Non-Random Sister Chromatid Segregation in Human Tissue Stem Cells


Abstract
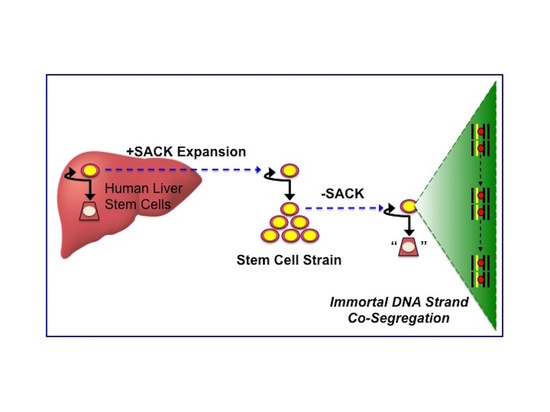
:
1. Introduction
2. Materials and Methods
2.1. SACK Expansion and Propagation of Human Hepatic Stem Cell Strains
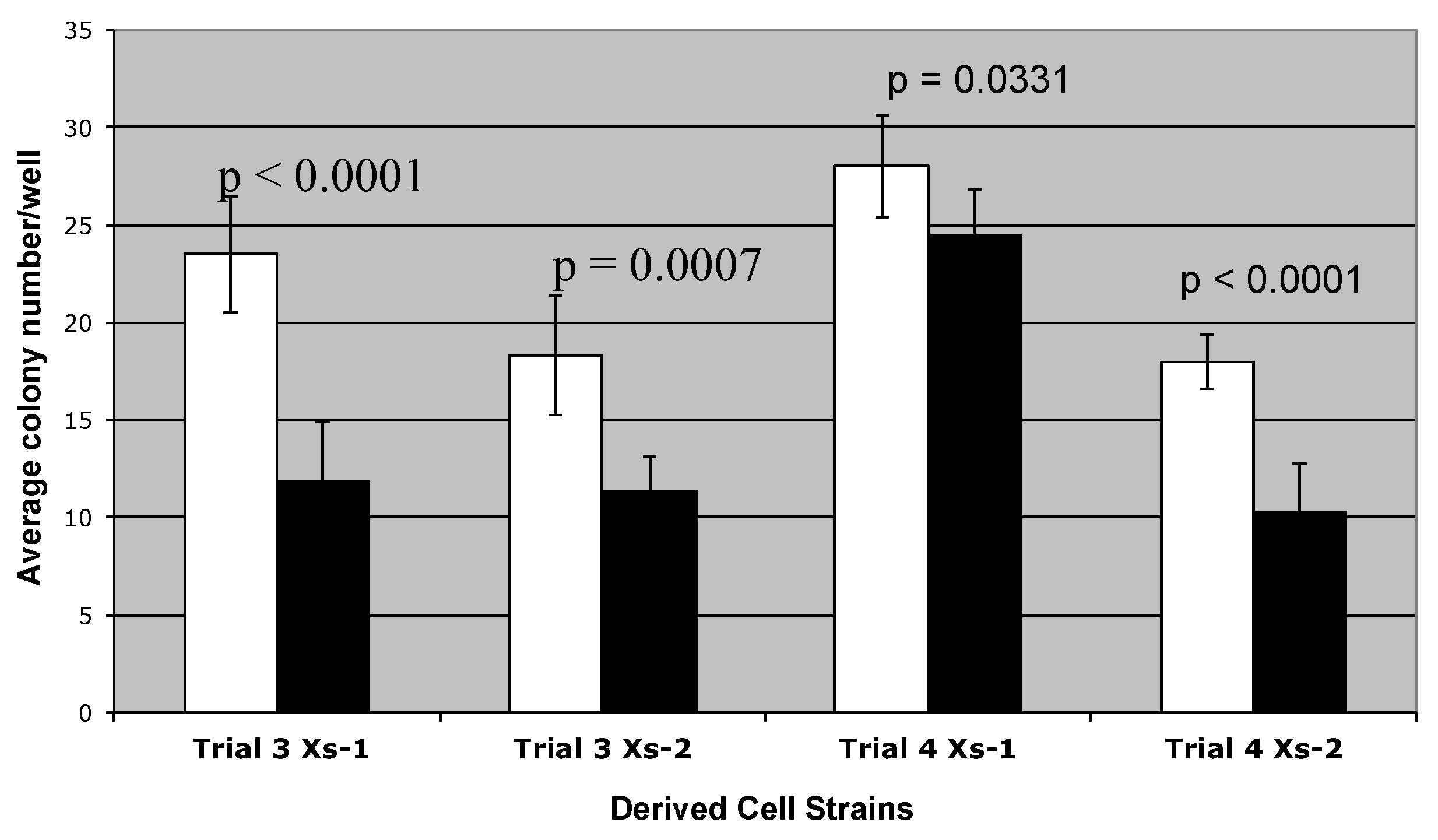
2.2. Colony Formation Studies
2.3. Colcemid Arrest Assay
2.4. Time-Lapse Microscopy Analyses
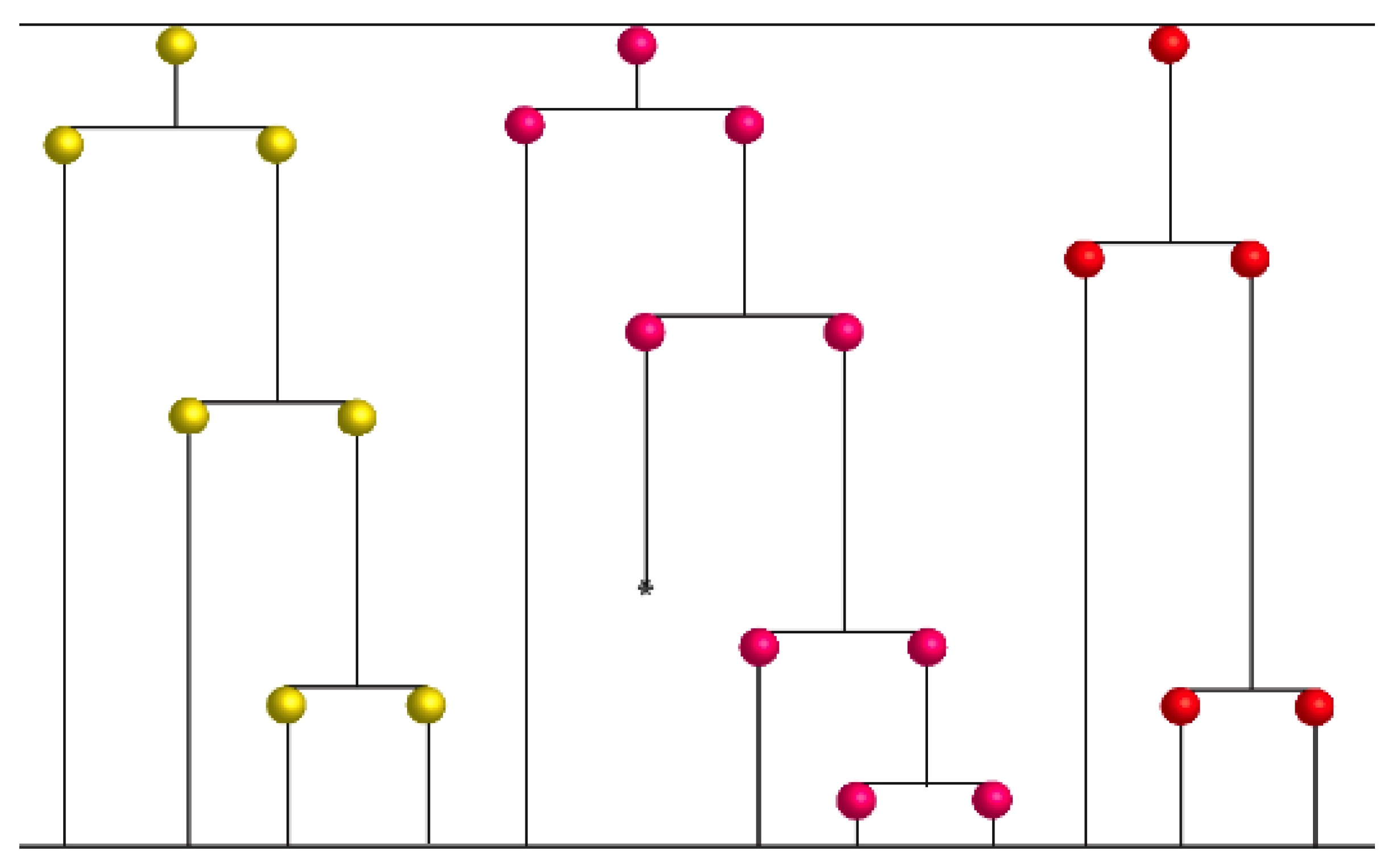
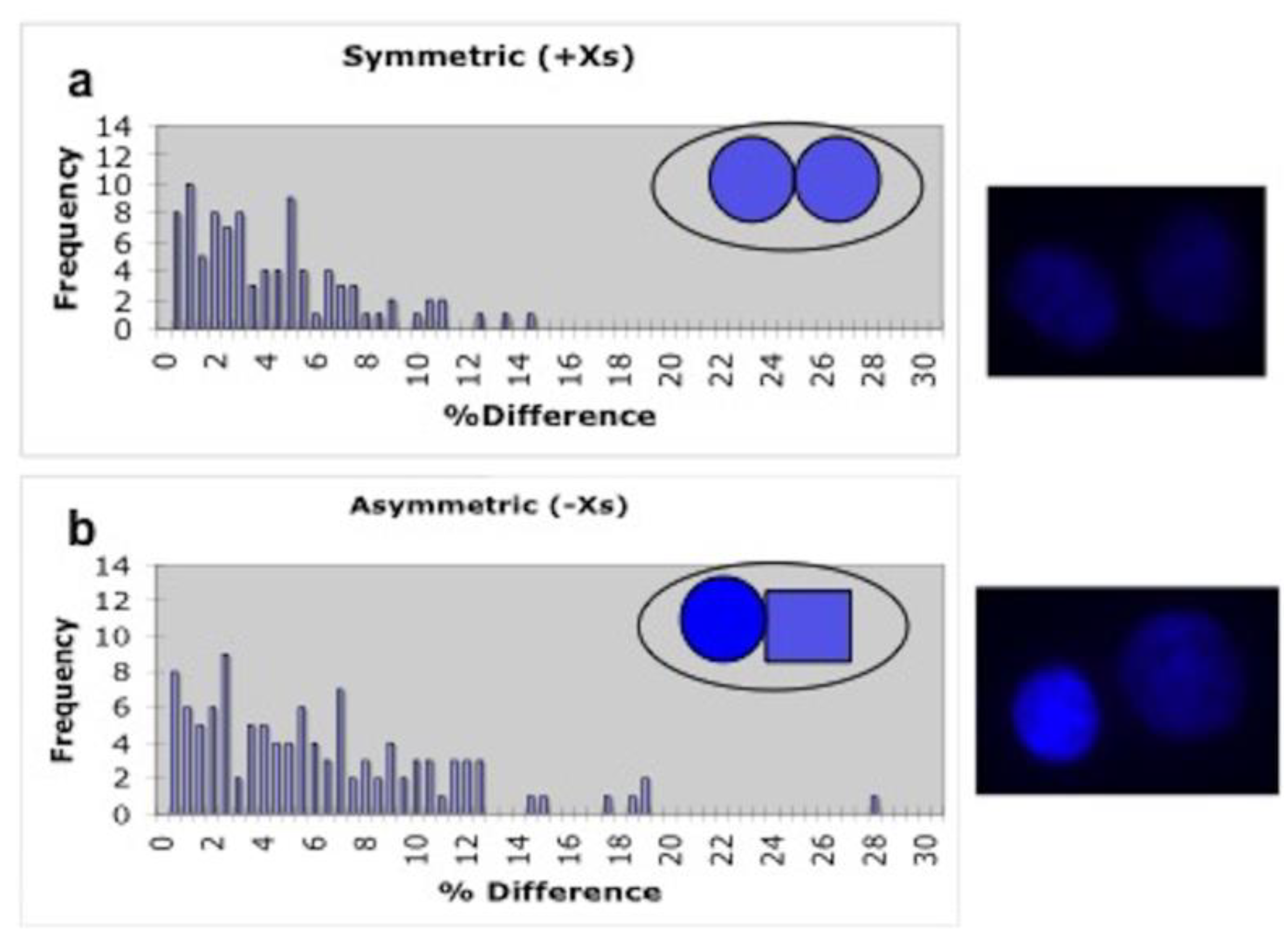
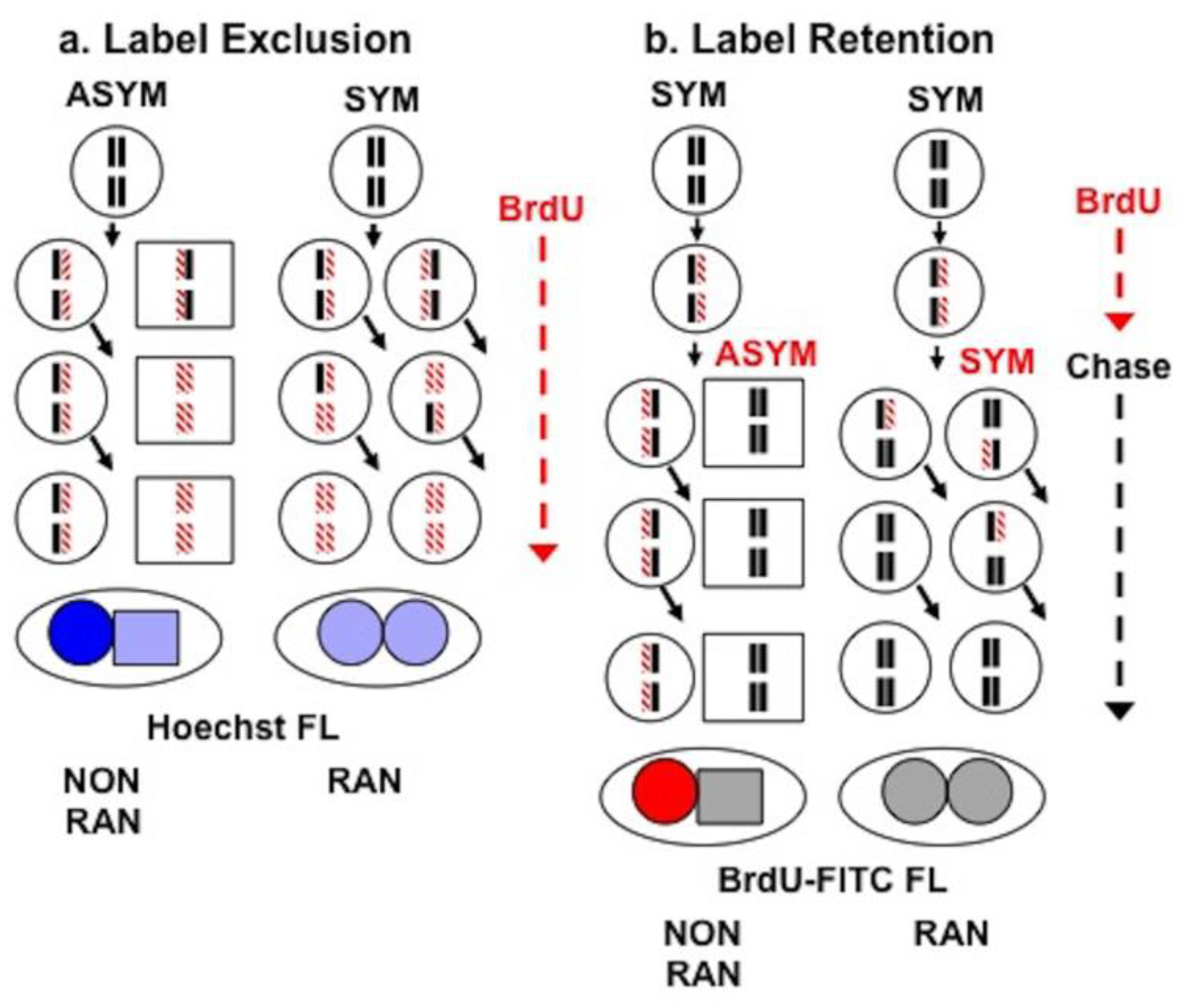
2.5. Sister Chromatid Segregation Analyses
2.6. Statistical Analyses
3. Results
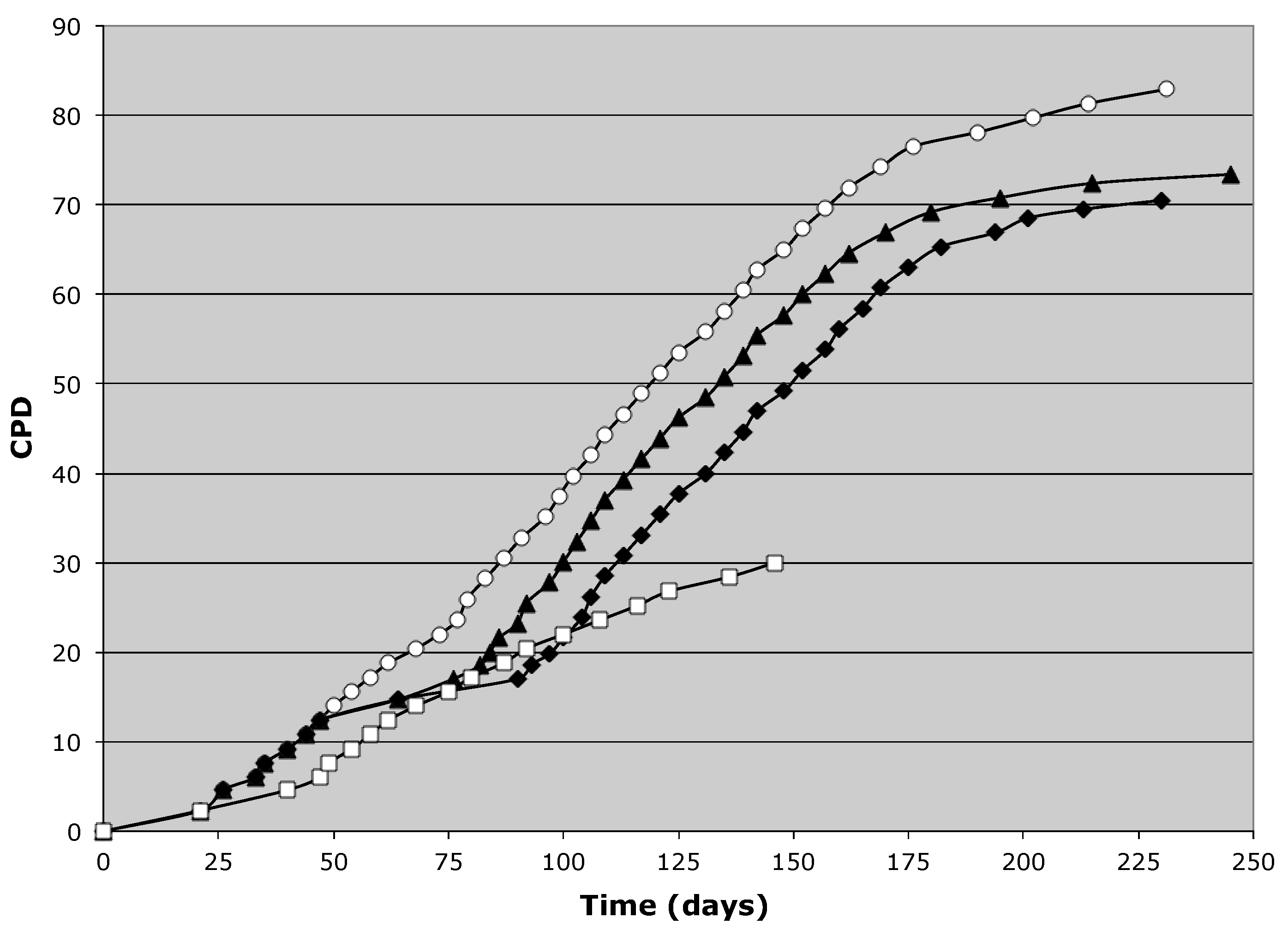
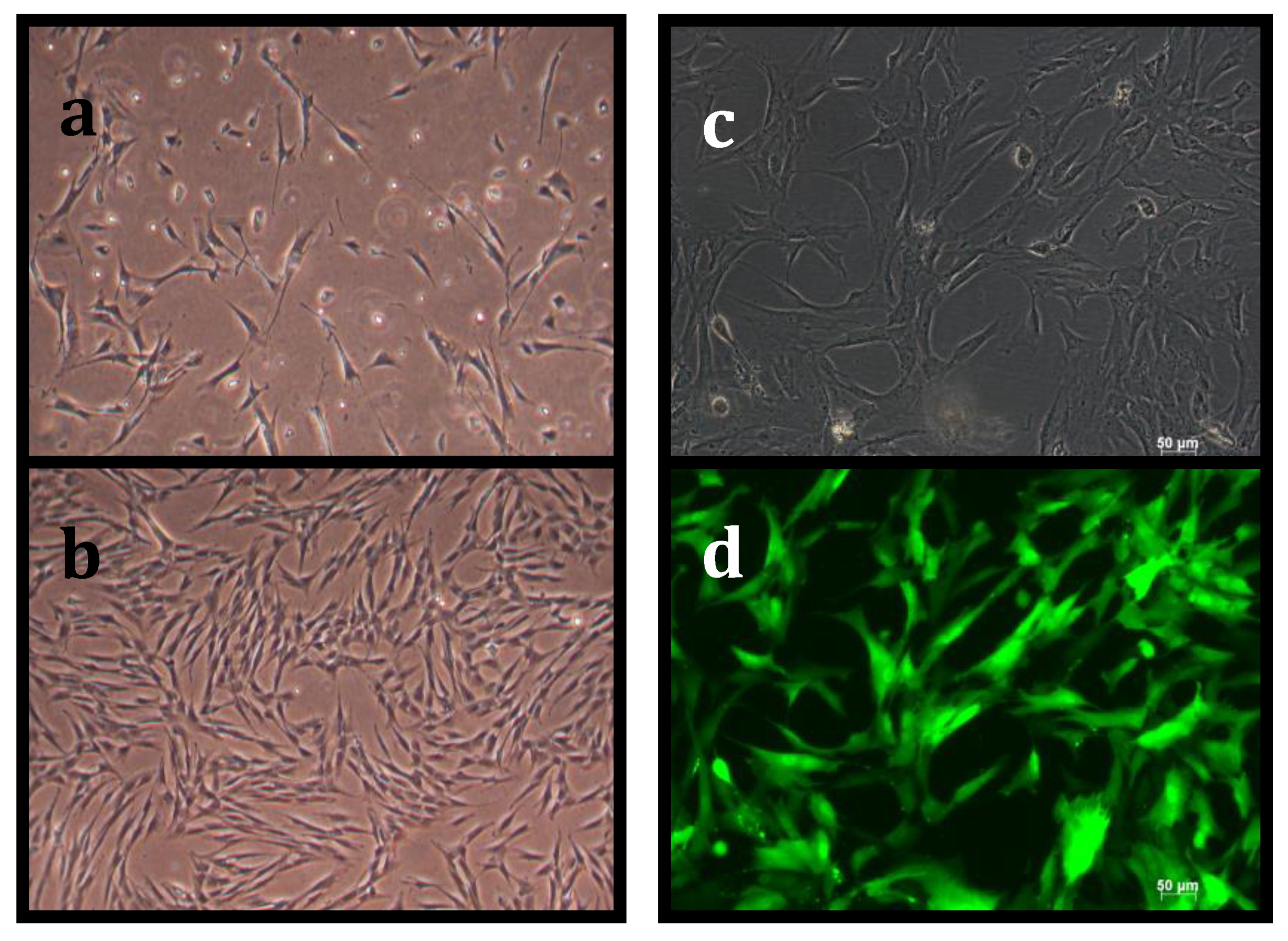
3.1. Suppression of Asymmetric Cell Kinetics (SACK) Derivation of Human Hepatic Cell Strain 12(3)
3.2. Tissue Stem Cell Kinetics Properties of Human Hepatic Cell Strain 12(3)
3.3. Detection of Non-Random Sister Chromatid Cosegregation by Human Hepatic Strain 12(3) Cells under Conditions that Promote Asymmetric Self-Renewal Kinetics
4. Discussion
5. Conclusions
6. Patents
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Merok, J.R.; Lansita, J.A.; Tunstead, J.R.; Sherley, J.L. Co-segregation of chromosomes containing immortal DNA strands in cells that cycle with asymmetric stem cell kinetics. Cancer Res. 2002, 62, 6791–6795. [Google Scholar] [PubMed]
- Lansita, J.A.; Merok, J.R.; Sherley, J.L. Physicochemical demonstration of immortal DNA strand segregation. In Mechanisms of Toxicity, Carcinogenesis, Cancer Prevention and Cancer Therapy Abstracts of the Seventeenth Aspen Cancer Conference. Toxicol. Pathol. 2003, 31, 163–164. [Google Scholar]
- Merok, J.R.; Tunstead, J.R.; Lansita, J.A.; Sherley, J.L. Demonstration of an immortal DNA strand mechanism in cells that cycle with asymmetric stem cell kinetics-implications for mechanisms of human cancer and aging. In Mechanisms of Toxicity, Carcinogenesis, Cancer Prevention and Cancer Therapy: Abstracts of the Seventeenth Aspen Cancer Conference. Toxicol. Pathol. 2003, 31, 165–166. [Google Scholar]
