Characterization of HIV-2 Protease Structure by Studying Its Asymmetry at the Different Levels of Protein Description

 , and
, and
Abstract
:1. Introduction
2. Results & Discussion
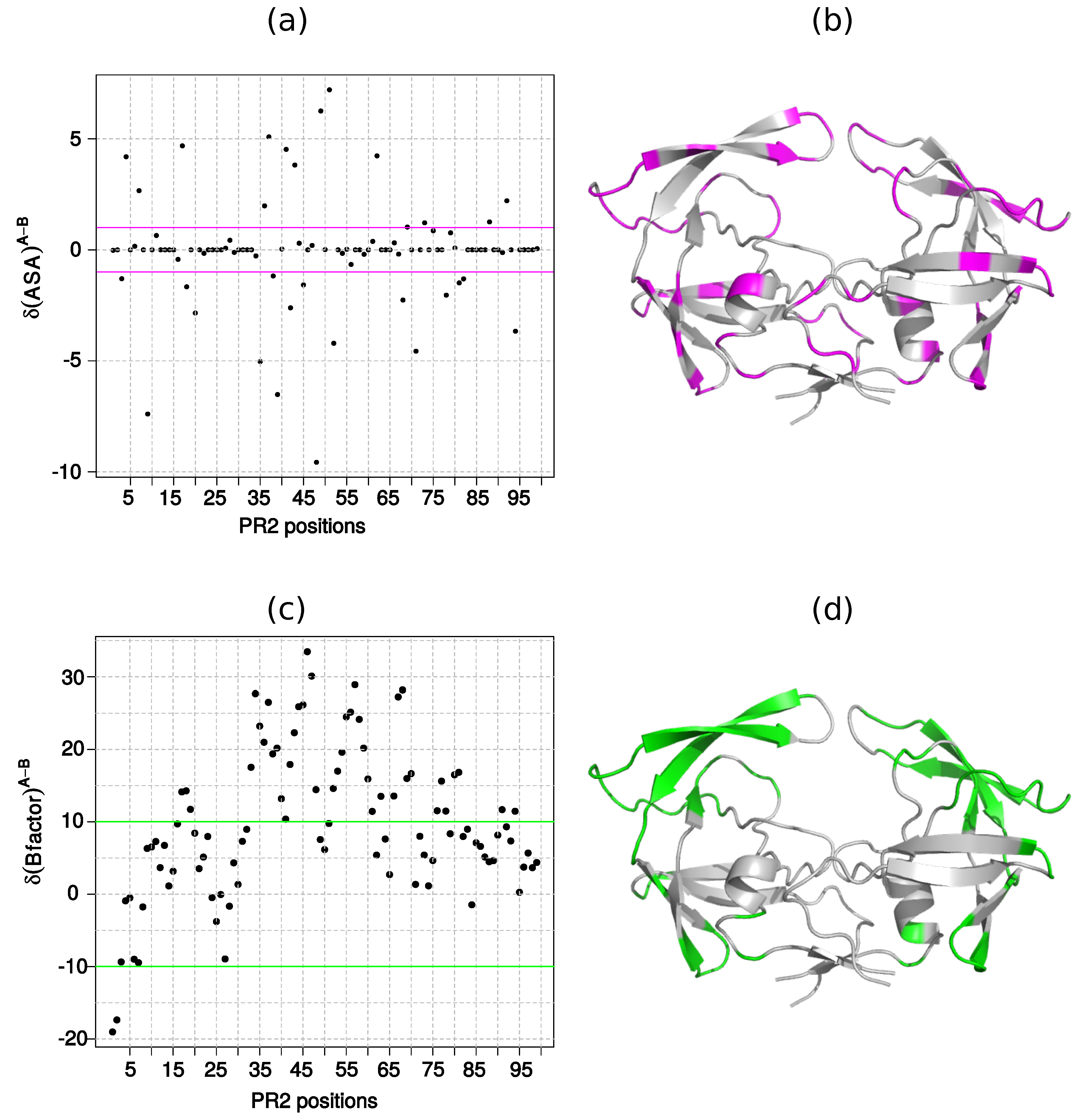
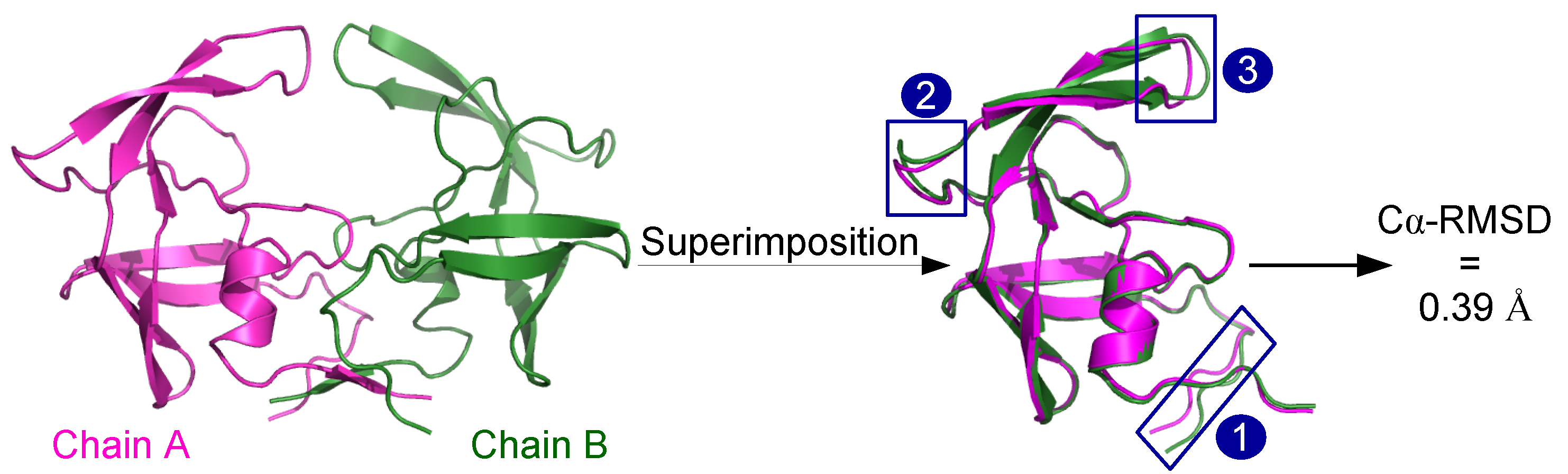
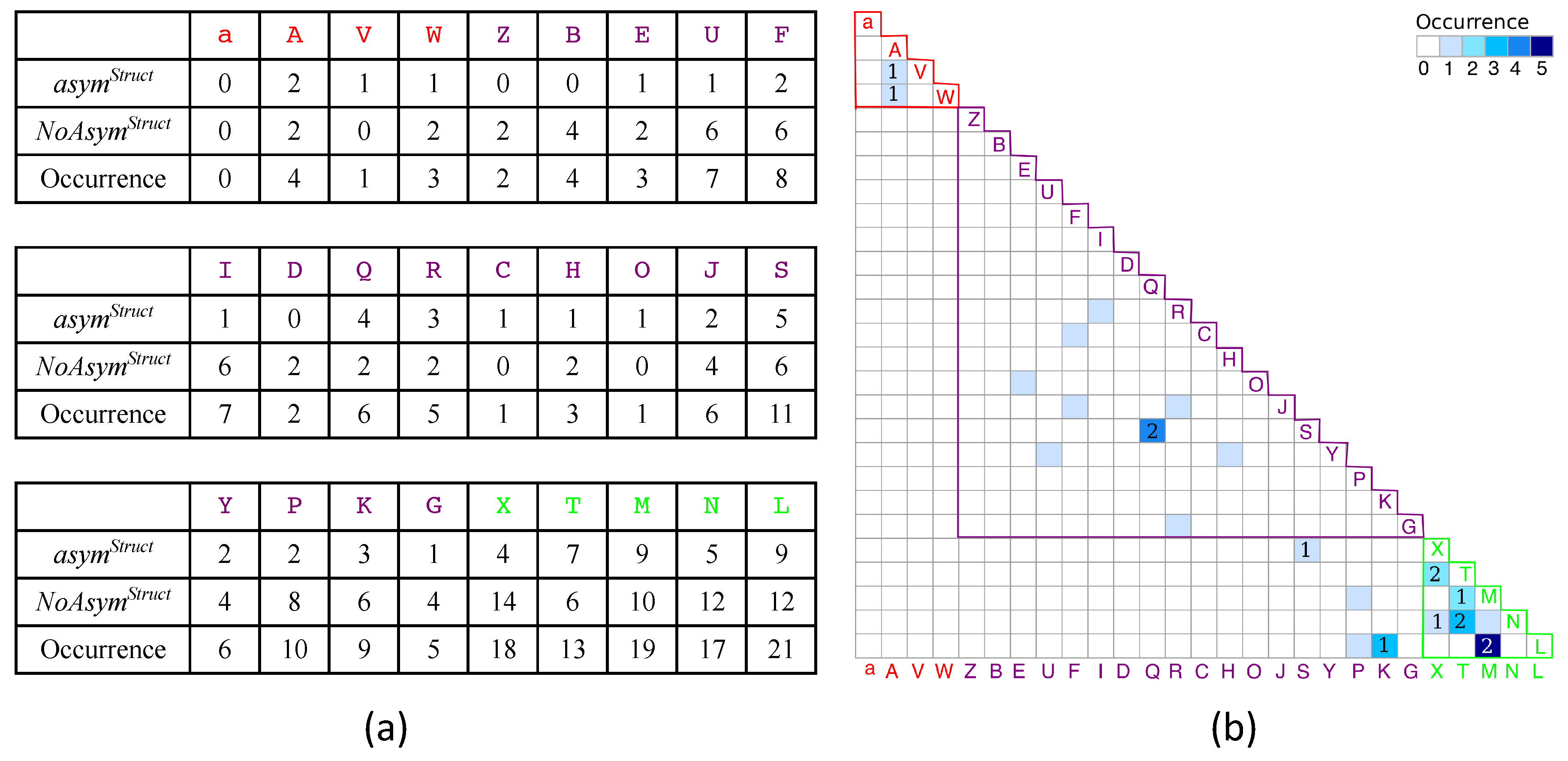
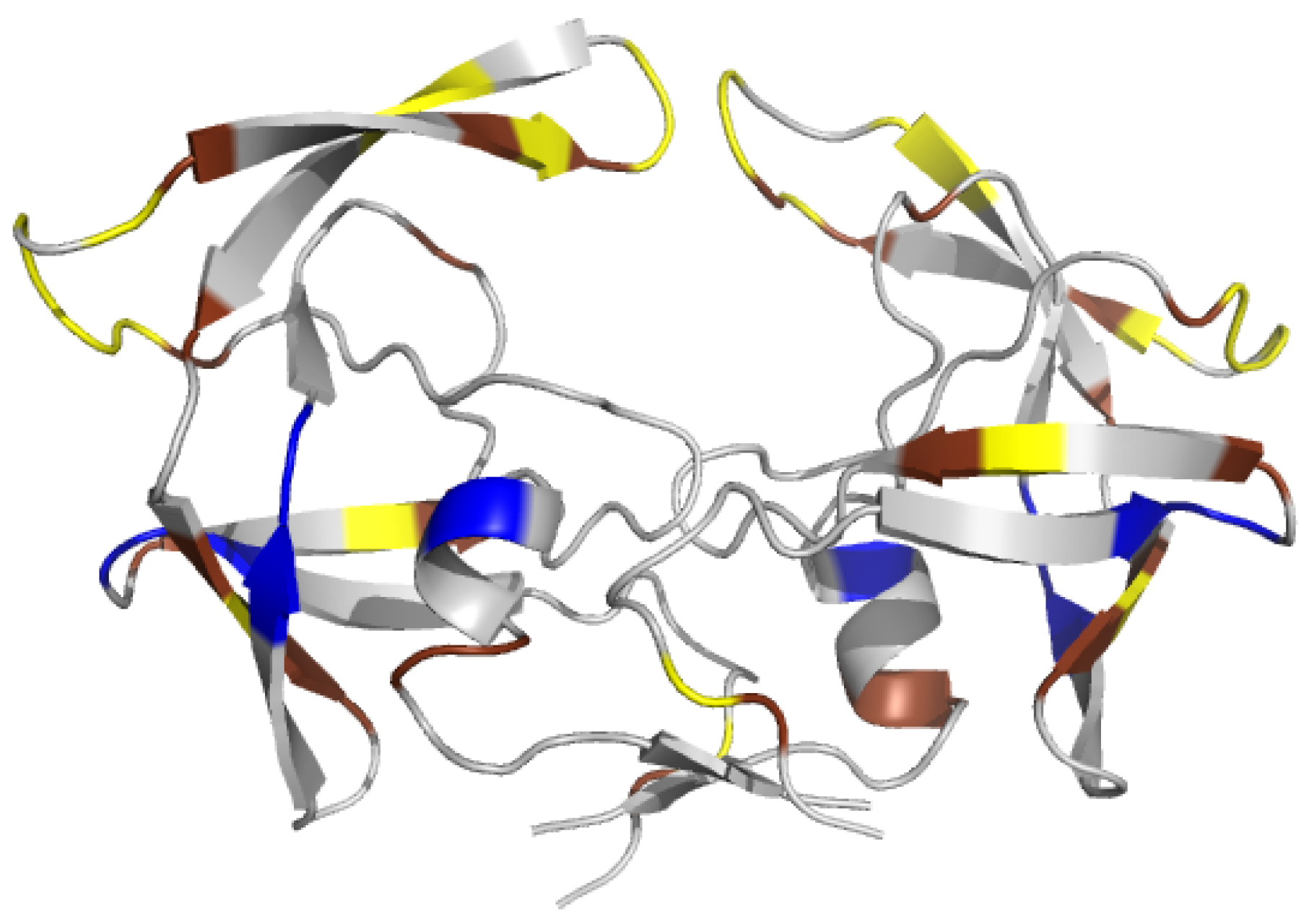
2.1. Localization of Asymmetry in the PR2 Structure
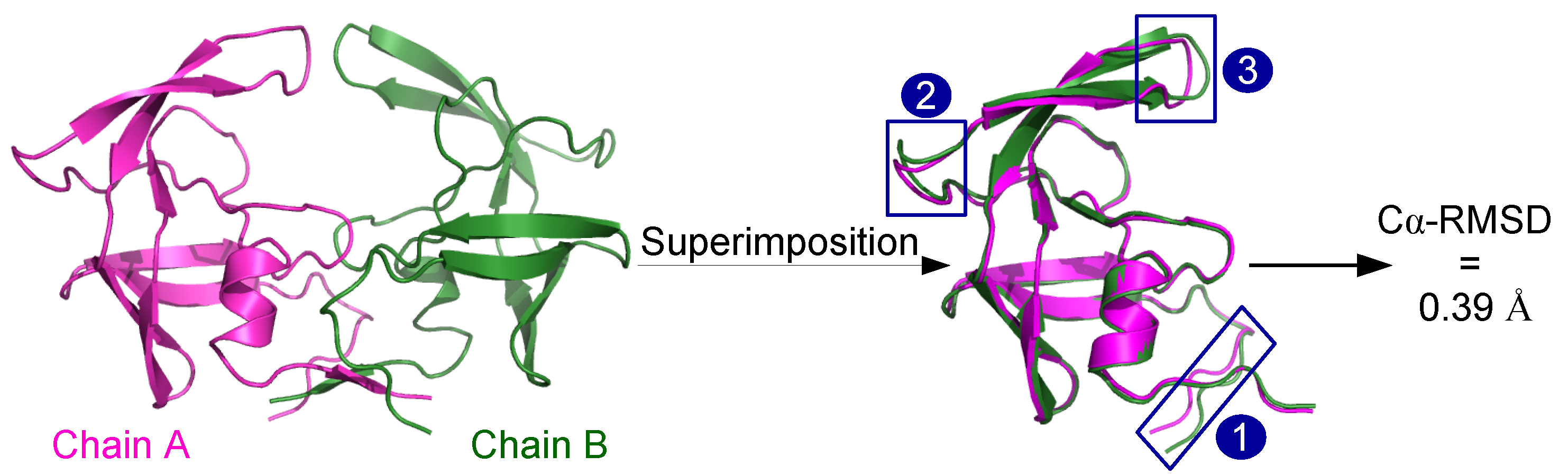
2.1.1. Structural Asymmetry in PR2
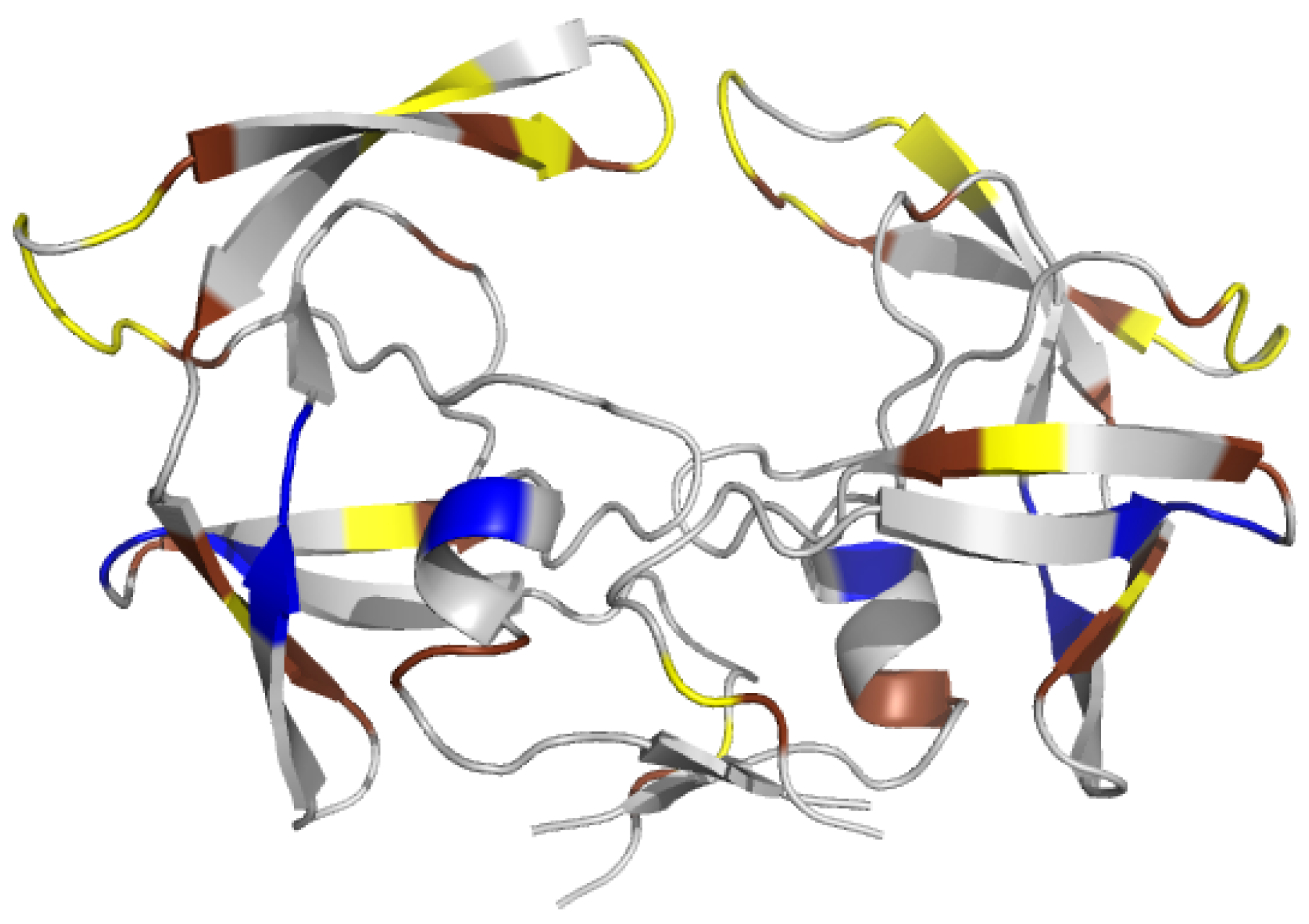
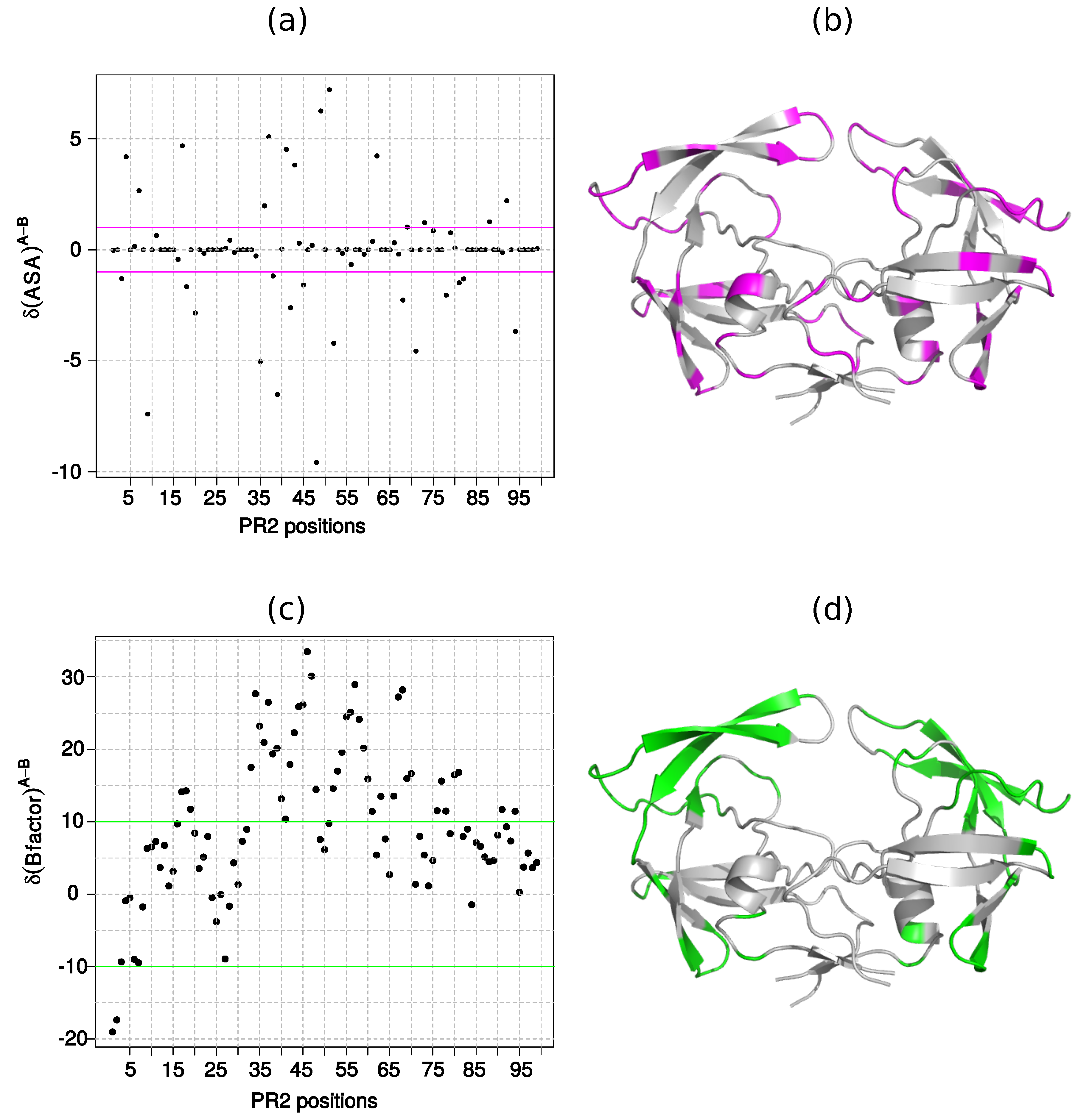
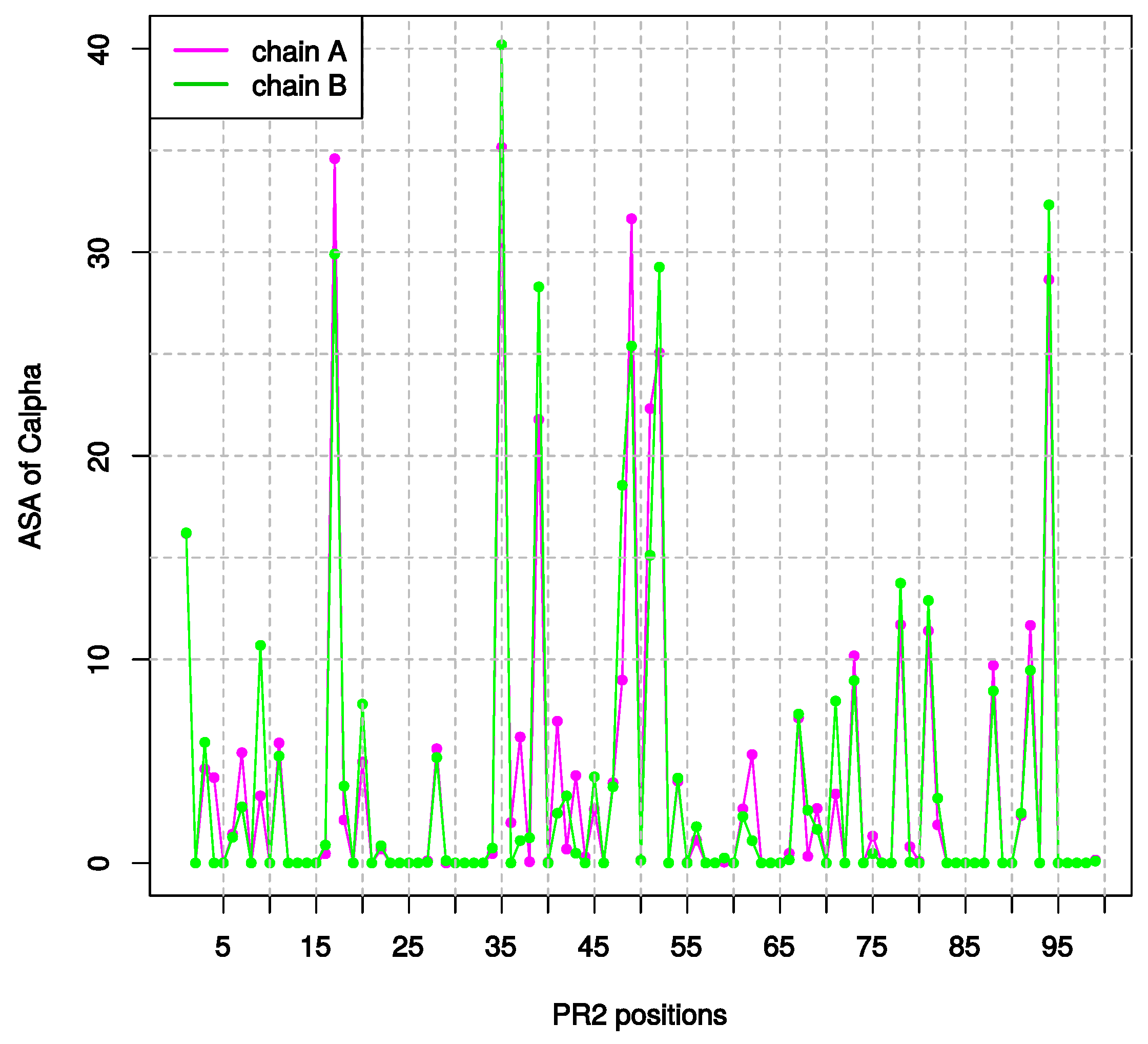
2.1.2. Asymmetry in Terms of Accessibility in PR2
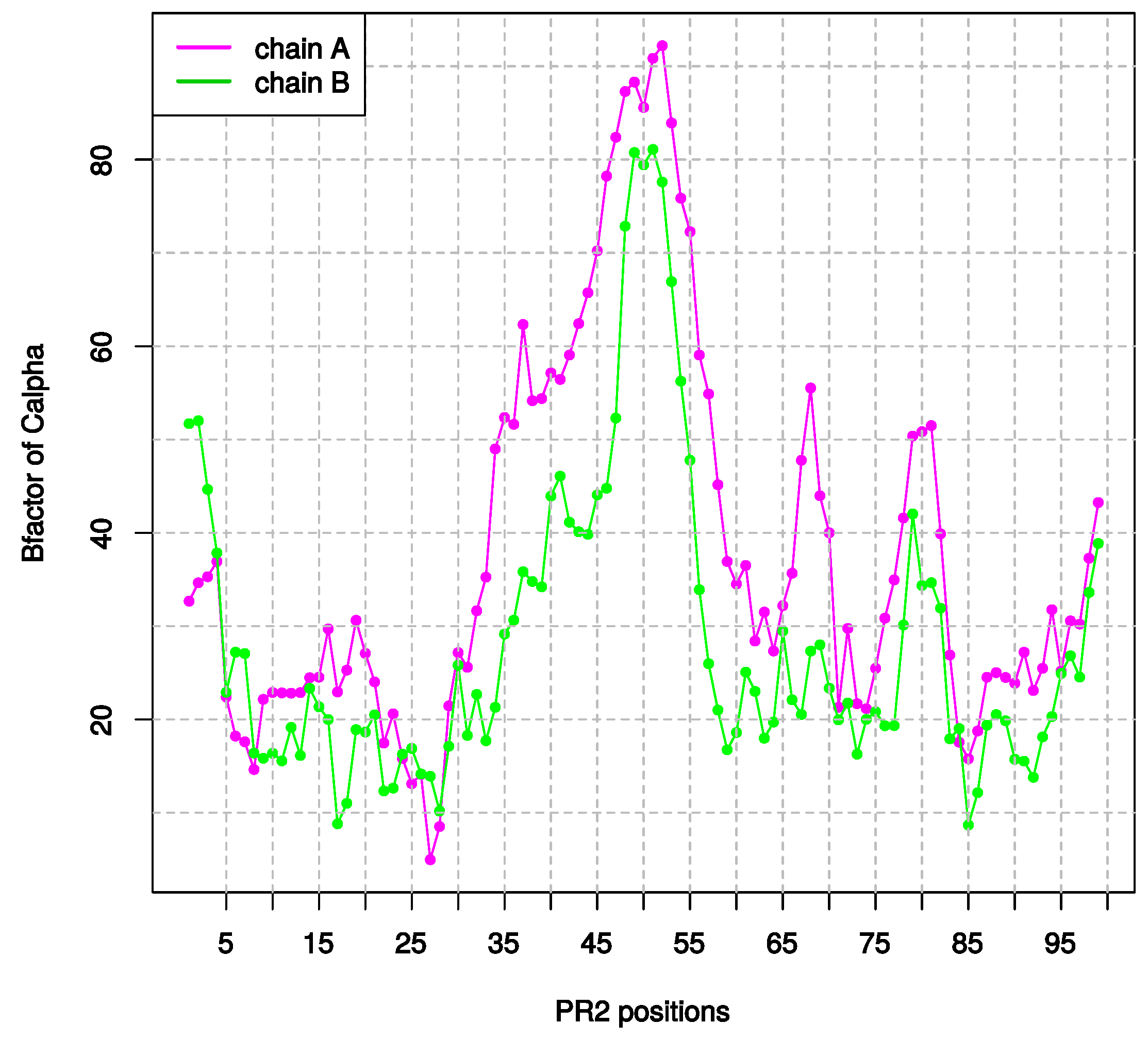
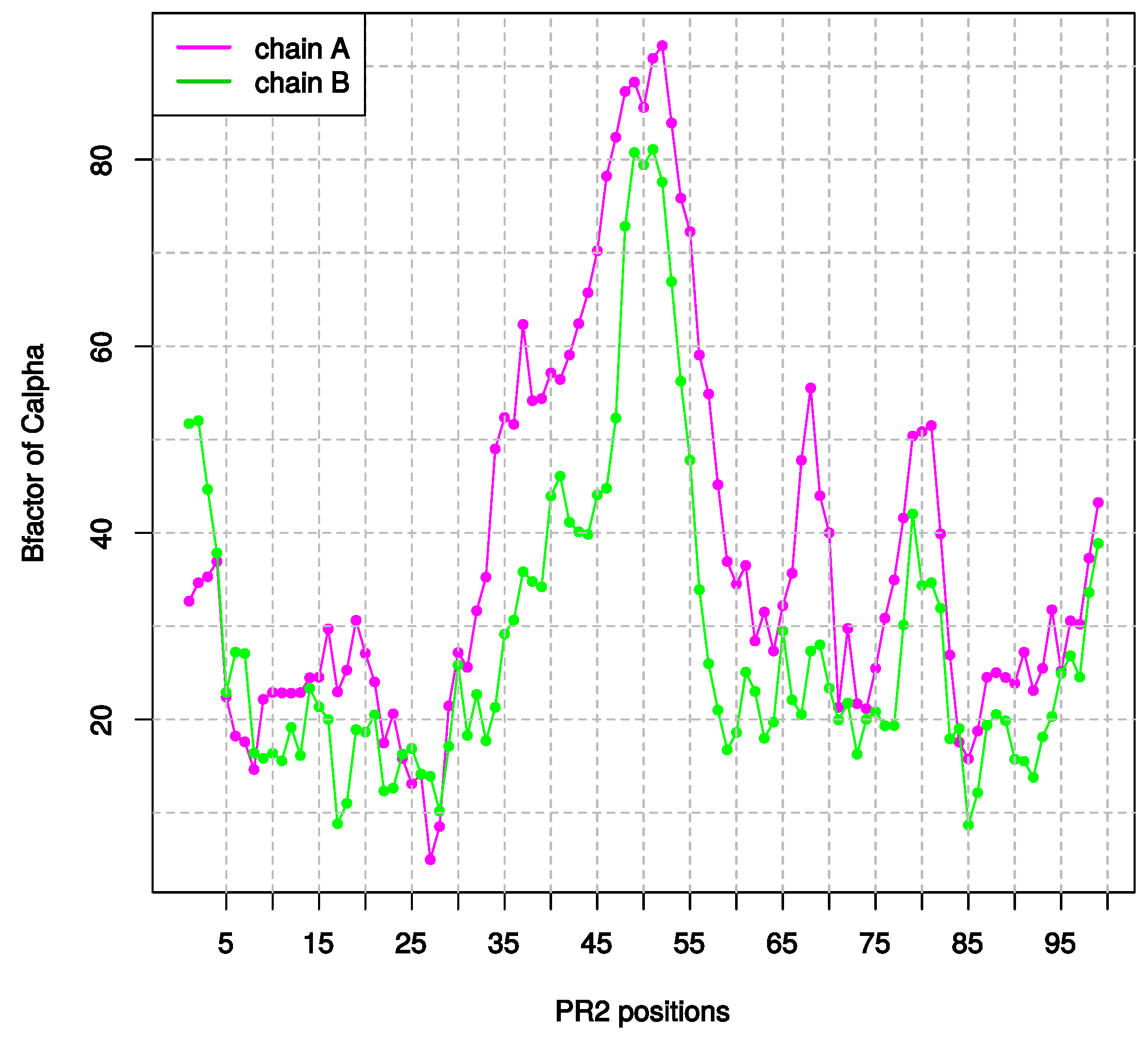
2.1.3. Asymmetry in Terms of Flexibility in PR2
2.2. Link between the Three Asymmetry Types
2.3. Link between Asymmetry and Crystal Packing
3. Materials and Methods
3.1. Presentation of the Data
3.1.1. Crystallographic Structure of the Unbound PR2
3.1.2. Generation of the Energetic-Minimized Structures of the Unbound PR2
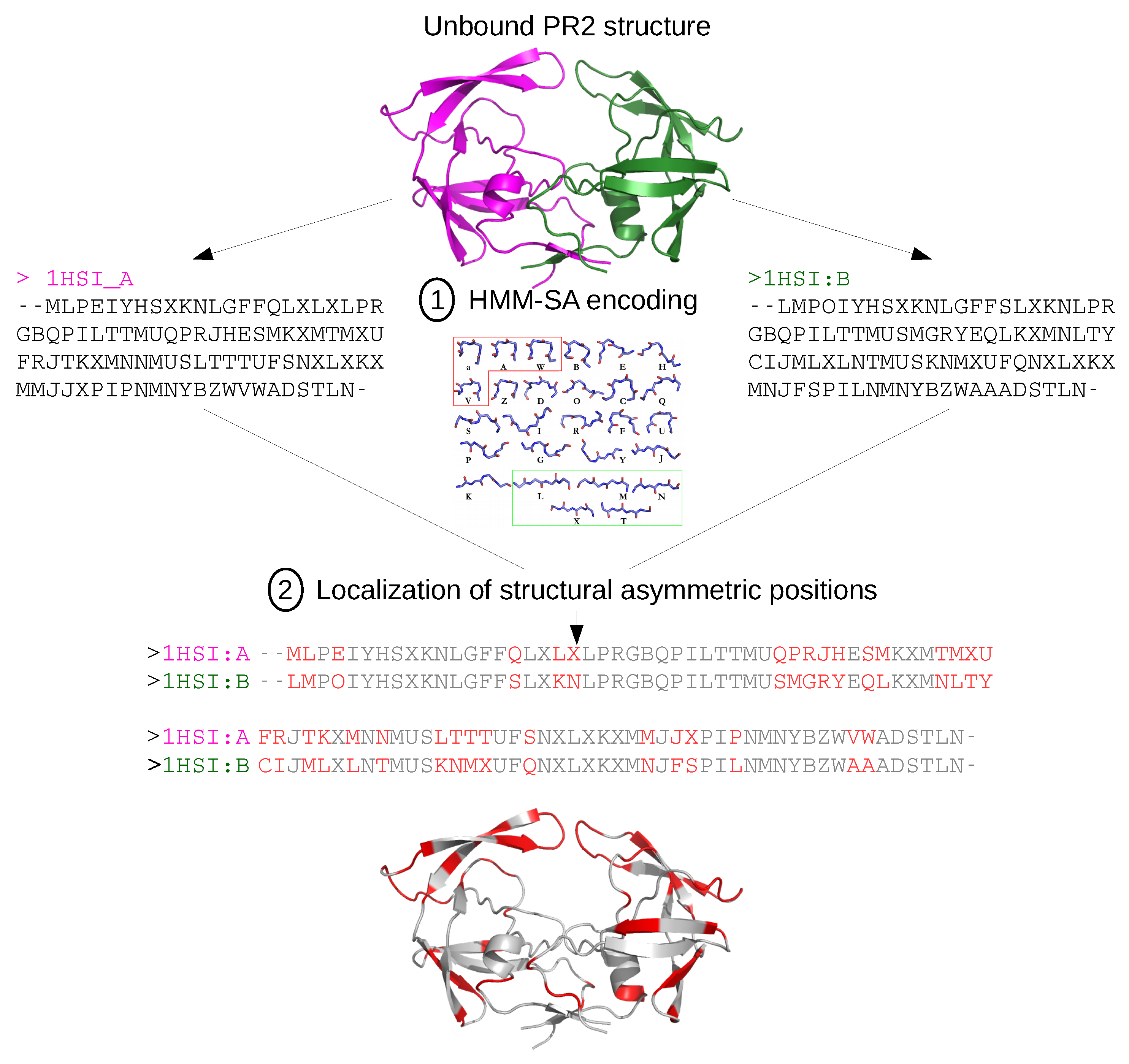
3.2. Detection of Asymmetry in PR2 Structures
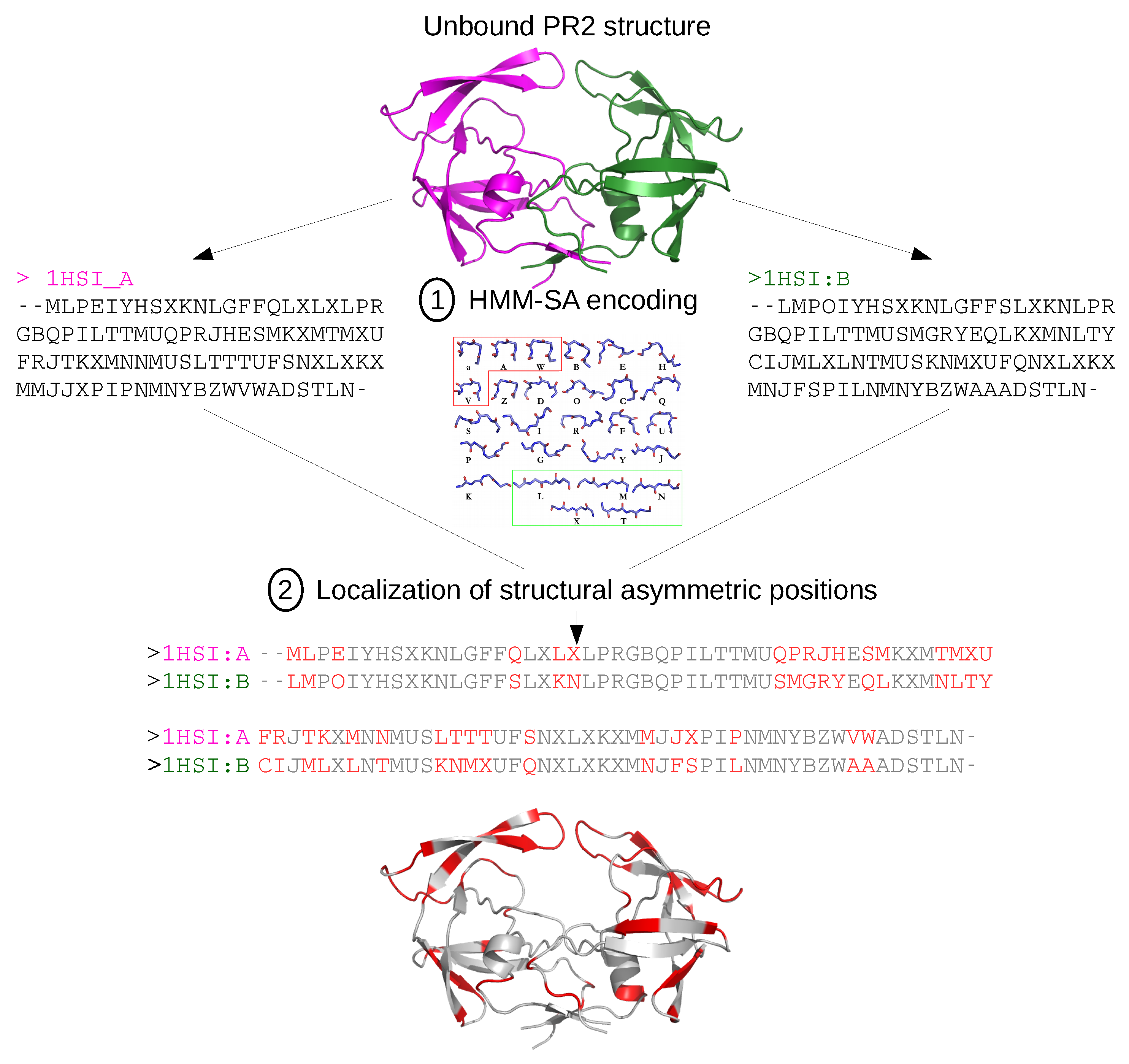
3.2.1. Structural Asymmetry
3.2.2. Accessibility Asymmetry
3.2.3. Flexibility Asymmetry
3.3. Analysis of the Link between the Three Asymmetry
3.4. Detection of Positions Putatively Involved in Crystal Packing
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AIDS | acquired immunodeficiency syndrome |
| ASA | accessibility surface area |
| C-RMSD | root mean square deviation computed on Carbons |
| HIV-1 | human Immunodeficiency virus of type 1 |
| HIV-2 | human Immunodeficiency virus of type 2 |
