Synthesis of a Novel Chiral Stationary Phase by (R)-1,1′-Binaphthol and the Study on Mechanism of Chiral Recognition


Abstract
:1. Introduction
2. Materials and Methods
2.1. General Experimental
2.2. High Perfomance Liquid Chromatography Instrumentation and Conditions
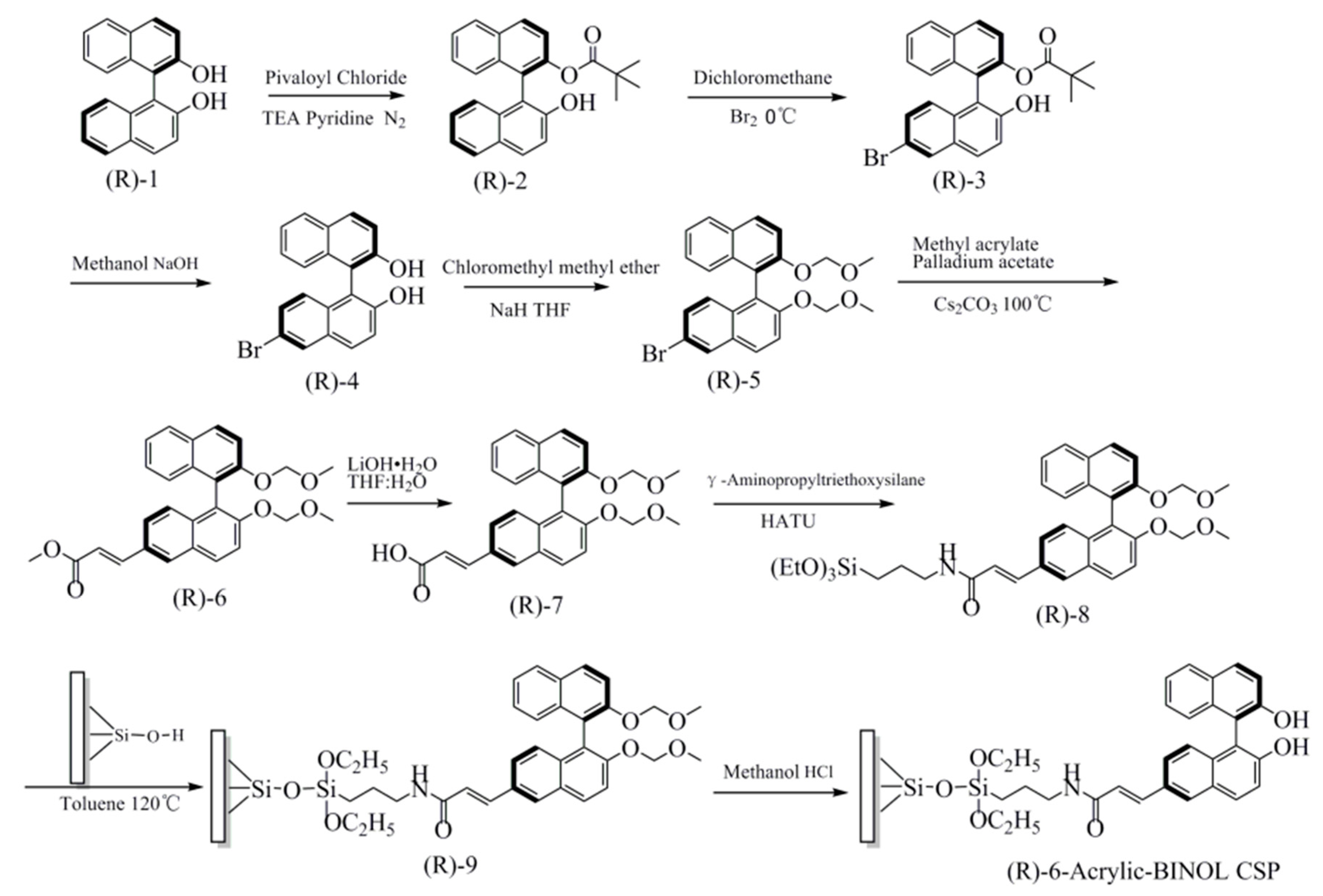
2.3. Synthesis
2.3.1. Synthesis of (R)-2
2.3.2. Synthesis of (R)-3
2.3.3. Synthesis of (R)-4
2.3.4. Synthesis of (R)-5
2.3.5. Synthesis of (R)-6
2.3.6. Synthesis of (R)-7
2.3.7. Synthesis of (R)-8
2.3.8. Preparation of the Chiral Stationary Phase
3. Results and Discussion
3.1. Infrared Spectrum
3.2. Elemental Analysis
3.3. Scanning Electron Microscope
3.4. Enantioseparation
3.4.1. Effect of the Mobile Phase
3.4.2. Temperature Effect
Effect of Temperature on Retention Factor
Effect of Temperature on Separation Factor
4. Conclusions
Supplementary Materials
Supplementary File 1Author Contributions
Funding
Conflicts of Interest
References
- Scriba, G.K.E. Chiral recognition in separation science—An update. J. Chromatogr. A 2016, 1467, 56–78. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Wang, C.; Traeger, S.C.; Discenza, L.; Obermeier, M.T.; Tymiak, A.A.; Zhang, Y. The role of chromatographic and chiroptical spectroscopic techniques and methodologies in support of drug discovery for atropisomeric drug inhibitors of Bruton’s tyrosine kinase. J. Chromatogr. A 2017, 1487, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, A.R.; Maia, A.S.; Cass, Q.B.; Tiritan, M.E. Enantioseparation of chiral pharmaceuticals in biomedical and environmental analyses by liquid chromatography: An overview. J. Chromatogr. B 2014, 968, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Tang, J.; Tang, W.H. Recent development of cationic cyclodextrins for chiral separation. TrAC Trends Anal. Chem. 2015, 65, 22–29. [Google Scholar] [CrossRef]
- Xiao, Y.; Ng, S.C.; Tan, T.T.Y.; Wang, Y. Recent development of cyclodextrin chiral stationary phases and their applications in chromatography. J. Chromatogr. A 2012, 1269, 52–68. [Google Scholar] [CrossRef] [PubMed]
- Rocco, A.; Aturki, Z.; Fanali, S. Chiral separations in food analysis. TrAC Trends Anal. Chem. 2013, 52, 206–225. [Google Scholar] [CrossRef]
- Tang, M.L.; Zhang, J.; Zhuang, S.L.; Liu, W.P. Development of chiral stationary phases for high-performance liquid chromatographic separation. TrAC Trends Anal. Chem. 2012, 39, 180–194. [Google Scholar] [CrossRef]
- Cavazzini, A.; Pasti, L.; Massi, A.; Marchetti, N.; Dondi, F. Recent applications in chiral high performance liquid chromatography: A review. Anal. Chim. Acta 2011, 706, 205–222. [Google Scholar] [CrossRef]
- Shen, J.; Ikai, T.; Okamoto, Y. Synthesis and application of immobilized polysaccharide-based chiral stationary phases for enantioseparation by high-performance liquid chromatography. J. Chromatogr. A 2014, 1363, 51–61. [Google Scholar] [CrossRef]