- Rambhatla, L.; Ram-Mohan, S.; Cheng, J.J.; Sherley, J.L. Immortal DNA strand co-segregation requires p53/IMPDH-dependent asymmetric self-renewal associated with adult stem cells. Cancer Res. 2005, 65, 3155–3161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huh, Y.H.; Sherley, J.L. Molecular Cloaking of H2A.Z on Mortal DNA Chromosomes During Nonrandom Segregation. STEM Cells 2011, 29, 1620–1627. [Google Scholar] [CrossRef] [PubMed]
- Huh, Y.H.; King, J.; Cohen, J.; Sherley, J.L. SACK-Expanded Hair Follicle Stem Cells Display Asymmetric Nuclear Lgr5 Expression with Non-Random Sister Chromatid Segregation. Sci. Rep. 2011, 1, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huh, Y.H.; Cohen, J.; Sherley, J.P.J. Higher 5-hydroxymethylcytosine identifies immortal DNA strand chromosomes in asymmetrically self-renewing distributed stem cells. Proc. Natl. Acad. Sci. USA 2013, 110, 16862–16867. [Google Scholar] [CrossRef] [Green Version]
- Huh, Y.H.; Sherley, J.P.J. Decreased H3K27 and H3K4 trimethylation on mortal chromosomes in distributed stem cells. Cell Death Dis. 2014, 5, e1554. [Google Scholar] [CrossRef] [Green Version]
- Huh, Y.H.; Noh, M.; Burden, F.; Chen, J.C.; Winkler, D.A.; Sherley, J.L. Use of sparse feature bioinformatics to identify a novel pattern-specific biomarker for counting asymmetrically self-renewing distributed stem cells. Stem Cell Res. 2015, 14, 144–154. [Google Scholar] [CrossRef] [Green Version]
- Panchalingam, K.; Noh, M.; Huh, Y.H.; Sherley, J.L. Distributed Stem Cell Kinetotoxicity: A New Concept to Account for the Human Carcinogenicity of Non-Genotoxic Toxicants. In Human Stem Cell Toxicology, Issues in Toxicology No. 29; Sherley, J.L., Ed.; Royal Society of Chemistry: London, UK, 2016; pp. 250–279. [Google Scholar]