| HMM-SA | hidden Markov model - structural alphabet |
| PR | protease |
| PR1 | HIV-1 protease |
| PR2 | HIV-2 protease |
Appendix A. Accessibility of Each Residue

Appendix B. Flexibility of Each Residue
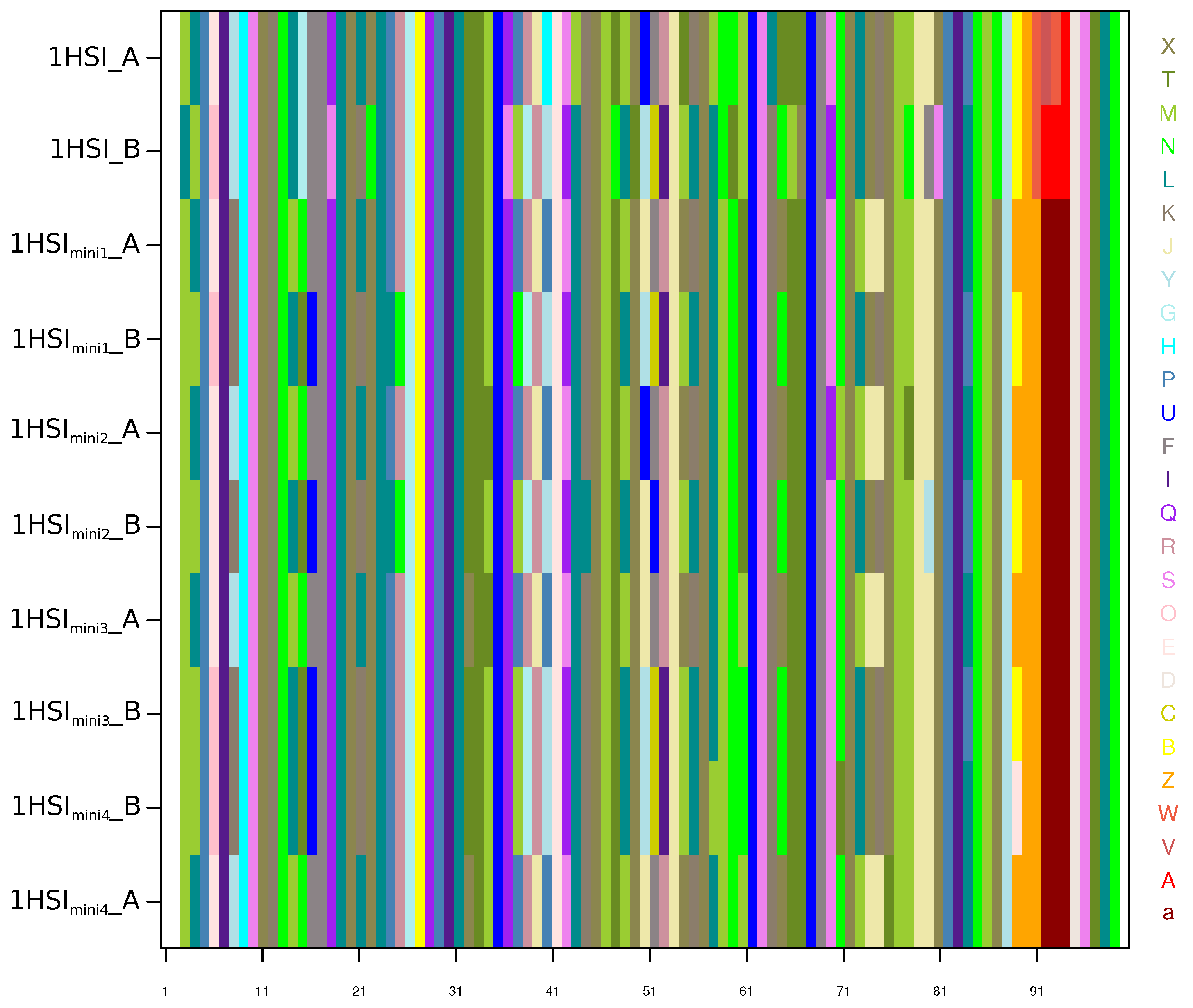
Appendix C. Structural-Letter Sequences of 1HSI and 1HSImini Structures

Appendix D. Comparison of the Structural Asymmetry in 1HSI and 1HSImini Structures
References
- Masse, S.; Lu, X.; Dekhtyar, T.; Lu, L.; Koev, G.; Gao, F.; Mo, H.; Kempf, D.; Bernstein, B.; Hanna, G.; et al. In vitro selection and characterization of human immunodeficiency virus type 2 with decreased susceptibility to lopinavir. Antimicrob. Agents Chemother. 2007, 51, 3075–3080. [Google Scholar] [CrossRef] [PubMed]
- Rodés, B.; Sheldon, J.; Toro, C.; Jiménez, V.; Alvarez, M.; Soriano, V. Susceptibility to protease inhibitors in HIV-2 primary isolates from patients failing antiretroviral therapy. Antimicrob. Agents Chemother. 2006, 57, 709–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brower, E.; Bacha, U.M.; Kawasaki, Y.; Freire, E. Inhibition of HIV-2 protease by HIV-1 protease inhibitors in clinical use. Chem. Biol. Drug Des. 2008, 71, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Raugi, D.; Smith, R.; Ba, S.; Toure, M.; Traore, F.; Sall, F.; Pan, C.; Blankenship, L.; Montano, A.; Olson, J.; et al. Complex patterns of protease inhibitor resistance among antiretroviral treatment-experienced HIV-2 patients from senegal: Implications for second-line therapy. Antimicrob. Agents Chemother. 2013, 57, 2751–2760. [Google Scholar] [CrossRef] [PubMed]
