Abstract
Experimental and theoretical results are presented based on vibrational spectra and motional dynamics of 1,8-bis(dimethylamino)naphthalene (DMAN) and its protonated forms (DMANH+ and the DMANH+ HSO4− complex). The studies of these compounds have been performed in the gas phase and solid-state. Spectroscopic investigations were carried out by infrared spectroscopy (IR), Raman, and incoherent inelastic neutron scattering (IINS) experimental methods. Density functional theory (DFT) and Car–Parrinello molecular dynamics (CPMD) methods were applied to support our experimental findings. The fundamental investigations of hydrogen bridge vibrations were accomplished on the basis of isotopic substitutions (NH → ND). Special attention was paid to the bridged proton dynamics in the DMANH+ complex, which was found to be affected by interactions with the HSO4− anion.
1. Introduction
In the last few years, considerable attention has been devoted to the symmetry of hydrogen bonding [1,2,3,4,5,6,7,8,9,10]. Hydrogen maleate, hydrogen phthalate anions, and proton sponges have been studied intensively due to their physicochemical features that are derived from the presence of an intramolecular hydrogen bond [2,4,5,7,8,11,12,13,14]. The significance of proton sponges, the main object of the current study, is demonstrated by their high presence in the literature (e.g., [15,16,17,18,19,20,21]). The discussion concerning the symmetry of the hydrogen bridge has been the subject of many experimental and theoretical studies. The X-ray, neutron diffraction, incoherent inelastic neutron scattering (IINS), infrared spectroscopy (IR), and nuclear magnetic resonance (NMR) methods of Limbach et al. [22,23,24,25] and Perrin et al. [1,7] have been applied to investigate the isotopic perturbation of the equilibrium [26] to distinguish between symmetric and asymmetric intramolecular hydrogen bonds.
Starting with the paper by Alder [27], where the first synthesis of 1,8-bis(dimethylamino)naphthalene (DMAN; naphthalene proton sponges) was published, proton sponges have raised a great interest on the side of researchers [28,29,30,31,32,33]. The most promising feature of proton sponges is their significant basicity, which has been investigated in a series of papers [34,35,36,37,38,39]. The next unique trait of proton sponges is their extremely strong hydrogen bonding, already analyzed by structural and spectroscopic methods [40,41,42,43,44,45,46,47,48,49,50]. Remarkably, the protonation process can occur by the direct proton transfer from a strong acid to between two nitrogen atoms. However, in [51,52,53,54], an elaborate out–in protonation mechanism is outlined. Such rotational mechanism rests upon the intermolecular protonation of one of the amino groups by a strong acid, with the following reorientation (isomerization) of this group, and formation of the intramolecular hydrogen bond NHN. The scientific value/attractiveness of proton sponges also lies in the possibility of double protonation of the hydrogen bridge [37,55].
The localization of the proton in a very short hydrogen bridge in proton sponges seems to be a very interesting scientific challenge. A solution to such a challenge has been suggested in a number of structural as well as spectroscopic studies [38,39,40,41,42,43,44,45]. Theoretical studies of proton dynamics in a hydrogen bridge are of particular interest [55,56,57,58,59,60,61] because they provide information on the time-evolution of metric parameters involved in hydrogen bridge formation, therefore giving an insight into the dynamical features of the studied system. It is very hard to get similar information only by the construction and analysis of standard/static models. These papers show the dynamics of the bridged proton in proton sponges by the Car–Parrinello molecular dynamics (CPMD) method. In [62,63,64,65,66,67], the authors investigated a strong influence of steric squeezing of bulky groups in Positions 2 and 7 on the hydrogen bridge in proton sponges. In the recent paper by Pozharskii et al. [68], the authors opened up a synthetic route to new proton sponges, exhibiting the strong buttressing effect mentioned above. The expected result, which is currently being verified experimentally, is an extremely low energy barrier on proton transfer (ΔE ~ 0.1 kcal/mol, which effectively means the barrier’s absence) in these new proton sponges, which are also expected to possess extremely short NHN bridges [68].
It should be noted that proton sponges are interesting not only in the area of fundamental research. Studies have shown the potential use of the investigated proton sponges in cardiology and dye fields [69,70].
The main aim of this study is the experimental and computational investigation of the symmetry/asymmetry of the NHN hydrogen bridge in proton sponges (Figure 1). This requires a detailed structural and spectroscopic insight into hydrogen bridge properties. For this purpose, the paper presents two synthesized complexes—DMANH+ HSO4− and DMAND+ DSO4−—for the consequent studies of the isotopic effects on spectral parameters. As a part of these studies, IINS and IR spectra have been measured in the wide spectral range of the synthesized complexes. Here, for the first time, we present the IINS results for the discussed complex and their comparison with data obtained by Car–Parrinello molecular dynamics [71]. The analysis of the experimental and calculated vibrational spectra was completed; the studies of the bridged proton dynamics and molecular skeleton symmetry were performed using the Car–Parrinello molecular dynamics (CPMD) method [71].
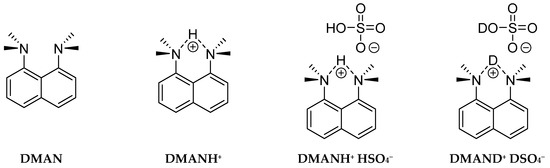
Figure 1.
Chemical structures of studied molecules DMAN and DMANH+ and complexes DMANH+ HSO4 and DMAND+ DSO4−.
2. Materials and Methods
2.1. Compound and Synthesis
The studied compound and solvents were purchased from Sigma-Aldrich and used without further purification. Melting points were determined in glass capillaries on a Stuart SMP30 device and are uncorrected.
Bis(dimethylamino)naphthalene Sulfate
To the vigorously stirred solution of 1,8-bis(dimethylamino)naphthalene (10 g, 47 mmol) in 200 mL of diethyl ether, a 98% aqueous solution of H2SO4 (2.7 mL, 50 mmol) was added dropwise. The resulting suspension was additionally stirred for 15 min and filtrated. The precipitate was dried over P2O5 in vacuo (2 torr) for 2 h to turn the product into white crystalline powder with a 90% yield (13.2 g); melting point (mp) 177–178 °C. 1H NMR (400 MHz, DMSO-d6) δ 18.30 (s, 1H), 8.15–8.07 (m, 4H), 7.74 (t, J = 7.9 Hz, 2H), 3.15 (d, J = 2.6 Hz, 12H). 13C NMR (100 MHz, DMSO-d6) δ 145.17, 135.29, 129.33, 127.43, 122.29, 119.45, 46.23.
8-Bis(dimethylamino)naphthalene Sulfate-d2
To the vigorously stirred solution of 1,8-bis(dimethylamino)naphthalene (10 g, 47 mmol) in 200 mL of absolute diethyl ether, a 98% solution of D2SO4 in D2O (2.8 mL, 50 mmol) was added dropwise. The resulting suspension was additionally stirred for 15 min and filtrated. The precipitate was dried over P2O5 in vacuo (2 torr) for 2 h to turn the product into white crystalline powder with a 95% yield (14 g); mp 178–179 °C. 1H NMR (400 MHz, D2O) δ 7.84–7.74 (m, 4H), 7.55 (t, J = 8.0 Hz, 2H), 3.00 (s, 12H). 13C NMR (100 MHz, D2O) δ 143.79, 135.04, 129.08, 126.89, 121.20, 118.63, 45.58. 2H NMR (500 MHz, solid-state) δ 19.47, 14.20. The solid-state magic angle spinning (MAS) 2H NMR (76.7 MHz, 18 kHz spinning, 3.2 mm rotor) confirmed the successful deuteration: two isotropic signals were observed at ca. 19.5 ppm (the NDN bridging deuterons) and ca. 14.2 ppm (the remaining DSO4– deuterons; see Figure A8).
2.2. Infrared Spectra Measurements
Fourier transform far- and middle-infrared (FT-FIR and FT-MIR) absorption measurements were performed using a Bruker Vertex 70v vacuum Fourier transform spectrometer. The transmission spectra were collected with a resolution of 2 cm−1 with 64 scans. The FT-FIR spectra (500–50 cm−1) were collected for the samples suspended in Apiezon N grease and placed on a polyethylene disc. The FT-MIR spectra were collected for the samples in a KBr pellet.
2.3. Incoherent Inelastic Neutron Scattering (IINS) Measurements
Inelastic neutron scattering spectra were measured using the time-of-flight inverted geometry spectrometer NERA (pulsed IBR-2 reactor in JINR (Dubna, Russia)) at 5 K temperature in the 5–1600 cm−1 spectral region. The spectra were converted from neutron per channel to S(Q, ω) function per energy transfer according to the scattering law [72].
The IINS and IR spectra of DMAN are presented in Figure A1.
2.4. NMR Measurements
Liquid-state NMR experiments were performed using a Bruker Avance III 400 spectrometer (400.13 MHz for 1H and 100.61 MHz for 13C) at the Centre for Magnetic Resonance, St. Petersburg State University Research Park. 1H and 13C NMR chemical shifts were referenced to tetramethylsilane (TMS) using the unified scale, according to IUPAC recommendations [73]. The number of scans varied between 128 and 256. The corresponding spectra are presented in Figure A2, Figure A3, Figure A4, Figure A5, Figure A6, Figure A7 and Figure A8. The NMR spectra and the line shape analysis were performed using MestReNova 8.1 software [74].