- Cairns, J. Mutation selection and the natural history of cancer. Nat. Cell Biol. 1975, 255, 197–200. [Google Scholar] [CrossRef]
- Cairns, J. Somatic stem cells and the kinetics of mutagenesis and carcinogenesis. Proc. Natl. Acad. Sci. USA 2002, 99, 10567–10570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cairns, J. Cancer and the Immortal Strand Hypothesis. Genet 2006, 174, 1069–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherley, J. Mechanisms of Genetic Fidelity in Mammalian Adult Stem Cells. In Tissue Stem Cells; Potten, C.S., Clarke, R.B., Wilson, J., Renehan, A.G., Eds.; Taylor Francis: New York, NY, USA, 2006; pp. 37–54. [Google Scholar]
- Sherley, J.L. New cancer diagnostics and therapeutics from a 9th “hallmark of cancer”: Symmetric self-renewal by mutated distributed stem cells. Expert Rev. Mol. Diagn. 2013, 13, 797–810. [Google Scholar] [CrossRef] [PubMed]
- Sherley, J.L. The p53 tumor suppressor gene as regulator of somatic stem cell renewal division. Cope 1996, 12, 9–10. [Google Scholar]
- Karpowicz, P.; Morshead, C.M.; Kam, A.; Jervis, E.; Ramunas, J.; Cheng, V.; Van Der Kooy, D. Support for the immortal strand hypothesis: Neural stem cells partition DNA asymmetrically in vitro. J. Cell Biol. 2005, 170, 721–732. [Google Scholar] [CrossRef] [Green Version]
- Sundararaman, B.; Avitabile, D.; Konstandin, M.H.; Cottage, C.T.; Gude, N.; Sussman, M.A. Asymmetric chromatid segregation in cardiac progenitor cells is enhanced by Pim-1 kinase. Circ. Res. 2012, 110, 1169–1173. [Google Scholar] [CrossRef] [Green Version]
- Potten, C.S.; Hume, W.J.; Reid, P.; Cairns, J. The segregation of DNA in epithelial stem cells. Cell 1978, 15, 899–906. [Google Scholar] [CrossRef]