- Cavaco-Silva, J.; Aleixo, M.; Van Laethem, K.; Faria, D.; Valadas, E.; Gonçalves, M.D.; Gomes, P.; Vandamme, A.; Cunha, C.; Camacho, R.J. Mutations selected in HIV-2-infected patients failing a regimen including atazanavir. Antimicrob. Agents Chemother. 2013, 68, 190–192. [Google Scholar] [CrossRef] [PubMed]
- Ntemgwa, M.; Toni, T.; Brenner, B.; Oliveira, M.; Asahchop, E.; Moisi, D.; Wainberg, M. Nucleoside and nucleotide analogs select in culture for different patterns of drug resistance in human immunodeficiency virus types 1 and 2. Antimicrob. Agents Chemother. 2009, 53, 708–715. [Google Scholar] [CrossRef] [PubMed]
- Menéndez-Arias, L.; Alvarez, M. Antiretroviral therapy and drug resistance in human immunodeficiency virus type 2 infection. Antiviral Res. 2014, 102, 70–86. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Singh, S.; Nanduri, B.; Awasthi, Y.; Zimniak, P.; Ji, X. Crystal structure of a murine glutathione S-transferase in complex with a glutathione conjugate of 4-hydroxynon, 2-enal in one subunit and glutathione in the other: Evidence of signaling across the dimer interface. Biochemistry 1999, 38, 11887–11894. [Google Scholar] [CrossRef] [PubMed]
- Cha, H.; Kopetzki, E.; Hubert, R.; Lanzendorfer, M.; Brandsteller, H. Structural basis of the adaptive molecular recognition by MMP9. J. Mol. Biol. 2002, 320, 1065–1079. [Google Scholar] [CrossRef]
- Jin, L.; Stec, B.; Lipscomb, W.N.; Kantrowitz, E.R. Insights into the mechanisms of catalysis and heterotropic regulation of Escherichia coli aspartate transcarbamoylase based upon a structure of the enzyme complexed with the bisubstrate analogue N-phosphonacetyl-L-aspartate at 2.1 AA. Proteins Struct. Funct. Genet. 1999, 37, 729–742. [Google Scholar] [CrossRef]