2.5. Static DFT and D3-DFT Calculations
The static density functional theory (DFT) calculations were accomplished for DMANH+ and the DMANH+ HSO4− complex using the Gaussian 16 Rev. C01 program [75], with the correlation of the Lee–Yang–Parr formula (B3LYP) [76,77]. The 6-311++G(d,p) basis set [78,79,80] was used. The atom pair-wise correction method for dispersion forces (DFT-D3) was applied [81]. The potential curves of proton transfer within the NHN intramolecular hydrogen bond were calculated using the stepwise elongation of the N–H distance by 0.05 Å gradient, with full optimization of the rest of the structural parameters. The calculations were performed for the gas phase and acetonitrile (ACN; the polarizable continuum model (PCM) approach [82] in an IEF-PCM formulation). The computed structures were shown using the MOLDEN program [83].
2.6. Symmetry-Adapted Perturbation Theory Calculations
Next, the investigation of the interaction energy components in the DMANH+ HSO4− complex was carried out within the framework of symmetry-adapted perturbation theory (SAPT) [84]. The simulations were performed at the SAPT2 level of theory [85] for the B3LYP/6-311++G(d,p) structure. The SAPT2 calculations for the DFT-optimized structures were carried out with the aug-cc-pVDZ basis set [86], employed for atomic orbital expansion. The interaction energy calculated at the SAPT2/aug-cc-pVDZ level was corrected for the basis set superposition error using the counterpoise approach, taking DMANH+ as the “monomer” and the HSO4− anion as the second “monomer” of the “dimer”. The SAPT calculations were performed using the Psi4 1.2.1 [87] program.
2.7. Car–Parrinello Molecular Dynamics Simulations
The first-principle molecular dynamics (FPMD) simulations were performed on the basis of the Car–Parrinello method [71]. The DMAN molecule and its protonated complex with HSO4− were investigated in the gas phase. The models for CPMD simulations were constructed based on DFT results for DMAN and the DMANH+ HSO4− complex. The computations were carried out using the CPMD 3.17.1 program [88]. Molecular dynamics trajectory postprocessing was achieved with the utilities developed onsite and with the VMD 1.9.3. program [89]. The Gnuplot graphics package [90] and the VMD 1.9.3. program [89] were used for the results’ graphical presentation. The DMAN molecule and its protonated complex were placed in cubic boxes with a = 16 Å and a = 18 Å, respectively. The weak interactions present in the investigated complex were reproduced with the empirical van der Waals protocol developed by Grimme [81]. The PBE exchange-correlation DFT functional [91] was used in both simulations. The Troullier–Martins norm-conserving pseudopotentials [92] substituted the core electrons, while the plane-wave Kohn–Sham orbitals were set to possess the maximum kinetic energy value of 80 Ry for DMAN and 100 Ry for the DMANH+ HSO4− complex. Hockney’s scheme of periodicity removal [93] ensured the simulation in the regime of the isolated system. The translational and rotational movements of the molecules, resulting from numerical rounding errors, were also removed at every 20 MD steps during the simulations. The dynamics of the orbital coefficients were carried out with the fictitious orbital mass of 400 a.u., and the timestep of the nuclear dynamics was 3 a.u. in the case of DMAN and 2 a.u. in the case of the studied complex. The CPMD simulations were performed with the equilibration of the simulation conditions and the subsequent trajectory collection for further metric and spectroscopic feature analyses. Equilibration was carried out by thermostatting the ionic temperature at 300 K for DMAN and 297 K for the complex using a separate Nosé–Hoover thermostat chain, with default settings for each degree of freedom (“massive” thermostatting) [94,95]. The data collection lasted for 21.8 ps for DMAN and 24.1 ps for the DMANH+ HSO4− complex. The NVE microcanonical ensemble was used, i.e., the thermostatting was discontinued during the production run simulations. Then, the obtained trajectories were used to extract information on the time-evolution of atoms forming the hydrogen bridge and of other metric parameters, e.g., the NC-NC dihedral angle, as well as to determine the vibrational features from the power spectra of the atomic velocity of the investigated molecules (Figure A9 and Figure A10).
3. Results and Discussion
3.1. SAPT Analysis of the DMANH+ HSO4− Complex
The SAPT scheme partitions the interaction energy of a dimer into diverse components [84,85]. For the purpose of this study, however, it is sufficient to use the most general scheme of grouping these components into electrostatic, exchange, polarization, and dispersion terms. The first one refers to the Coulombic interaction between the nuclei and electronic densities of the monomers DMANH+ and HSO4−, with the assumption that these monomers are in their original electronic states, that is, DMANH+ is not perturbed by HSO4− and vice versa. The same assumption holds for the exchange (Pauli repulsion) term. The effects of mutual polarization or induction, together with their impact on Pauli repulsion, are included in the third term. Finally, the last component gathers the instantaneous multipole attraction (dispersion forces) between the monomers. These four terms are composed of many contributions, some of them arising as the result of intramonomer electron correlation. Selecting the noncorrelated terms yields the Hartree–Fock interaction energy. All these contributions for the DMANH+ HSO4− complex are summarized in Table 1.

Table 1.
Interaction energy components (in kcal/mol) calculated at the SAPT2/aug-cc-pVDZ level of theory for the B3LYP/6-311++G(d,p) structure of the DMANH+ HSO4− complex in a vacuum.
A striking agreement is found between the electrostatics term and total SAPT2 interaction energy. The remaining three components of the intermolecular forces are close to zero when summed up. This fact is the manifestation of the dominant role of electrostatics in an ionic complex. Exchange, induction, and dispersion are important individually, but their combined effect is just one kcal/mol of destabilization. Interestingly, even for such a strongly bound complex, electron correlation strengthens the interaction by 9 kcal/mol, almost 10% of total SAPT2 energy. Nevertheless, the net interaction strength corresponds strongly to the purely Coulombic picture of the cation–anion pair. The impact of this strong electrostatic interaction is visible in the modified time scale of bridge proton dynamics, which is described in the next section.
3.2. Metric Parameter Time-Evolution Analysis Based on Car–Parrinello Molecular Dynamics
The CPMD simulations in the gas phase were carried out by us with a twofold aim: first, to identify the apparent symmetry of the gas-phase DMAN molecular skeleton, and second, to investigate in detail the behavior of the intramolecular hydrogen bridge. The timescale of over 20 ps was found sufficient to cover the characteristic features of the neutral and protonated DMAN. Firstly, we examined the relative arrangement of the aromatic rings, distorted by the sterically bulky substituents of the amine moieties. Figure 2 presents the time evolution of the relevant CN-CN dihedral angle between the C-N bonds (which effectively measures whether the two naphthalene rings are coplanar).
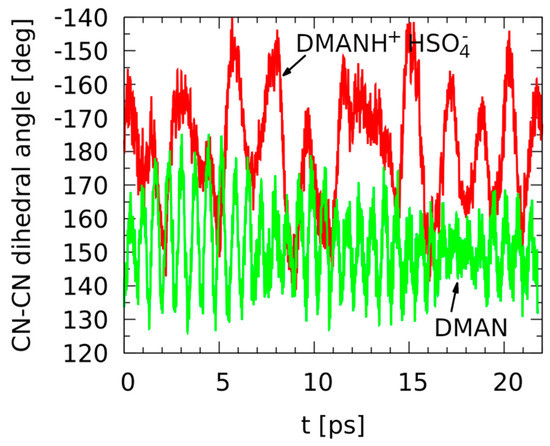
Figure 2.
Time evolution of the dihedral angle between the C-N bonds—results of the gas-phase Car–Parrinello molecular dynamics (CPMD) simulations for the neutral DMAN (green line) and DMANH+ HSO4− (red line).
The relatively slow motion of the aromatic rings is rather strongly harmonic in the nonprotonated DMAN, but the symmetry of the molecular skeleton is broken by the methyl groups, which are bulky enough to prevent the two rings from being coplanar. The average CN-CN dihedral angle is, for this case, 152.1°. When the DMANH+ HSO4− complex is studied, the average angle is 181.2°, much closer to the “ideal coplanarity” value. These findings are determined by the fact that in the nonprotonated DMAN, the lone pairs of the nitrogen atoms act, in addition to the size of the N(CH3)2 groups, as additional “steric-blocking” factors, and they do not allow the substituents of the naphthalene ring to cross the molecular plane. In effect, the movement of the -N(CH3)2 moieties is limited in its amplitude and centered at a value far from 180°. Additionally, the N···N distance is also larger in DMAN than in the protonated complex. This brings us to the role of the proton in the NHN hydrogen bridge. The proton connects the -N(CH3)2 moieties closely, bringing about a decrease in the N···N distance and increasing in the planarity of the compound. Additionally, the HSO4− group cannot be neglected while analyzing the results. The intermolecular dynamics of the DMANH+ HSO4− moieties are very slow in comparison to the intramolecular vibrations, leading to larger and less systematic amplitudes of the dihedral angle seen in Figure 2. Thus, within the 20 ps time scale, one might conclude that the cationic DMANH+, with the hydrogen bridge intramolecular linker, is planar but not necessarily more harmonic. It is visible that the trajectory of the dihedral angle is very harmonic for DMAN but much more rugged for DMANH+. This suggests that the dynamics of the hydrogen bridge in the complex might reveal strong anharmonicity (see Figure 3).