- Potten, C.S.; Owen, G.; Booth, D. Intestinal stem cells protect their genome by selective segregation of template DNA strands. J. Cell Sci. 2002, 115, 2381–2388. [Google Scholar]
- Smith, G.H. Label-retaining epithelial cells in mouse mammary gland divide asymmetrically and retain their template DNA strands. Development 2005, 132, 681–687. [Google Scholar] [CrossRef] [Green Version]
- Shinin, V.; Gayraud-Morel, B.; Gomès, D.; Tajbakhsh, S. Asymmetric division and cosegregation of template DNA strands in adult muscle satellite cells. Nat. Cell Biol. 2006, 8, 677–682. [Google Scholar] [CrossRef]
- Conboy, M.J.; Karasov, A.O.; Rando, T.A. High incidence of non-random template strand segregation and asymmetric fate determination in dividing stem cells and their progeny. PLoS ONE 2007, 5, e102. [Google Scholar]
- Kajstura, J.; Bai, Y.; Cappetta, N.; Kim, J.; Arranto, C.; Sanada, F.; D’Amario, M.; Matsuda, A.; Bardelli, S.; Ferreira-Martins, J.; et al. Tracking chromatid segregation to identify human cardiac stem cells that regenerate extensively the infarcted myocardium. Circ. Res. 2012, 111, 894–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pine, S.R.; Ryan, B.M.; Varticovski, L.; Robles, A.I.; Harris, C.C. Microenvironmental modulation of asymmetric cell division in human lung cancer cells. Proc. Natl. Acad. Sci. USA 2010, 107, 2195–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bussard, K.M.; Boulanger, C.A.; Kittrell, F.S.; Behbod, F.; Medina, D.; Smith, G.H. Immortalized, premalignant epithelial cell populations contain long-lived, label-retaining cells that asymmetrically divide and retain their template DNA. Breast Cancer Res. 2010, 12, R86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hari, D.; Xin, H.-W.; Jaiswal, K.; Wiegand, G.; Kim, B.