- Renatus, M.; Stennicke, H.R.; Scott, F.L.; Liddington, R.C.; Salvesen, G.S. Dimer formation drives the activation of the cell death protease caspase 9. Proc. Natl. Acad. Sci. USA 2001, 98, 14250–14255. [Google Scholar] [CrossRef] [PubMed]
- Prabu-jeyabalan, M.; Nalivaika, E.; King, N.; Schiffer, C. Viability of a drug-resistant human immunodeficiency virus type 1 protease variant: Structural insights for better antiviral therapy. Society 2003, 77, 1306–1315. [Google Scholar] [CrossRef]
- Prabu-Jeyabalan, M.; Nalivaika, E.; Schiffer, C.A. How does a symmetric dimer recognize an asymmetric substrate? A substrate complex of HIV-1 protease. J. Mol. Biol. 2000, 301, 1207–1220. [Google Scholar] [CrossRef] [PubMed]
- Mulichak, A.M.; Hui, J.; Tomasselli, A.; Heinrikson, R.; Curry, K.; Tomich, C.; Thaisrivongs, S.; Sawyer, T.; Watenpaugh, K. The crystallographic structure of the protease from human immunodeficiency virus type 2 with two synthetic peptidic transition state analog inhibitors. J. Biol. Chem. 1993, 268, 13103–13109. [Google Scholar] [PubMed]
- Tong, L.; Pav, S.; Pargellis, C.; Dô, F.; Lamarre, D.; Anderson, P. Crystal structure of human immunodeficiency virus (HIV) type 2 protease in complex with a reduced amide inhibitor and comparison with HIV-1 protease structures. Proc. Natl. Acad. Sci. USA 1993, 90, 8387–8391. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.; Pav, S.; Mui, S.; Lamarre, D.; Yoakim, C.; Beaulieu, P.; Anderson, P. Crystal structures of HIV-2 protease in complex with inhibitors containing the hydroxyethylamine dipeptide isostere. Structure 1995, 3, 33–40. [Google Scholar] [CrossRef]
