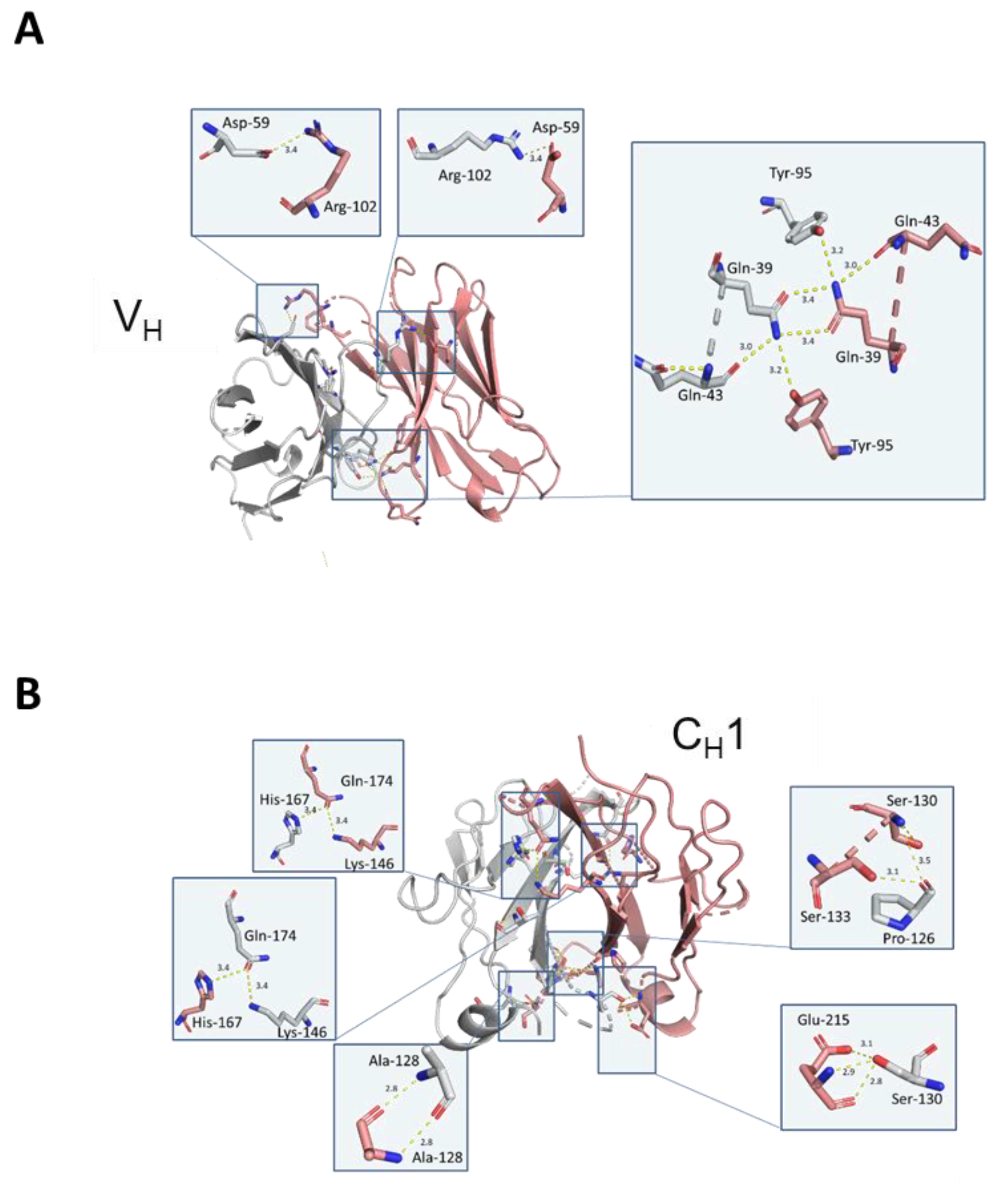
Structurally, the solved crystal structure of Fd2-A is remarkably symmetrical and involves many conserved residues in the homodimer interface that have similar or analogous interactions in the Fab HC-LC interface. Like Fabs, the CDR loops are facing outward and primed for and amenable to antigen binding. As aforementioned, our novel HC-only Fd2 complex superposes remarkably with a representative Fab structure (5vsi, See
Figure 6D), demonstrating that these molecules are quite similar to Fabs, and therefore have potential therapeutic utility. Although HC dimers A and B characterized in this study are not active against the same target antigens that the mAb/Fabs are (both in the sub-nM affinity range), this is fully expected since the entire LC is missing and further, the arrangement and conformation of the HC-CDR loops are very likely to differ between the HC2 and mAb forms. In the typical Fab structure, principal contacts exist between the HC-CDR3 and LC- CDR1. In this HC2 structure for molecule A, the HC-CDR3 is also primarily involved in interfacial contacts, with key contacts existing between the opposing HC-CDR2 chain, which apparently substitutes for the LC-CDR1 in mAbs. The HC2 homodimer structure is also highly symmetrical in nature, which may offer interesting binding and therapeutic properties, and may perhaps be amenable to engineering hetero-HC dimers (composed from two different HCs) that have functional utility. To accomplish this, HC-only domain libraries could be selected against a given target by yeast surface display, as many non-conventional antibodies and scaffold proteins have been engineered using this platform technology [
39]. Further, hetero-human-HC dimers may be engineered to associate orthogonally by utilizing interfacial knob-hole interactions, for instance. These approaches may facilitate multi-specific formats in the presence of conventional Fab domains.
Mutations designed to disrupt the Fd2 dimer interface were based on direct V
H contacts revealed by the crystal structure of Fd2-A. These mutations were made for mAb-A and to aligned residues in mAb-B. When these key, interfacial CDR residues present in Fd2-A are mutated to smaller residues or residues present in other non-HC dimer forming mAbs (C and D), HC dimer expression is significantly reduced for both A and B (See
Table 6). Likewise, the HC2-A and HC2-B molecules have many other similarities, such as both being linked by an intermolecular disulfide bond and having high thermal stability, indicating that the structures of HC2-A and HC2-B are likely similar and conserved. Additionally, as demonstrated by mutagenesis of interfacial dimer contacts, further engineering is achievable to modulate HC dimer formation through engineering or display technologies. Surely, other sequence or specificity determinants of HC2 formation likely exist and understanding these will further advance potential engineering. The use of engineered and/or antibody generated human heavy-chain dimers can have advantages such as being smaller in nature versus full-length antibodies as well as avoiding LC mispairing in multi-specific formats [
40]. We show herein these human HC dimers are highly stable, structurally well-ordered, crystallizable, and their likely conserved and modular nature presents a path forward towards potential engineering and development of these molecules into therapeutic modalities.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}