-K.; Ambe, C.; Burka, D.; Koizumi, T.; Ray, S.; Garfield, S.; et al. Isolation of Live Label-Retaining Cells and Cells Undergoing Asymmetric Cell Division via Nonrandom Chromosomal Cosegregation from Human Cancers. Stem Cells Dev. 2011, 20, 1649–1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, H.-W.; Hari, D.M.; Mullinax, J.E.; Ambe, C.M.; Koizumi, T.; Ray, S.; Anderson, A.J.; Wiegand, G.W.; Garfield, S.H.; Thorgeirsson, S.S.; et al. Tumor-Initiating Label-Retaining Cancer Cells in Human Gastrointestinal Cancers Undergo Asymmetric Cell Division. Stem Cells 2012, 30, 591–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Jeganathan, G.; Amiri, S.; Morgan, K.M.; Ryan, B.M.; Pine, S.R. Asymmetric segregation of template DNA strands in basal-like human breast cancer cell lines. Mol. Cancer 2013, 12, 139. [Google Scholar] [CrossRef] [Green Version]
- Rosenberger, R.F.; Kessel, M. Nonrandom Sister Chromatid Segregation and Nuclear Migration in Hyphae of Aspergillus nidulans. J. Bacteriol. 1968, 96, 1208–1213. [Google Scholar] [CrossRef] [Green Version]
- Karpowicz, P.; Pellikka, M.; Chea, E.; Godt, D.; Tepass, U.; Van Der Kooy, D. The germline stem cells of Drosophila melanogaster partition DNA non-randomly. Eur. J. Cell Biol. 2009, 88, 397–408. [Google Scholar] [CrossRef]
- Yadlapalli, S.; Yamashita, Y.M. Chromosome-specific nonrandom sister chromatid segregation during stem-cell division. Nat. Cell Biol. 2013, 498, 251–254. [Google Scholar] [CrossRef] [Green Version]
- Kiel, M.J.; He, S.; Ashkenazi, R.; Gentry, S.N.; Teta, M.; Kushner, J.A.; Jackson, T.L.; Morrison, S.J. Haematopoietic stem cells do not asymmetrically segregate chromosomes or retain BrdU. Nat. Cell Biol. 2007, 449, 238–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sotiropoulou, P.A.; Candi, A.; Blanpain, C. The Majority of Multipotent Epidermal Stem Cells Do Not Protect