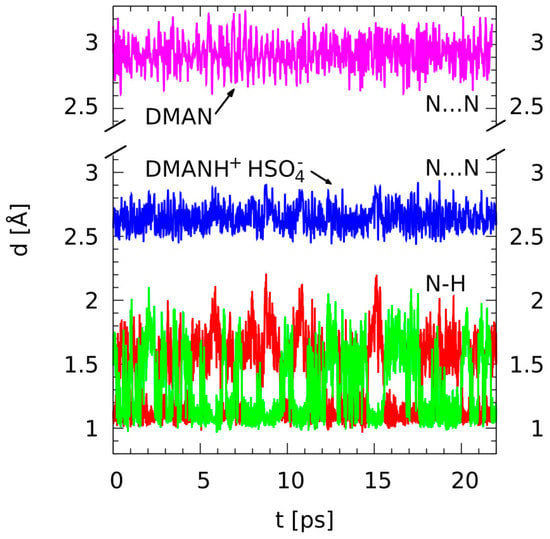
Figure 3.
Time evolution of selected metric parameters—results of the gas-phase CPMD simulations for the neutral DMAN (purple line: N···N distance) and DMANH+ HSO4− (blue line: N···N distance; red and green lines: bridge N-H distances).
Figure 3 is an example of the proton bridge dynamics studied by first-principle molecular dynamics methods. Time evolution of the important metric parameters (N-H, H···N, and N···N distances) is presented. The time scale available for the CPMD simulation is such that we could observe the high mobility of the bridged proton; the proton moves between the donor and acceptor atoms, which is indicated by the red and green lines crossing continually. For clarity, the data for the nonprotonated DMAN (the N···N distance) is presented as the inset above the data for the protonated complex. Figure 3 shows clearly how strongly the bridged proton is able to force the two nitrogen atoms closer. The N···N distance averaged over the CPMD run is 2.929 Å for the neutral DMAN and only 2.646 Å for the DMAN HSO4− complex. The hydrogen bridge is not totally symmetric within the 20 ps time scale: the averages of the two N-H distances are 1.346 and 1.396 Å. We do not expect this lack of symmetry in the bridge to result from any significant physical phenomenon because the molecular structure of DMANH+ does not contain any substituents that could break the symmetry of the proton potential function. We attribute the imperfect averaging of two N-H distances to the effect of the slow motion of the heavier HSO4− anion in the neighborhood of the hydrogen bond. It is visible that the proton resides at a given nitrogen atom for a period of ca. 1 picosecond, and then it abruptly switches to the other nitrogen site. This residence period of ca. 1 picosecond is much longer than the time of 0.3 ps that was observed in the CPMD simulations of gas-phase DMANH+ [56]. SAPT analysis has shown the absolutely dominant role of the electrostatic interactions, and this fact means that the structures with localized charges are preferred. In turn, this means that the transfer of the bridged proton over the hydrogen bond center displaces the location of the positive charge and is accompanied by the motion of the heavier HSO4- anion as well. Moreover, the central-symmetric structures with two equal N-H distances are not favored because such a structure would have the most symmetric charge distribution and would provide less than optimal electrostatic interaction.
3.3. DFT and D3-DFT Analysis of PES
Part of the presented studies covers the investigation of the potential curves of the proton transfer in DMANH+ and DMANH+ HSO4−. The studies are based on the calculations of the potential curves by the B3LYP/611++G(d,p) method, with Grimme correction (D3-DFT) [81] and without it (DFT), for the gas phase and involve the influence of the solvent (acetonitrile; the PCM model). The calculations with Grimme correction exposed a lower potential barrier of the proton transfer in the studied molecules in the gas phase and in the polar medium (Figure 4A,B) than the calculation without this correction. This conclusion is absolutely reliable since it is in accordance with the approximation suggested by Grimme [96]. Taking into account that dispersive interactions must show a stronger interrelation in the hydrogen bridge, as a consequence, there is a decrease of the potential barrier.
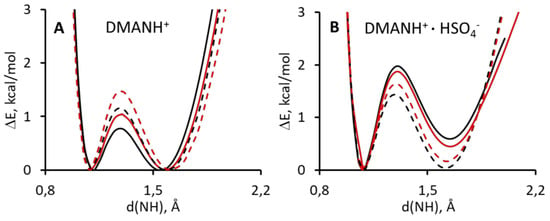
Figure 4.
Calculated density functional theory (DFT) and atom pair-wise correction method for dispersion force (DFT-D3) potential energy functions for gradual proton displacement in the intramolecular hydrogen bond of DMANH+ (A) and DMANH+ HSO4− (B). Left panel: black solid line, DMANH+ with Grimme approach for gas phase; red solid line, DMANH+ for gas phase; black dashed line, DMANH+ with Grimme approach for ACN; red dashed line, DMANH+ ACN. Right panel: black solid line, DMANH+ HSO4− with Grimme approach for gas phase; red solid line, DMANH+ HSO4− for gas phase; black dashed line, DMANH+ HSO4− with Grimme approach for ACN; red dashed line, DMANH+ HSO4− for ACN.
The analysis of the influence of the polar environment (the transition from the gas phase to acetonitrile) reveals the difference between DMANH+ and DMANH+ HSO4−. Notably, the specific interactions (intermolecular hydrogen bond between DMANH+ and HSO4−) increase the value of the potential barrier for the proton transfer in the intramolecular hydrogen bridge due to its weakening by bifurcation. In the case of DMANH+, the growth of environment polarity also brings about the increase of the barrier for proton transfer. This result, obtained by DFT and D3-DFT calculations, proves that the polarity increase leads to the weakening of the symmetric NHN hydrogen-bonding in DMANH+ under the absence of outside specific interactions (HSO4−). This trend is consistent with the generally accepted concepts of hydrogen bonds presented in [97,98]. Nevertheless, reverse dependence is observed in the presence of such specific interactions. For the complex DMANH+ HSO4−, the growth of the polar environment evokes the decrease of the barrier for the proton transfer. It is important to note that the calculation of the complex DMANH+ HSO4− for the gas phase does not present symmetric potential, which seemed to be expected because of the reorientation of the HSO4− anion under the proton transfer in the NHN bridge. However, the calculation of the complex DMANH+ HSO4− involving solvent influence has potential, with two minima of equal depth (Figure 4B).
The whole picture of the calculated potential curves enables us to conclude that the hydrogen bonding in DMANH+ HSO4− is symmetric but with the height of the barrier for proton transfer equal to 1.5 kcal/mol.
3.4. Comparison of Experimental IINS, IR, and Calculated CPMD Spectra
The spectroscopic measurements and theoretical calculations of molecular properties are complementary tools to gain insight into the nature of the studied systems. In particular, static DFT and dynamical CPMD simulations have determined the symmetry/asymmetry of the proton potential function in the bridge. There are, however, some points in which the experiment and calculations can be directly conjugated. Thus, this part of the paper is concerned with the analysis of the spectral bands assigned to the vibrations of bridged proton and methyl groups, performed by means of CPMD and DFT calculations (Figure 5 and Figure 6, Table A1 and Table A2). Moreover, these studies verify the reliability of the CPMD simulations. As well-known [99,100,101,102,103], in IINS spectra, the most intensive are the vibration bands of hydrogen atoms, especially torsional vibrations of the methyl groups [102,103,104,105,106,107,108,109,110]. Therefore, we conducted the measurements of IINS spectra and the calculation of the vibrations of all the hydrogen atoms of the investigated DMAN complex by the CPMD method (Figure A9 and Figure A10). According to the performed CPMD calculations, the bands of the deformational and torsional vibrations are located at 1380, 1100, 1030, 970, and 230 cm−1. Notably, the CPMD calculation gives a picture of more expanded bands, which is closer to reality (see spectra and Figure 5A and Figure 6C,D). There is a very good agreement between the calculated vibrations of the methyl groups and the measured positions of the bands in the IINS spectrum <1300 cm−1 (red arrows on Figure 5). The bands at 1380, 1100, 1030, and 970 cm−1 are assigned to δ, γ deformational, and τ torsional vibrations of the methyl groups, respectively. Interestingly, only the IINS spectrum features all the mentioned bands; meanwhile, IR and Raman spectra are characterized by just two bands (1050 and 330 cm−1). This fact clearly points out the prevalence of the IINS method over traditional IR and Raman methods when it comes to the studies of the methyl group vibrations.
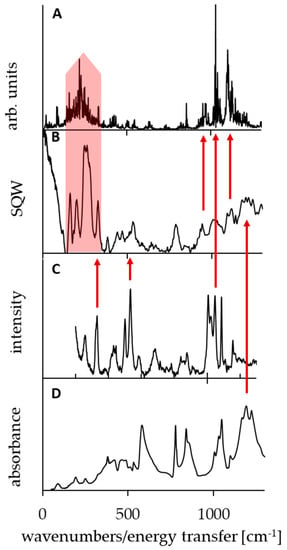
Figure 5.
Simulated power spectrum of the torsional methyl group modes by the CPMD method (panel (A)) and experimental incoherent inelastic neutron scattering (IINS; panel (B)), Raman (panel (C)), and infrared spectroscopy (IR) spectra (panel (D)) of DMANH+ HSO4−. The red arrows show the best fit between the calculated and experimental bands.
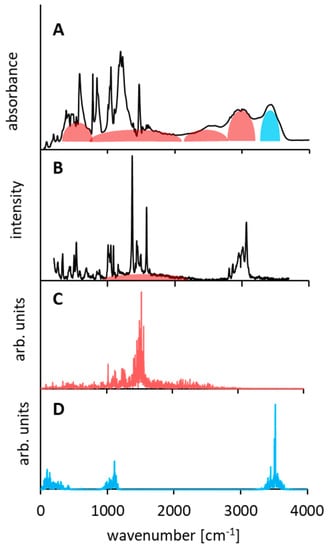
Figure 6.
Experimental IR (panel (A)), Raman (panel (B)) and atomic velocity power spectra for the hydrogen-bonded protons of the DMANH+ HSO4− complex (results of the CPMD simulation in the gas phase; panels (C) and (D) for NHN and OH protons, respectively).