- Wang, H.S. Enantioseparation on Ligand-exchange-based Restricted Access Stationary Phase Prepared via Atom Transfer Radical Polymerization. Chem. Lett. 2016, 45, 661–663. [Google Scholar] [CrossRef]
- Younes, A.A.; Ates, H.; Mangelings, D.; Heyden, Y.V. A separation strategy combining three HPLC modes and polysaccharide-based chiral stationary phases. J. Pharm. Biomed. Anal. 2013, 75, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.S.; Vitharana, D.; Pu, L. An efficient and practical direct resolution of racemic 1,1′-bi-2-naphthol to both of its pure enantiomers. Tetrahedron Asymmetry 1995, 6, 2123–2126. [Google Scholar] [CrossRef]
- Hu, X.Y.; Shan, Z.X.; Chang, Q. An improved and practical approach to essentially enantiopure BINOLs: Enantioselective inclusion complexation of (S)-proline. Tetrahedron Asymmetry 2012, 23, 1327–1331. [Google Scholar] [CrossRef]
- Nájera, C.; Sansano, J.M.; Saá, J.M. BifunctionalBinols: Chiral 3,3′-Bis(aminomethyl)-1,1′-bi-2-naphthols (Binolams) in Asymmetric Catalysis. Eur. J. Org. Chem. 2009, 2009, 2385–2400. [Google Scholar] [CrossRef]
- Pu, L. Enantioselective fluorescent sensors: A tale of BINOL. Acc. Chem. Res. 2012, 45, 150–163. [Google Scholar] [CrossRef] [PubMed]
- Shockravi, A.; Javadi, A.; Abouzari-Lotf, E. Binaphthyl-based macromolecules: A review. RSC Adv. 2013, 3, 6717–6746. [Google Scholar] [CrossRef]
- Yu, S.S.; Pu, L. Recent progress on using BINOLs in enantioselective molecular recognition. Tetrahedron 2015, 71, 745–772. [Google Scholar] [CrossRef]
- Sousa, L.R.; Sogah, G.D.Y.; Hoffman, D.H.; Cram, D.J. Host-guest complexation. 12. total optical resolution of amine and amino ester salts by chromatography. J. Am. Chem. Soc. 1978, 100, 4569–4576. [Google Scholar] [CrossRef]
- Sogah, G.D.Y.; Cram, D.J. Host-guest complexation. 14. Host covalently bound to polystyrene resin for chromatographic resolution of enantiomers of amino acid and ester salts. J. Am. Chem. Soc. 1979, 101, 3035–3042. [Google Scholar] [CrossRef]
- Wang, H.S.; Tian, X.Y.; Yang, D.; Pan, Y.M.; Wu, Q.; He, C.H. Synthesis and enantiomeric recognition ability of 22-crown-6 ethers derived from rosin acid and BINOL. Tetrahedron Asymmetry 2011, 22, 381–386. [Google Scholar] [CrossRef]
- Sudo, Y.; Yamaguchi, T.; Shinbo, T. Preparation and enantioselectivity of (S)-binaphthol-bonded phase for high-performance liquid chromatography. J. Chromatogr. A 1996, 736, 39–49. [Google Scholar] [CrossRef]
- Sudo, Y.; Yamaguchi, T.; Shinbo, T. Preparation and chiral recognition of (S)-binaphthol derivative-bonded phase for high-performance liquid chromatography. J. Chromatogr. A 1998, 813, 35–45. [Google Scholar] [CrossRef]