Their Genome by Asymmetrical Chromosome Segregation. Stem Cells 2008, 26, 2964–2973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waghmare, S.K.; Bansal, R.; Lee, J.; Zhang, Y.V.; McDermitt, D.J.; Tumbar, T. Quantitative proliferation dynamics and random chromosome segregation of hair follicle stem cells. EMBO J. 2008, 27, 1309–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escobar, M.; Nicolas, P.; Sangar, F.; Laurent-Chabalier, S.; Clair, P.; Joubert, D.; Jay, P.; Legraverend, C. Intestinal epithelial stem cells do not protect their genome by asymmetric chromosome segregation. Nat. Commun. 2011, 2, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadlapalli, S.; Cheng, J.; Yamashita, Y.M. Drosophila male germline stem cells do not asymmetrically segregate chromosome strands. J. Cell Sci. 2011, 124, 933–939. [Google Scholar] [CrossRef] [Green Version]
- Steinhauser, M.L.; Bailey, A.P.; Senyo, S.E.; Guillermier, C.; Perlstein, T.S.; Gould, A.P.; Lee, R.T.; Lechene, C.P. Multi-isotope imaging mass spectrometry quantifies stem cell division and metabolism. Nat. Cell Biol. 2012, 481, 516–519. [Google Scholar] [CrossRef] [Green Version]
- Lark, K.G. Nonrandom Segregation of Sister Chromatids. In Genetics & Developmental Biology; Teas, H.J., Ed.; University of Kentucky Press: Lexington, KY, USA, 1969; pp. 8–24. [Google Scholar]
- Lark, K.G. Discovering non-random segregation of sister chromatids: The naïve treatment of a premature discovery. Front. Oncol. 2013, 2, 211. [Google Scholar] [CrossRef] [Green Version]
- Lark, K.G.; Consigli, R.A.; Minocha, H.C. Segregation of Sister Chromatids in Mammalian Cells. Science 1966, 154, 1202–1205. [Google Scholar] [CrossRef]