As for the analysis of the bridged proton (NHN) vibrations, the CPMD calculation revealed the presence of only one wide intensive band covering a spectral region of 1000–2500 cm−1 and centered at 1500 cm−1 (Figure 6C). This calculated band corresponds to the wide band of a weak intensity in the Raman spectrum (Figure 6B). A weak intensity of the bands in the Raman spectrum, assigned to the hydrogen bridge vibrations, is quite reasonable and complies with the commonly accepted rule [111,112]. However, in the infrared spectrum of the studied complex (Figure 6A), one can observe a wide band (300–3000 cm−1) called Zundel’s continuum absorption, which is an indicator of extra strong hydrogen bonds [113,114]. The CPMD calculation, which complies with the experimental IR data, states that 3500, 1100–1000, and 300–80 cm−1 bands correspond to hydroxyl group vibrations (corresponding, respectively, to the ν(OH), δ(OH), γ(OH), and τ(OH) modes).
For a more precise analysis of the bands in the IINS spectra of the studied complex, we have also completed the studies of its deutero-substituted derivative (NHN → NDN). The degree and reliability of the synthesized deutero-substituted complex are presented by means of NMR measurements (Figure A5, Figure A6, Figure A7 and Figure A8). The comparison of two IINS spectra of the protonated and deuterated complexes (Figure A11) showed a decrease of the band intensity at 530 and 262 cm−1. These bands are assigned to the NHN and NHO vibrations of the hydrogen bridges. A similar interpretation was earlier performed in [115,116].
4. Conclusions
In this study, we present the experimental investigation of 1,8-bis(dimethylamino)naphthalene sulfate (DMANH+ HSO4−, as well as its deuterated isotopolog DMAND+ DSO4−; protonated proton sponges) in solid-state by means of inelastic incoherent neutron scattering (IINS), IR, and Raman spectroscopy, supplemented by the theoretical investigation of isolated DMANH+ and (DMANH+ HSO4−) salt using DFT, D3-DFT, and CPMD methods.
We show that the strong intramolecular NHN hydrogen bond in DMANH+ moiety is asymmetric and that the bridged proton is moving in a double-well potential. However, the energy barrier for the proton transfer is very low, ca. 1.5 kcal/mol, which determines the short residence time of the proton close to one of the nitrogen atoms of about 1 ps or 0.3 ps (depending on whether the presence of the HSO4− anion and its motion are taken into account or not). The HSO4− anion interacts with the cation by forming additional intermolecular OH···N hydrogen bonds and occasionally by forming bifurcated hydrogen bonds with the NHN proton; nevertheless, the energy of the anion–cation interaction remains predominantly electrostatic.
We show that the polarity of the surrounding medium has an effect on the proton transfer barrier height: for isolated DMANH+, higher polarity increases the barrier (as it destabilizes the central-symmetric structure with the most delocalized positive charge); in contrast, for DMANH+ HSO4−, higher polarity decreases the barrier height because of the higher stabilization of the more isolated HSO4− anion in the central-symmetric transition state structure.
The shortness of the NHN bridge leads to the extreme broadening of the NH stretching band in vibrational spectra (a Zundel continuum covering the 300–3000 cm−1 range is observed in the IR spectra). Thus, the intramolecular hydrogen bond in DMANH+ could be classified as a low-barrier hydrogen bond (LBHB) or a short strong hydrogen bond (SSHB), but its symmetry is present only on average. In the majority of instances, the symmetry is broken due to the asymmetric placement of the anion, whose motion determines the dynamics and drives the bridged proton transfer.
In conclusion, it is worth emphasizing that the CPMD calculation can quite realistically describe some aspects of experimental neutron scattering spectra.
Author Contributions
Conceptualization, A.J. and A.F.; methodology, P.P., A.J., J.J.P., E.A.G., A.F.P., A.S.A., P.M.T., and A.F.; software, P.P., A.J., J.J.P., E.A.G., A.F.P., A.S.A., P.M.T., and A.F.; validation, P.P., A.J., J.J.P., E.A.G., A.F.P., A.S.A., P.M.T., and A.F.; formal analysis, P.P., A.J., J.J.P., E.A.G., A.F.P., A.S.A., P.M.T., and A.F.; investigation, P.P., A.J., J.J.P., E.A.G., A.F.P., A.S.A., P.M.T., and A.F.; resources, P.P., A.J., J.J.P., E.A.G., A.F.P., A.S.A., P.M.T., and A.F.; data curation, P.P., A.J., J.J.P., E.A.G., A.F.P., A.S.A., P.M.T., and A.F.; writing—original draft preparation, A.J, J.P., and A.F.; writing—review and editing, P.P., A.J., J.J.P., E.A.G., A.F.P., A.S.A., P.M.T., and A.F.; visualization, P.P., A.J., J.J.P., E.A.G., A.F.P., A.S.A., P.M.T., and A.F.; supervision, A.J. and A.F.; project administration, A.J. and A.F.; funding acquisition, A.J. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Russian Science Foundation (RSF; grant no. 18-13-00050) and the Polish Government Plenipotentiary for JINR in Dubna (75/24/2020; p. 75; data 03.02.2020).
Acknowledgments
The authors gratefully acknowledge the Wrocław Center for Networking and Supercomputing (WCSS) for generous grants of CPU time and technical support.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A

Figure A1.
The IINS and IR spectra for 1,8-bis(dimethylamino)naphthalene.
Figure A1.
The IINS and IR spectra for 1,8-bis(dimethylamino)naphthalene.


Figure A2.
1H NMR spectrum for 1,8-bis(dimethylamino)naphthalene sulfate (400 MHz, DMSO-d6).
Figure A2.
1H NMR spectrum for 1,8-bis(dimethylamino)naphthalene sulfate (400 MHz, DMSO-d6).


Figure A3.
13C NMR spectrum for 1,8-bis(dimethylamino)naphthalene sulfate (100 MHz, DMSO-d6).
Figure A3.
13C NMR spectrum for 1,8-bis(dimethylamino)naphthalene sulfate (100 MHz, DMSO-d6).


Figure A4.
13C DEPT135 NMR spectrum for 1,8-bis(dimethylamino)naphthalene sulfate (100 MHz, DMSO-d6).
Figure A4.
13C DEPT135 NMR spectrum for 1,8-bis(dimethylamino)naphthalene sulfate (100 MHz, DMSO-d6).


Figure A5.
1H NMR spectrum for 1,8-bis(dimethylamino)naphthalene sulfate-d2 (400 MHz, D2O).
Figure A5.
1H NMR spectrum for 1,8-bis(dimethylamino)naphthalene sulfate-d2 (400 MHz, D2O).


Figure A6.
13C NMR spectrum for 1,8-bis(dimethylamino)naphthalene sulfate-d2 (100 MHz, D2O).
Figure A6.
13C NMR spectrum for 1,8-bis(dimethylamino)naphthalene sulfate-d2 (100 MHz, D2O).


Figure A7.
13C DEPT135 NMR spectrum for 1,8-bis(dimethylamino)naphthalene sulfate-d2 (100 MHz, D2O).
Figure A7.
13C DEPT135 NMR spectrum for 1,8-bis(dimethylamino)naphthalene sulfate-d2 (100 MHz, D2O).


Figure A8.
Solid-state 2H NMR spectrum for 1,8-bis(dimethylamino)naphthalene sulfate-d2 (76.7 MHz, MAS at 18 kHz).
Figure A8.
Solid-state 2H NMR spectrum for 1,8-bis(dimethylamino)naphthalene sulfate-d2 (76.7 MHz, MAS at 18 kHz).


Figure A9.
Calculated spectra of atomic velocity—results of the CPMD run for DMAN.
Figure A9.
Calculated spectra of atomic velocity—results of the CPMD run for DMAN.


Figure A10.
Calculated spectra of atomic velocity—results of the CPMD run for DMANH+ HSO4−.
Figure A10.
Calculated spectra of atomic velocity—results of the CPMD run for DMANH+ HSO4−.


Figure A11.
The IINS spectra of DMANH+ HSO4− (black line) and its derivative DMAND+ DSO4− (red line).
Figure A11.
The IINS spectra of DMANH+ HSO4− (black line) and its derivative DMAND+ DSO4− (red line).

Table A1.
Experimental IR, IINS, and calculated (B3LYP/6-311++G(d,p)) spectral data of DMAN.
Table A1.