- Priestle, J.; Fässler, A.; Rösel, J.; Tintelnot-Blomley, M.; Strop, P.; Grütter, M. Comparative analysis of the X-ray structures of HIV-1 and HIV-2 proteases in complex with CGP 53820, a novel pseudosymmetric inhibitor. Structure 1995, 3, 381–389. [Google Scholar] [CrossRef]
- Triki, D.; Cano Contreras, M.; Flatters, D.; Visseaux, B.; Descamps, D.; Camproux, A.; Regad, L. Analysis of the HIV-2 protease’s adaptation to various ligands: characterization of backbone asymmetry using a structural alphabet. Sci. Rep. 2018, 8, 710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camproux, A.; Gautier, R.; Tufféry, P. A hidden Markov model derived structural alphabet for proteins. J. Mol. Biol. 2004, 339, 591–605. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.; Dill, K. Origins of structure in globular proteins. Proc. Natl. Acad. Sci. USA 1990, 87, 6388–6392. [Google Scholar] [CrossRef] [PubMed]
- Karplus, P.; Schulz, G. Prediction of chain flexibility in proteins. Naturwissenschaften 1985, 72, 212–213. [Google Scholar] [CrossRef]
- Chen, J.; Liang, Z.; Wang, W.; Yi, C.; Zhang, S.; Zhang, Q. Revealing origin of decrease in potency of darunavir and amprenavir against HIV-2 relative to HIV-1 protease by molecular dynamics simulations. Sci. Rep. 2014, 4, 6872. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Li, Y.; Chen, E.; Hall, D.; Darke, P.; Culberson, C.; Shafer, J.; Kuo, L. Crystal structure at 1.9-A resolution of human immunodeficiency virus (HIV) II protease complexed with L-735,524, an orally bioavailable inhibitor of the HIV proteases. J. Biol. Chem. 1994, 269, 26344–26348. [Google Scholar] [PubMed]