- Lark, K.G. Nonrandom segregation of sister chromatids in Vicia faba and Triticum boeoticum. Proc. Natl. Acad. Sci. USA 1967, 58, 352–359. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-S.; Crane, G.G.; Merok, J.R.; Tunstead, J.R.; Hatch, N.L.; Panchalingam, K.; Powers, M.J.; Griffith, L.G.; Sherley, J.L. Clonal expansion of adult rat hepatic stem cell lines by suppression of asymmetric cell kinetics (SACK). Biotechnol. Bioeng. 2003, 83, 760–771. [Google Scholar] [CrossRef]
- Paré, J.; Sherley, J.L. Biological Principles for Ex Vivo Adult Stem Cell Expansion. In Current Topics in Developmental Biology; Schatten, G., Ed.; Elsevier, Inc.: San Diego, CA, USA, 2006; Volume 73, pp. 141–171. [Google Scholar]
- Paré, J.-F.; Sherley, J.L. Culture Environment-Induced Pluripotency of SACK-Expanded Tissue Stem Cells. J. Biomed. Biotechnol. 2011, 2011, 312457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherley, J.P.J. Ex vivo Expansion of Human Adult Pancreatic Cells with Properties of Distributed Stem Cells by Suppression of Asymmetric Cell Kinetics. J. Stem Cell Res. Ther. 2013, 3, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherley, J.L.; Panchalingam, K. Methods for Ex Vivo Propagation of Adult Hepatic Stem Cells. U.S. Patent No. 7,824,912 B2, 2 November 2010. [Google Scholar]
- Rambhatla, L.; Bohn, S.A.; Stadler, P.B.; Boyd, J.T.; Coss, R.A.; Sherley, J.L. Cellular senescence: Ex vivo p53-dependent asymmetric cell kinetics. J. Biomed. Biotechnol. 2001, 1, 28–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taghizadeh, R.R.; Sherley, J.L. CFP and YFP, but Not GFP, Provide Stable Fluorescent Marking of Rat Hepatic Adult Stem Cells. J. Biomed. Biotechnol. 2008, 2008, 453590. [Google Scholar] [CrossRef]
- Sherley, J.L. Asymmetric self-renewal: The mark of the adult stem cell. In Stem Cell Repair and Regeneration; Habib, N.A., Gordon, M.Y., Levicar, N., Jiao, L., Thomas-Black, G., Eds.; Imperial College Press: London, UK, 2005; pp. 21–28. [Google Scholar]