Experimental IR, IINS, and calculated (B3LYP/6-311++G(d,p)) spectral data of DMAN.
| Mode № | IR | IINS | Wavenumber [cm−1] | Intensities [km/mol] |
|---|---|---|---|---|
| exp. | exp. | calc. | calc. | |
| tr | 9, 14, 35, 46 | |||
| 1 | 56 | 51.74 | 0.52 | |
| 2 | 72 | |||
| 3 | 84 | 80 | ||
| 4 | 93 | 91 | 99.49 | 0.23 |
| 5 | 115.46 | 3.64 | ||
| 6 | 124 | 122 | 122.23 | 0.28 |
| 7 | 126.25 | 1.04 | ||
| 8 | 135 | 141 | 151.23 | 0.59 |
| 9 | 162 | 163 | 172.50 | 0.95 |
| 10 | 193 | 190 | ||
| 11 | 200 | 211.02 | 0.02 | |
| 12 | 243 | 222 | 231.19 | 0.68 |
| 13 | 263 | 259 | 261.03 | 4.05 |
| 14 | 286 | 277 | 295.65 | 1.52 |
| 15 | 304 | 296 | 303.45 | 1.05 |
| 16 | 310 | 312.48 | 4.87 | |
| 17 | 334 | 325.28 | 0.29 | |
| 18 | 351 | 351 | 350.55 | 1.38 |
| 19 | 363 | 361 | 367.12 | 4.21 |
| 20 | 382 | 385 | 385.57 | 3.15 |
| 21 | 443 | 446 | 442.71 | 1.12 |
| 22 | 468 | 474 | 478.21 | 5.77 |
| 23 | 484.85 | 2.56 | ||
| 24 | 525 | 528 | 535.68 | 5.75 |
| 25 | 536.21 | 1.67 | ||
| 26 | 538.78 | 0.01 | ||
| 27 | 577 | 551.01 | 1.67 | |
| 28 | 643 | 620 | 632.15 | 2.95 |
| 29 | 661 | 649 | 654.22 | 8.74 |
| 30 | 677.28 | 7.42 | ||
| 31 | 752 | 766 | 766.11 | 0.11 |
| 32 | 779 | 771.44 | 0.69 | |
| 33 | 775.50 | 79.16 | ||
| 34 | 793.09 | 3.25 | ||
| 35 | 818 | 839.55 | 27.81 | |
| 36 | 870 | 876.68 | 0.14 | |
| 37 | 887 | 892.78 | 1.88 | |
| 38 | 897.72 | 2.49 | ||
| 39 | 937 | 947.92 | 40.51 | |
| 40 | 956 | 971 | 973.02 | 0.65 |
| 41 | 980.44 | 0.31 | ||
| 42 | 1032 | 1041.07 | 95.25 | |
| 43 | 1056 | 1053.03 | 25.44 | |
| 44 | 1072.89 | 23.97 | ||
| 45 | 1092 | 1073.11 | 9.06 | |
| 46 | 1108.97 | 12.30 | ||
| 47 | 1113 | 1112.14 | 4.67 | |
| 48 | 1122.56 | 0.38 | ||
| 49 | 1136 | 1131.23 | 4.10 | |
| 50 | 1152 | 1158.87 | 0.66 | |
| 51 | 1162.50 | 44.36 | ||
| 52 | 1185 | 1178.07 | 1.90 | |
| 53 | 1197 | 1188.05 | 2.11 | |
| 54 | 1200 b | 1205.20 | 27.08 | |
| 55 | 1220.59 | 6.34 | ||
| 56 | 1226 | 1224.03 | 4.29 | |
| 57 | 1242.37 | 18.36 | ||
| 58 | 1248.75 | 14.89 | ||
| 59 | 1324 | 1333.17 | 15.30 | |
| 60 | 1343 | 1348.38 | 47.07 | |
| 61 | 1360.88 | 23.49 | ||
| 62 | 1384 | 1369.77 | 22.02 | |
| 63 | 1420 | 1407.98 | 74.73 | |
| 64 | 1448 | 1443.03 | 0.43 | |
| 65 | 1443.45 | 0.39 | ||
| 66 | 1456.69 | 21.91 | ||
| 67 | 1463 | 1466 b | 1464.24 | 5.54 |
| 68 | 1473.32 | 1.03 | ||
| 69 | 1479 | 1477.99 | 0.62 | |
| 70 | 1481.23 | 3.35 | ||
| 71 | 1485.18 | 9.10 | ||
| 72 | 1492.70 | 20.46 | ||
| 73 | 1493.08 | 30.41 | ||
| 74 | 1500.92 | 2.48 | ||
| 75 | 1511 | 1502.60 | 16.87 | |
| 76 | 1521.37 | 11.25 | ||
| 77 | 1524.98 | 22.79 | ||
| 78 | 1576 | 1542.86 | 22.63 | |
| 79 | 1605 | 1608.92 | 191.50 | |
| 80 | 1722 | 1632.70 | 1.17 | |
| 81 | 1641.10 | 11.45 | ||
| 82 | 2926.35 | 160.99 | ||
| 83 | 2777 | 2926.67 | 11.27 | |
| 84 | 2824 | 2939.86 | 267.58 | |
| 85 | 2852 | 2941.71 | 37.29 | |
| 86 | 2928 | 3051.65 | 27.31 | |
| 87 | 2971 | 3051.81 | 17.97 | |
| 88 | 3013 | 3056.51 | 12.52 | |
| 89 | 3048 | 3057.11 | 96.53 | |
| 90 | 3093 | 3101.58 | 16.29 | |
| 91 | 3101.61 | 24.04 | ||
| 92 | 3158.93 | 4.96 | ||
| 93 | 3159.18 | 17.51 | ||
| 94 | 3159.31 | 0.06 | ||
| 95 | 3161.34 | 1.11 | ||
| 96 | 3176.94 | 10.44 | ||
| 97 | 3180.00 | 43.23 | ||
| 98 | 3204.39 | 11.51 | ||
| 99 | 3204.41 | 8.22 |
Table A2.
Experimental IR, IINS, Raman, and calculated (B3LYP/6-311++G(d,p)) spectral data of DMANH+ HSO4−.
Table A2.
Experimental IR, IINS, Raman, and calculated (B3LYP/6-311++G(d,p)) spectral data of DMANH+ HSO4−.
| № | IR | IINS | Raman | Wavenumber [cm−1] | Intensities [km/mol] |
|---|---|---|---|---|---|
| exp. | exp. | exp. | calc. | calc. | |
| 1 | 14.57 | 0.03 | |||
| 2 | 17.59 | 1.10 | |||
| 3 | 36.81 | 1.37 | |||
| 4 | 42.72 | 0.88 | |||
| 5 | 60.97 | 1.86 | |||
| 6 | 83.84 | 25.22 | |||
| 7 | 90 | 90.41 | 5.08 | ||
| 8 | 93.29 | 0.17 | |||
| 9 | 147.02 | 43.61 | |||
| 10 | 152.75 | 31.05 | |||
| 11 | 162 | 161.14 | 3.03 | ||
| 12 | 185 | 185.56 | 9.32 | ||
| 13 | 185.81 | 15.51 | |||
| 14 | 194 | 198 | 198.61 | 0.12 | |
| 15 | 242.02 | 3.42 | |||
| 16 | 243.48 | 2.73 | |||
| 17 | 249 | 252.75 | 10.81 | ||
| 18 | 256.74 | 0.23 | |||
| 19 | 262 | 262 | 263.71 | 0.50 | |
| 20 | 274 | 288.84 | 3.22 | ||
| 21 | 325 | 325.96 | 0.01 | ||
| 22 | 329 | 332.45 | 8.04 | ||
| 23 | 378.53 | 3.30 | |||
| 24 | 382 | 383 | 384.15 | 6.38 | |
| 25 | 399.35 | 13.15 | |||
| 26 | 419 | 422.01 | 12.28 | ||
| 27 | 440 | 439 | 441.72 | 3.17 | |
| 28 | 466 | 470 | 481.60 | 0.24 | |
| 29 | 503 | 498.35 | 10.20 | ||
| 30 | 511 | 525.17 | 5.77 | ||
| 31 | 525.65 | 52.86 | |||
| 32 | 533 | 533 | 534 | 536.22 | 9.97 |
| 33 | 539.01 | 0.17 | |||
| 34 | 540.81 | 3.02 | |||
| 35 | 545.32 | 31.75 | |||
| 36 | 584 | 585 | 593 | 583.61 | 12.63 |
| 37 | 642 | 649.80 | 0.00 | ||
| 38 | 676 | 674.12 | 35.07 | ||
| 39 | 687 | 678.86 | 0.26 | ||
| 40 | 732.25 | 299.92 | |||
| 41 | 776.12 | 0.93 | |||
| 42 | 779 | 781.77 | 1.30 | ||
| 43 | 787 | 789.15 | 71.35 | ||
| 44 | 796.76 | 0.78 | |||
| 45 | 841 | 844 | 853.32 | 15.88 | |
| 46 | 846 | 877 | 866.23 | 13.02 | |
| 47 | 908.56 | 22.33 | |||
| 48 | 922.62 | 0.27 | |||
| 49 | 949 | 938.58 | 1.60 | ||
| 50 | 989.33 | 3.63 | |||
| 51 | 995.53 | 4.04 | |||
| 52 | 1002.12 | 1.26 | |||
| 53 | 1004.20 | 115.91 | |||
| 54 | 1007 | 1013 | 1007 | 1004.66 | 102.37 |
| 55 | 1031 | 1025 | 1047.32 | 17.60 | |
| 56 | 1049 | 1049 | 1047.85 | 18.21 | |
| 57 | 1087 | 1088.53 | 0.09 | ||
| 58 | 1099 | 1104.66 | 212.72 | ||
| 59 | 1108.80 | 8.72 | |||
| 60 | 1112 | 1110.39 | 10.96 | ||
| 61 | 1114 | 1114.95 | 4.13 | ||
| 62 | 1155 | 1170.56 | 30.47 | ||
| 63 | 1173 | 1175.17 | 65.76 | ||
| 64 | 1181.23 | 21.65 | |||
| 65 | 1193 | 1195.65 | 20.18 | ||
| 66 | 1196.71 | 26.24 | |||
| 67 | 1199.61 | 175.54 | |||
| 68 | 1200.30 | 166.15 | |||
| 69 | 1210 | 1217.08 | 170.92 | ||
| 70 | 1223 | 1228.08 | 9.32 | ||
| 71 | 1239.64 | 13.43 | |||
| 72 | 1245.05 | 26.67 | |||
| 73 | 1264.43 | 4.50 | |||
| 74 | 1305.30 | 11.88 | |||
| 75 | 1354.70 | 48.76 | |||
| 76 | 1365 | 1365.64 | 3.30 | ||
| 77 | 1379 | 1386.47 | 15.33 | ||
| 78 | 1414 | 1415 | 1436.10 | 1.65 | |
| 79 | 1431 | 1438.56 | 79.01 | ||
| 80 | 1443 | 1451.21 | 3.09 | ||
| 81 | 1469 | 1465.77 | 1.48 | ||
| 82 | 1480.31 | 7.45 | |||
| 83 | 1482.26 | 0.71 | |||
| 84 | 1488.20 | 15.25 | |||
| 85 | 1488.98 | 0.97 | |||
| 86 | 1490 | 1497.56 | 0.69 | ||
| 87 | 1495 | 1498.85 | 20.69 | ||
| 88 | 1503.20 | 8.53 | |||
| 89 | 1505.92 | 5.75 | |||
| 90 | 1511.50 | 82.43 | |||
| 91 | 1518 | 1514.96 | 23.30 | ||
| 92 | 1521.21 | 24.25 | |||
| 93 | 1578 | 1580 | 1551.47 | 22.12 | |
| 94 | 1603 | 1612.85 | 5.43 | ||
| 95 | 1626 | 1627 | 1617.33 | 8.56 | |
| 96 | 1644.36 | 9.08 | |||
| 97 | 1664.89 | 16.75 | |||
| 98 | 2600 | 2529.78 | 977.93 | ||
| 99 | 2809 | 2984.75 | 30.16 | ||