- Hyun, M.H. Liquid chromatographic enantioseparations on crown ether-based chiral stationary phases. J. Chromatogr. A 2016, 1467, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Hu, Y.; Galella, E.; Tomasella, F.P.; Fish, W.P. Separation of atropisomers by chiral liquid chromatography and thermodynamic analysis of separation mechanism. J. Pharm. Anal. 2017, 7, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.W.; Larsen, R.D.; Reider, P.J. Efficient synthesis of 6-mono-bromo-1,1′-bi-2-naphthol. Tetrahedron Lett. 2002, 43, 4055–4057. [Google Scholar] [CrossRef]
- Recsei, C.; McErlean, C.S.P. Synthesis of modified binol-phosphoramidites. Tetrahedron 2012, 68, 464–480. [Google Scholar] [CrossRef]
- Hocke, H.; Uozumi, Y. A simple synthetic approach to homochiral 6- and 6′-substituted 1,1′-binaphthyl derivatives. Tetrahedron 2003, 59, 619–630. [Google Scholar] [CrossRef]
- Shen, B.C.; Zhang, D.T.; Yuan, J.Y.; Xu, B.J.; Xu, X.Z. Evaluation and Comparison of a 3,5-Dimethylphenyl IsocyanateTeicoplanin and Phenyl IsocyanateTeicoplanin Chiral Stationary Phases. Chin. J. Chem. 2009, 27, 628–632. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | k1’ | α |
|---|---|---|
| (1) a | 5.96 | 1.111 |
| (2) b | 7.18 | 1.122 |
| (3) a | 9.09 | 1.057 |
| (4) a | 0.69 | 1.062 |
| (5) a | 2.41 | 1.033 |
| (6) c | 19.92 | 1.048 |
| Compound | ∆H (kJ/mol) | ∆(∆H) (kJ/mol) | ∆(∆S) (J/mol * K) | Tiso (K) | |
|---|---|---|---|---|---|
| (R)-1,1′-Binaphthol | −12.95 | −3.80 | −0.56 | −1.13 | 495.57 |
| (S)-1,1′-Binaphthol | −13.36 | −3.87 | |||
| (R)-3,5-dinitro-N-(1-phenylethyl)benzamide | −5.58 | −0.32 | −1.17 | −3.27 | 357.80 |
| (S)-3,5-dinitro-N-(1-phenylethyl)benzamide | −4.41 | 0.07 | |||
| (1)-5-methoxy flavanone | −9.70 | −1.97 | −0.43 | −1.08 | 398.15 |
| (2)-5-methoxy flavanone | −10.17 | −2.11 | |||
| (1)-6-methoxy flavanone | −7.14 | −3.46 | −0.96 | −2.93 | 327.65 |
| (2)-6-methoxy flavanone | −6.79 | −3.25 | |||
| (1)-2,2,2-trifluoro-1-(9-anthryl)-ethanol | −5.10 | −1.33 | −0.12 | −0.15 | 800 |
| (2)-2,2,2-trifluoro-1-(9-anthryl)-ethanol | −5.21 | −1.34 | |||
| (1)-thalidomide | −14.11 | −3.11 | −0.27 | −0.57 | 473.68 |
| (2)-thalidomide | −14.36 | −3.16 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Liu, D.; Zhang, Y.; Tang, Y.; Zhao, J.; Shen, B. Synthesis of a Novel Chiral Stationary Phase by (R)-1,1′-Binaphthol and the Study on Mechanism of Chiral Recognition. Symmetry 2018, 10, 704. https://doi.org/10.3390/sym10120704
Wang Y, Liu D, Zhang Y, Tang Y, Zhao J, Shen B. Synthesis of a Novel Chiral Stationary Phase by (R)-1,1′-Binaphthol and the Study on Mechanism of Chiral Recognition. Symmetry. 2018; 10(12):704. https://doi.org/10.3390/sym10120704
Chicago/Turabian StyleWang, Yongxi, Dandan Liu, Yili Zhang, Yanmei Tang, Jingfeng Zhao, and Baochun Shen. 2018. "Synthesis of a Novel Chiral Stationary Phase by (R)-1,1′-Binaphthol and the Study on Mechanism of Chiral Recognition" Symmetry 10, no. 12: 704. https://doi.org/10.3390/sym10120704