- Regad, L.; Guyon, F.; Maupetit, J.; Tufféry, P.; Camproux, A.C. A Hidden Markov Model applied to the protein 3D structure analysis. Comput. Stat. Data Anal. 2008, 52, 3198–3207. [Google Scholar] [CrossRef]
- Rost, B.; Sander, C. Conservation and prediction of solvent accessibility in protein families. Proteins 1994, 20, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Pollastri, G.; Baldi, P.; Fariselli, P.; Casadio, R. Prediction of coordination number and relative solvent accessibility in proteins. Proteins 2002, 47, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Kar, P.; Knecht, V.J. Origin of decrease in potency of darunavir and two related antiviral inhibitors against HIV-2 compared to HIV-1 protease. Phys. Chem. B 2012, 116, 2605–2614. [Google Scholar] [CrossRef] [PubMed]
- Goodsell, D.; Olson, A.J. Structural symmetry and protein function. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 105. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.H. Breaking symmetry in protein dimers: Designs and functions. Protein Sci. 2006, 15, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, M.; Murtolad, T.; Schulz, R.; Pálla, S.; Smith, J.; Hess, B.; Lindahl, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Regad, L.; Chéron, J.; Triki, D.; Senac, C.; Flatters, D.; Camproux, A. Exploring the potential of a structural alphabet-based tool for mining multiple target conformations and target flexibility insight. PLoS ONE 2017, 12, e0182972. [Google Scholar] [CrossRef] [PubMed]