| 100 | 2940 | 2866 | 2989.37 | 66.45 | |
| 101 | 2952 | 3056.91 | 10.25 | ||
| 102 | 3015 | 3060.71 | 59.04 | ||
| 103 | 3065 | 3081.84 | 19.34 | ||
| 104 | 3082.70 | 19.25 | |||
| 105 | 3134.13 | 0.65 | |||
| 106 | 3137.65 | 8.34 | |||
| 107 | 3148.81 | 1.78 | |||
| 108 | 3150.19 | 9.61 | |||
| 109 | 3154.58 | 2.99 | |||
| 110 | 3160.22 | 69.10 | |||
| 111 | 3168.92 | 0.48 | |||
| 112 | 3172.93 | 0.06 | |||
| 113 | 3181.07 | 9.40 | |||
| 114 | 3186.35 | 13.84 | |||
| 115 | 3192.70 | 10.19 | |||
| 116 | 3198.99 | 4.40 | |||
| 117 | 3794.06 | 87.38 |
References
- Perrin, C.L. Symmetries of hydrogen bonds in solution. Science 1994, 266, 1665. [Google Scholar] [CrossRef] [PubMed]
- Perrin, C.L.; Nielson, J.B. Asymmetry of Hydrogen Bonds in Solutions of Monoanions of Dicarboxylic Acids. J. Am. Chem. Soc. 1997, 119, 12734–12741. [Google Scholar] [CrossRef]
- Chambron, J.-C.; Meyer, M. The ins and outs of proton complexation. Chem. Soc. Rev. 2009, 38, 1663–1673. [Google Scholar] [CrossRef] [PubMed]
- Mavri, J.; Hodošček, M.; Hadži, D. Ab initio SCF and Møller-Plesset calculations on the hydrogen bond in hydrogen malonate: Effects of neighbour ions and polarizable medium. J. Mol. Struct. 1990, 209, 421–431. [Google Scholar] [CrossRef]
- Mavri, J.; Hadži, D. Influence of solvation on the hydrogen bond in hydrogen malonate an ab initio and semiempirical study. J. Mol. Struct. THEOCHEM 1998, 432, 257–262. [Google Scholar] [CrossRef]
- Staab, H.A.; Kriege, C.; Hieber, G.; Oberdorf, K. 1,8-Bis(dimethylamino)4,5-dihydroxynaphthalene, a Natural, Intramolecularly Protonated “Proton Sponge” with Zwitterionic Structure. Angew. Chem. Int. Ed. Engl. 1997, 36, 1884–1886. [Google Scholar] [CrossRef]
- Perrin, C.L.; Thoburn, J.D. Symmetries of hydrogen bonds in monoanions of dicarboxylic acids. J. Am. Chem. Soc. 1992, 114, 8559–8565. [Google Scholar] [CrossRef]
- Perrin, C.L.; Kim, Y.J. Symmetry of the Hydrogen Bond in Malonaldehyde Enol in Solution. J. Am. Chem. Soc. 1998, 120, 12641–12645. [Google Scholar] [CrossRef]
- Guo, J.; Tolstoy, P.M.; Koeppe, B.; Denisov, G.S.; Limbach, H.-H. NMR Study of Conformational Exchange and Geometries of Intramolecular Hydrogen Bonds in Monoanions of Succinic Acid and Derivatives. J. Phys. Chem. A 2011, 115, 9828–9836. [Google Scholar] [CrossRef]
- Guo, J.; Tolstoy, P.M.; Koeppe, B.; Golubev, N.S.; Denisov, G.S.; Smirnov, S.N.; Limbach, H.-H. Hydrogen Bond Geometries and Proton Tautomerism of Homo-Conjugated Anions of Carboxylic Acids Studied via H/D Isotope Effects on 13C NMR Chemical Shifts. J. Phys. Chem. A 2012, 116, 11180–11188. [Google Scholar] [CrossRef]
- Chmielewski, P.; Ozeryanskii, V.A.; Sobczyk, L.; Pozharskii, A.F. Primary 1 H/2 H isotope effect in the NMR chemical shift of HClO4 salts of 1,8-bis(dimethylamino)naphthalene derivatives. J. Phys. Org. Chem. 2007, 20, 643–648. [Google Scholar] [CrossRef]
- Zhou, S.; Wang, L. Symmetry and 1 H NMR chemical shifts of short hydrogen bonds: Impact of electronic and nuclear quantum effects. Phys. Chem. Chem. Phys. 2020, 22, 4884–4895. [Google Scholar] [CrossRef] [PubMed]
- White, P.B.; Hong, M. 15N and 1 H Solid-State NMR Investigation of a Canonical Low-Barrier Hydrogen-Bond Compound: 1,8-Bis(dimethylamino)naphthalene. J. Phys. Chem. B 2015, 119, 11581–11589. [Google Scholar] [CrossRef] [PubMed]
- Perrin, C.L.; Ohta, B.K. Symmetry of N–H–N Hydrogen Bonds in 1,8-Bis(dimethylamino)naphthalene·H+ and 2,7-Dimethoxy-1,8-bis(dimethylamino)naphthalene·H+. J. Am. Chem. Soc. 2001, 123, 6520–6526. [Google Scholar] [CrossRef] [PubMed]
- Woźniak, K.; Wilson, C.C.; Knight, K.S.; Jones, W.; Grech, E. Neutron Diffraction of a Complex of 1,8-Bis(dimethylamino)naphthalene with 1,2-Dichloromaleic Acid. Acta Cryst. B 1996, 52, 691–696. [Google Scholar] [CrossRef]
- Jones, A.O.F.; Kallay, A.A.; Lloyd, H.; McIntyre, G.J.; Wilson, C.C.; Thomas, L.H. The Effect of Local Crystalline Environment on Hydrogen Atom Behavior in Molecular Complexes of a Proton Sponge. Cryst. Growth Des. 2016, 16, 2123–2129. [Google Scholar] [CrossRef]
- Gregorovic, A.; Apih, T.; Zagara, V.; Seliger, J. 14N NQR spectroscopy reveals the proton position in N–H N bonds: A case study with proton sponges. Phys. Chem. Chem. Phys. 2019, 21, 306–313. [Google Scholar] [CrossRef]
- Sabet-Sarvestani, H.; Izadyar, M.; Eshghi, H.; Noroozi-Shad, N.; Bakavoli, M. Proton sponge as a new efficient catalyst for carbon dioxide transformation to methanol: Theoretical approach. Fuel 2018, 221, 491–500. [Google Scholar] [CrossRef]
- Pawlukojć, A.; Natkaniec, I.; Grech, E.; Baran, J.; Malarski, Z.; Sobczyk, L. Incoherent inelastic neutron scattering, Raman and IR absorption studies on 1,8-bis(dimethylamino)naphthalene and its protonated forms. Spectrochim. Acta A 1998, 54, 439–448. [Google Scholar] [CrossRef]
- Brzeziński, B.; Głowiak, T.; Grech, E.; Malarski, Z.; Sobczyk, L. Structure and IR spectra of protonated 1,8-bis(dimethylamino)naphthalene proton sponge. Croat. Chem. Acta 1992, 65, 101–108. [Google Scholar]
- Lee, J.; Cheong, I.; Lee, S.-Y. Successful application of a neutral organic base, 1,8-bis(tetramethylguanidino)naphthalene (TMGN), for the radiosynthesis of [11C]raclopride. Appl. Rad. Isotop. 2016, 118, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Lopez, C.; Lorente, P.; Claramunt, R.M.; Marin, J.; Foces-Foces, C.; Llamas-Saiz, A.L.; Elguero, J.; Limbach, H.H. Localization of Hydrogen Bond Deuterons in Proton Sponges by Dipolar Solid State N-15 NMR Spectroscopy. Ber. Bunsenges. Phys. Chem. Chem. Phys. 1998, 102, 414–418. [Google Scholar] [CrossRef]
- Pietrzak, M.; Wehling, J.P.; Kong, S.; Tolstoy, P.M.; Shenderovich, I.G.; Lopez, C.; Claramunt, R.M.; Elguero, J.; Denisov, G.S.; Limbach, H.-H. Symmetrization of Cationic Hydrogen Bridges of Protonated Sponges Induced by Solvent and Counteranion Interactions as Revealed by NMR Spectroscopy. Chem. Eur. J. 2010, 16, 1679–1690. [Google Scholar] [CrossRef] [PubMed]
- Pietrzak, M.; Wehling, J.; Limbach, H.-H.; Golubev, N.S.; Lopez, C.; Claramunt, R.M.; Elguero, J. 13C Detected Scalar Nitrogen-Nitrogen Couplings Across the Intramolecular Symmetric NHN Hydrogen Bond of Proton Sponge. J. Am. Chem. Soc. 2001, 123, 4338–4339. [Google Scholar] [CrossRef] [PubMed]
- Lopez, C.; Claramunt, R.M.; Llamas-Saiz, A.L.; Foces-Foces, C.; Elguero, J.; Sobrados, I.; Aguilar-Parrilla, F.; Limbach, H.H. X-ray diffraction and solid state NMR studies of 1,8-bis(dimethylamino)naphthalene and its complexes with picric and hexafluorophosphoric acids. New J. Chem. 1996, 20, 523–536. [Google Scholar]
- Saunders, M.; Telkowski, L.; Kates, M.R. NMR isotope shifts as a probe of electronic structure. J. Am. Chem. Soc. 1977, 99, 8070–8071. [Google Scholar] [CrossRef]