- Dutton, R.; Abdi, F.; Minnetyan, L.; Sherley, J.L.; Asymmetrex, L. A Computational Simulation Technology for Specific Counting of Perinatal and Postnatal Human Tissue Stem Cells for Transplantation Medicine. OBM Transplant. 2020, 4, 1–24. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trial No. | Age (Years) | Gender | Purine | CPD |
|---|---|---|---|---|
| 1 | 6 | M | Control-1 | 24 |
| Control-2 | 10 | |||
| Xn | 38 | |||
| Hx | 29 | |||
| Xs | 36 | |||
| 2 | 14 | F | Control | 10 |
| Hx | 26 | |||
| 3 | 1 | M | Control | 22 |
| Xn | 10 | |||
| Hx-1 | 30 | |||
| Hx-2 | 14 | |||
| Xs-1 | 77 | |||
| Xs-2 | 21 | |||
| 4 | 24 | M | Control | 16 |
| Xs-1 | 26 | |||
| Xs-2 | 13 |
| Time-Lapse Culture Condition | ||
|---|---|---|
| Pedigree Type | Xs-Free | Xs-Supplemented |
| Asymmetric | 7% | 9% |
| Terminal Cell | 23% | 37% |
| Terminal Division | 13% | 11% |
| Symmetric | 33% | 17% |
| Mixed | 23% | 26% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panchalingam, K.; Jacox, L.; Cappiello, B.D.; Sherley, J.L. Non-Random Sister Chromatid Segregation in Human Tissue Stem Cells. Symmetry 2020, 12, 1868. https://doi.org/10.3390/sym12111868
Panchalingam K, Jacox L, Cappiello BD, Sherley JL. Non-Random Sister Chromatid Segregation in Human Tissue Stem Cells. Symmetry. 2020; 12(11):1868. https://doi.org/10.3390/sym12111868
Chicago/Turabian StylePanchalingam, Krishnanchali, Laura Jacox, Benjamin D. Cappiello, and James L. Sherley. 2020. "Non-Random Sister Chromatid Segregation in Human Tissue Stem Cells" Symmetry 12, no. 11: 1868. https://doi.org/10.3390/sym12111868
APA StylePanchalingam, K., Jacox, L., Cappiello, B. D., & Sherley, J. L. (2020). Non-Random Sister Chromatid Segregation in Human Tissue Stem Cells. Symmetry, 12(11), 1868. https://doi.org/10.3390/sym12111868