- Sadiq, S.K.; de Fabritiis, G. Explicit solvent dynamics and energetics of HIV-1 protease flap opening and closing. Proteins Struct. Funct. Bioinform. 2010, 78, 2873–2885. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Robertson, A.; Jensen, J.H. Very fast empirical prediction and rationalization of protein pK a values. Proteins Struct. Funct. Genet. 2005, 61, 704–721. [Google Scholar] [CrossRef] [PubMed]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, version 1.8; Schrödinger, LLC.: San Carlos, CA, USA, 2002.
- Hubbard, S.; Thornton, J. NACCESS; Computer Program; Department of Biochemistry and Molecular Biology, University College London: London, UK, 1993. [Google Scholar]
- Parthasarathy, S.; Murthy, M. Analysis of temperature factor distribution in high-resolution protein structures. Protein Sci. 1997, 6, 2561–2567. [Google Scholar] [CrossRef] [PubMed]
- Abdi, H.; Valentin, D. Encyclopedia of measurement and statistics. In Multiple Correspondence Analysis; Sage: Thousand Oaks, CA, USA, 2007; pp. 651–657. [Google Scholar]
- Lê, S.; Rennes, A.; Josse, J.; Husson, F. FactoMineR: An R Package for Multivariate Analysis. J. Stat. Softw. 2008, 25, 18677. [Google Scholar] [CrossRef]
- Carugo, O.; Djinovic-Carugo, K. How many packing contacts are observed in protein crystals? J. Struct. Biol. 2012, 180, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Damm, K.; Ung, P.; Quintero, J.; Gestwicki, J.; Carlson, H. A poke in the eye: Inhibiting HIV-1 protease through its flap-recognition pocket. Biopolymers 2008, 89, 643–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perryman, A.; Zhang, Q.; Soutter, H.; Rosenfeld, R.; McRee, D.; Olson, A.; Elder, J.; Stout, C. Analysis of temperature factor distribution in high-resolution protein structures. Chem. Biol. Drug Des. 2010, 75, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Ung, P.; Dunbar, J.; Gestwicki, J.; Carlson, H. An Allosteric Modulator of HIV-1 Protease Shows Equipotent Inhibition of Wild-Type and Drug-Resistant Proteases. J. Med. Chem. 2014, 57, 6468–6478. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structures | 1HSI | 1HSImini1 | 1HSImini2 | 1HSImini3 | 1HSImini4 |
|---|---|---|---|---|---|
| C-RMSD (Å) | 0.386 | 0.429 | 0.454 | 0.431 | 0.416 |
| Number of positions | 34 | 25 | 30 | 27 | 28 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ollitrault, G.; Fartek, S.; Descamps, D.; Camproux, A.-C.; Visseaux, B.; Regad, L. Characterization of HIV-2 Protease Structure by Studying Its Asymmetry at the Different Levels of Protein Description. Symmetry 2018, 10, 644. https://doi.org/10.3390/sym10110644
Ollitrault G, Fartek S, Descamps D, Camproux A-C, Visseaux B, Regad L. Characterization of HIV-2 Protease Structure by Studying Its Asymmetry at the Different Levels of Protein Description. Symmetry. 2018; 10(11):644. https://doi.org/10.3390/sym10110644
Chicago/Turabian StyleOllitrault, Guillaume, Sandrine Fartek, Diane Descamps, Anne-Claude Camproux, Benoît Visseaux, and Leslie Regad. 2018. "Characterization of HIV-2 Protease Structure by Studying Its Asymmetry at the Different Levels of Protein Description" Symmetry 10, no. 11: 644. https://doi.org/10.3390/sym10110644
APA StyleOllitrault, G., Fartek, S., Descamps, D., Camproux, A.-C., Visseaux, B., & Regad, L. (2018). Characterization of HIV-2 Protease Structure by Studying Its Asymmetry at the Different Levels of Protein Description. Symmetry, 10(11), 644. https://doi.org/10.3390/sym10110644