- Alder, R.W.; Bowman, P.S.; Steele, W.R.S.; Winterman, D.R. The remarkable basicity of 1,8-bis(dimethylamino)naphthalene. J. Chem. Soc. Chem. Commun. 1968, 723–724. [Google Scholar] [CrossRef]
- Alder, R.W.; East, S.P. In/Out Isomerism. Chem. Rev. 1996, 96, 2097–2111. [Google Scholar] [CrossRef]
- Staab, H.A.; Saupe, T. “Proton Sponges” and the Geometry of Hydrogen Bonds: Aromatic Nitrogen Bases with Exceptional Basicities. Angew. Chem. Int. Ed. Engl. 1988, 27, 865–879. [Google Scholar] [CrossRef]
- Pozharskii, A.F. Naphthalene “Proton Sponges”. Russ. Chem. Rev. 1998, 67, 1–27. [Google Scholar] [CrossRef]
- Pozharskii, A.F.; Ozeryanskii, V.A. Proton sponges. In The Chemistry of Anilines; Rappoport, Z., Ed.; J. Wiley & Sons: Chichester, UK, 2007; pp. 931–1026. [Google Scholar]
- Sobczyk, L. The specificity of the [NHN]+ hydrogen bonds in protonated naphthalene proton sponges. J. Mol. Struct. 2010, 972, 59–63. [Google Scholar] [CrossRef]
- Llamas-Saiz, A.L.; Foces-Foces, C.; Elguero, J. Proton sponges. J. Mol. Struct. 1994, 328, 297–323. [Google Scholar] [CrossRef]
- Kaljurand, I.; Saame, J.; Rodima, T.; Koppel, I.; Koppel, I.A.; Kögel, J.F.; Sundermeyer, J.; Köhn, U.; Coles, M.P.; Leito, I. Experimental Basicities of Phosphazene, Guanidinophosphazene, and Proton Sponge Superbases in the Gas Phase and Solution. J. Phys. Chem. A 2016, 120, 2591–2604. [Google Scholar] [CrossRef] [PubMed]
- Kçgel, J.F.; Xie, X.; Baal, E.; Gesevičius, D.; Oelkers, B.; Kovačević, B.; Sundermeyer, J. Superbasic Alkyl-Substituted Bisphosphazene Proton Sponges: Synthesis, Structural Features, Thermodynamic and Kinetic Basicity, Nucleophilicity and Coordination Chemistry. Chem. Eur. J. 2014, 20, 7670–7685. [Google Scholar] [CrossRef]
- Korzhenevskaya, N.G.; Schroeder, G.; Brzezinski, B.; Rybachenko, V.I. Concept of Superbasicity of 1,8-Bis(dialkylamino)naphthalenes ([Proton Sponges]). Rus. J. Org. Chem. 2001, 37, 1603–1610. [Google Scholar] [CrossRef]
- Raab, V.; Kipke, J.; Gschwind, R.M.; Sundermeyer, J. 1,8-Bis(tetramethylguanidino)naphthalene (TMGN): A New, Superbasic and Kinetically Active “Proton Sponge”. Chem. Eur. J. 2002, 8, 1682–1693. [Google Scholar] [CrossRef]
- Hibbert, F.; Robbins, H.J. Base-catalyzed proton transfer from an intramolecularly hydrogen-bonded naphthylammonium ion in 70% dimethyl sulfoxide-water (v/v). J. Am. Chem. Soc. 1978, 100, 8239−8244. [Google Scholar] [CrossRef]
- Hibbert, F.; Simpson, G.R. Acid–base properties of highly substituted diaminonaphthalenes. J. Chem. Soc. Perkin Trans. 1987, 2, 243–246. [Google Scholar] [CrossRef]
- Woźniak, K.; He, H.Y.; Klinowski, J.; Barr, T.L.; Milart, P. ESCA and Solid-State NMR Studies of Ionic Complexes of 1,8- Bis(dimethylamino)naphthalene. J. Phys. Chem. 1996, 100, 11420–11426. [Google Scholar] [CrossRef]
- Parkin, A.; Woźniak, K.; Wilson, C.C. From Proton Disorder to Proton Migration: A Continuum in the Hydrogen Bond of a Proton Sponge in the Solid State. Cryst. Grow. Des. 2007, 7, 1393–1398. [Google Scholar] [CrossRef]
- Hoser, A.A.; Dobrzycki, Ł.; Gutmann, M.J.; Woźniak, K. Charge Densities of Two Polymorphs of Hydrated 1,8-Bis(dimethylamino)naphthalene Hydrochloride; Similarities and Differences. Cryst. Grow. Des. 2010, 10, 5092–5104. [Google Scholar] [CrossRef]
- Mallinson, P.R.; Smith, G.T.; Wilson, C.C.; Grech, E.; Wozniak, K. From Weak Interactions to Covalent Bonds: A Continuum in the Complexes of 1,8-Bis(dimethylamino)naphthalene. J. Am. Chem. Soc. 2003, 125, 4259–4270. [Google Scholar] [CrossRef] [PubMed]
- Woźniak, K.; Krygowski, T.M.; Kariuki, B.; Jones, W.; Grech, E. Crystallographic Studies on Sterically Affected Chemical-Species 0.2. Molecular and Crystal-Structure of 1,8-Bis(Dimethylamino)-Naphthalene Tetrafluoroborate—Analysis of Distortion of Geometry in the Aromatic Part Due to Intramolecular Hydrogen-Bonding. J. Mol. Struct. 1990, 240, 111–118. [Google Scholar] [CrossRef]
- Grech, E.; Klimkiewicz, J.; Nowicka-Scheibe, J.; Pietrzak, M.; Schilf, W.; Pozharski, A.F.; Ozeryanskii, V.A.; Bolvig, S.; Abildgaard, J.; Hansen, P.E. Deuterium isotope effects on 15N, 13C and 1 H chemical shifts of proton sponges. J. Mol. Struct. 2002, 615, 121–140. [Google Scholar] [CrossRef]
- Pawełka, Z.; Zeegers-Huyskens, T. The strange behaviour of the hydrogen bond complexes of 1,8-bis(dimethylamino) naphthalene in solution. J. Mol. Struct. THEOCHEM 1989, 200, 565–573. [Google Scholar] [CrossRef]
- Ozeryanskii, V.A.; Pozharskii, A.F.; Bieńko, A.J.; Sawka-Dobrowolska, W.; Sobczyk, L. [NHN]+ Hydrogen Bonding in Protonated 1,8-Bis(dimethylamino)-2,7-dimethoxynaphthalene. X-ray Diffraction, Infrared, and Theoretical ab Initio and DFT Studies. J. Phys. Chem. A 2005, 109, 1637–1642. [Google Scholar] [CrossRef]
- Ozeryanskii, V.A.; Pozharskii, A.F.; Głowiak, T.; Majerz, I.; Sobczyk, L.; Grech, I.; Nowicka-Szajbe, J. X-ray diffraction and IR-spectroscopic studies on protonated 4-amino-1,8-bis(dimethyloamino)naphthalene. J. Mol. Struct. 2002, 607, 1–8. [Google Scholar] [CrossRef]
- Brzezinski, B.; Schroeder, G.; Jarczewski, A.; Grech, E.; Nowicka-Scheibe, J.; Stefaniak, L.; Klimkiewicz, J. Proton transfer reactions from N-H acid to proton sponges in acetonitrile. Part 2. J. Mol. Struct. 1996, 377, 149–154. [Google Scholar]
- Baran, J.; Pawlukojc, A.; Majerz, I.; Malarski, Z.; Sobczyk, L.; Grech, E. Vibrational spectra of the adduct of 1,8-bis(dimethylamino)naphthalene with dichloromaleic acid (DMAN*DCM). Spectrochim. Acta A 2000, 56, 1801–1812. [Google Scholar] [CrossRef]
- Belding, L.; Stoyanov, P.; Dudding, T. Synthesis, Theoretical Analysis, and Experimental pKa Determination of a Fluorescent, Nonsymmetric, In−Out Proton Sponge. J. Org. Chem. 2016, 81, 6–13. [Google Scholar] [CrossRef]
- Antonov, A.S.; Pozharskii, A.F.; Tolstoy, P.M.; Filarowski, A.; Khoroshilova, O.V. 1,8-Bis(dimethylamino)naphthyl-2-ketimines: Inside vs outside protonation. Beilstein J. Org. Chem. 2018, 14, 2940–2948. [Google Scholar] [CrossRef] [PubMed]
- Ozeryanskii, V.A.; Pozharskii, A.F.; Antonov, A.S.; Filarowski, A. Out-Basicity of 1,8-bis(dimethylamino)naphthalene: The experimental and theoretical challenge. Org. Biomol. Chem. 2014, 12, 2360–2369. [Google Scholar] [CrossRef] [PubMed]
- Pozharskii, A.F.; Degtyarev, A.V.; Ryabtsova, O.V.; Ozeryanskii, V.A.; Kletskii, M.E.; Starikova, Z.A.; Sobczyk, L.; Filarowski, A. 2-α-hydroxyalkyl- and 2,7-di(α-hydroxyalkyl)-1,8-bis(dimethylamino)naphthalenes: Stabilization of nonconventional in/out conformers of “proton sponges” via N···H-O intramolecular hydrogen bonding. A remarkable kind of tandem nitrogen inversion. J. Org. Chem. 2007, 72, 3006–3019. [Google Scholar] [CrossRef] [PubMed]
- Belding, L.; Dudding, T. Synthesis and Theoretical Investigation of a 1,8-Bis(bis(diisopropylamino)cyclopropeniminyl)naphthalene Proton Sponge Derivative. Chem. Eur. J. 2014, 20, 1032–1037. [Google Scholar] [CrossRef] [PubMed]
- Jezierska, A.; Panek, J.J. Theoretical study of intramolecular hydrogen bond in selected symmetric “proton sponges” on the basis of DFT and CPMD methods. J. Mol. Model. 2020, 26, 37. [Google Scholar] [CrossRef]
- Jezierska, A.; Panek, J.J. “Zwitterionic proton sponge” hydrogen bonding investigations on the basis of Car-Parrinello molecular dynamics. J. Chem. Inf. Model. 2015, 55, 1148–1157. [Google Scholar] [CrossRef]
- Masuda, Y.; Mori, Y.; Sakurai, K. Effects of Counterion and Solvent on Proton Location and Proton Transfer Dynamics of N−H···N Hydrogen Bond of Monoprotonated 1,8-Bis(dimethylamino)naphthalene. J. Phys. Chem. A 2013, 117, 10576–10587. [Google Scholar] [CrossRef]
- Majerz, I.; Olovsson, I. Proton transfer in the intramolecular NHN+ bonds in proton sponges with different hydrogen bridge flexibility. Phys. Chem. Chem. Phys. 2009, 11, 1297–1302. [Google Scholar] [CrossRef]
- Majerz, I.; Olovsson, I. The shape of the potential energy curves for NHN+ hydrogen bonds and the influence of non-linearity. Phys. Chem. Chem. Phys. 2008, 10, 3043–3051. [Google Scholar] [CrossRef]
- Horbatenko, Y.; Vyboishchikov, S.F. Hydrogen Motion in Proton Sponge Cations: A Theoretical Study. ChemPhysChem 2011, 12, 1118–1129. [Google Scholar] [CrossRef]
- Pozharskii, A.F.; Ryabtsova, O.V.; Ozeryanskii, V.A.; Degtyarev, A.V.; Kazheva, O.N.; Alexandrov, G.G.; Dyachenko, O.A. Organometallic Synthesis, Molecular Structure, and Coloration of 2,7-Disubstituted 1,8-Bis(dimethylamino)naphthalenes. How Significant Is the Influence of “Buttressing Effect” on Their Basicity? J. Org. Chem. 2003, 68, 10109–10122. [Google Scholar] [CrossRef]
- Alder, R.W.; Orpen, A.G.; Sessions, R.B. The structure of 1,6-diazabicyclo[4.4.4]tetradecane and of its inside protonated ion. J. Chem. Soc. Chem. Commun. 1983, 999–1000. [Google Scholar] [CrossRef]
- Alder, R.W.; Bryce, M.R.; Goode, N.C.; Miller, N.; Owen, J. Preparation of a range of NNN′N′-tetrasubstituted 1,8-diaminonaphthalenes. J. Chem. Soc. Perkin Trans. 1981, 1, 2840–2847. [Google Scholar] [CrossRef]
- Degtyarev, A.V.; Ryabtsova, O.V.; Pozharskii, A.F.; Ozeryanskii, V.A.; Starikova, Z.A.; Sobczyk, L.; Filarowski, A. 2,7-disubstituted proton sponges as borderline systems for investigating barrier-free intramolecular hydrogen bonds. Protonated 2,7-bis(trimethylsilyl)- and 2,7-di(hydroxymethyl)-1,8-bis(dimethylamino)naphthalenes. Tetrahedron 2008, 64, 6209–6214. [Google Scholar] [CrossRef]
- Pozharskii, A.F.; Ozeryanskii, V.A.; Starikova, Z.A. Molecular structure of 5,6-bis(dimethylamino)acenaphthene, 5,6- bis(dimethylamino)acenaphthylene, and their monohydrobromides: A comparison with some naphthalene proton sponges. J. Chem. Soc. Perkin Trans. 2002, 2, 318–322. [Google Scholar] [CrossRef]
- Barnett, G.H.; Hibbert, F. General base catalysis, isotope effects, activation parameters, and the mechanism of removal of the hydrogen-bonded proton from protonated 1,8-bis(diethylamino)-2,7-dimethoxynaphthalene. J. Am. Chem. Soc. 1984, 106, 2080–2084. [Google Scholar] [CrossRef]
- Filatova, E.A.; Gulevskaya, A.V.; Pozharskii, A.F.; Ermolenko, E.A.; Ozeryanskii, V.A.; Misharev, A.D. Synthesis of 2-Aryl- and 2,7-Diaryl-1,8-bis(dimethylamino)naphthalenes. Overview of the “Buttressing effect” in 2,7-Disubstituted Proton Sponges. ChemistrySelect 2020, 5, 9932–9945. [Google Scholar] [CrossRef]
- Randolph, C.E.; Fabijanczuk, K.C.; Blanksby, S.J.; McLuckey, S.A. Proton Transfer Reactions for the Gas-Phase Separation, Concentration, and Identification of Cardiolipins. Anal. Chem. 2020, 92, 10847–10855. [Google Scholar] [CrossRef]
- Swor, C.D.; Zakharov, L.N.; Tyler, D.R. A Colorimetric Proton Sponge. J. Org. Chem. 2010, 75, 6977–6979. [Google Scholar] [CrossRef]
- Car, R.; Parrinello, M. Unified Approach for Molecular Dynamics and Density-Functional Theory. Phys. Rev. Lett. 1985, 55, 2471–2474. [Google Scholar] [CrossRef]
- Mitchell, P.C.H.; Parker, S.F.; Ramirez-Cuesta, A.J.; Tomkinson, J. Series on Neutron Techniques and Applications, Vibrational Spectroscopy with Neutrons; World Scientific Publishing Co. Pte. Ltd.: Singapore, 2005. [Google Scholar]
- Harris, R.K.; Becker, E.D.; Cabral de Menezes, S.M.; Goodfellow, R.; Granger, P. NMR nomenclature. Nuclear spin properties and conventions for chemical shifts (IUPAC recommendations 2001). Pure Appl. Chem. 2001, 73, 1795–1818. [Google Scholar] [CrossRef]
- Mestrelab Research, S.L. Available online: https://mestrelab.com/software/mnova (accessed on 12 October 2020).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H. et al. Gaussian 16, Revision С.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11-18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type Density Functional Constructed with a Long-Range Dispersion Correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Soc. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
- Schaftenaar, G.; Noordik, J.H. Molden: A pre- and post-processing program for molecular and electronic structures. J. Comput. Aided Mol. Design. 2000, 14, 123–134. [Google Scholar] [CrossRef]
- Jeziorski, B.; Moszyński, R.; Szalewicz, K. Perturbation Theory Approach to Intermolecular Potential Energy Surfaces of van der Waals Complexes. Chem. Rev. 1994, 7, 1887–1930. [Google Scholar] [CrossRef]
- Hohenstein, E.G.; Sherrill, C.D. Density fitting of intramonomer correlation effects in symmetry-adapted perturbation theory. J. Chem. Phys. 2010, 133, 014101. [Google Scholar] [CrossRef] [PubMed]
- Kendall, R.A.; Dunning, T.H., Jr.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Parrish, R.M.; Burns, L.A.; Smith, D.G.A.; Simmonett, A.C.; DePrince, A.E.; Hohenstein, E.G.; Bozkaya, U.; Sokolov, A.Y.; Di Remigio, R.; Richard, R.M.; et al. Psi4 1.1: An Open-Source Electronic Structure Program Emphasizing Automation, Advanced Libraries, and Interoperability. J. Chem. Theory Comput. 2017, 13, 3185–3197. [Google Scholar] [CrossRef]
- CPMD 3.17.1; IBM Corp: 1990–2004; MPI für Festkörperforschung Stuttgart, 1997–2001.
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Gnuplot 4.2; Thomas Williams, Colin Kelley, 1986–1993, 1998, 2004, 2007.
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Troullier, N.; Martins, J.L. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. 1991, B43, 1993–2006. [Google Scholar] [CrossRef]
- Hockney, R.W. The potential calculation and some applications. Meth. Comput. Phys. 1970, 9, 136–211. [Google Scholar]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. 1985, A31, 1695–1697. [Google Scholar] [CrossRef]
- Grimme, S.; Hansen, A.; Brandenburg, J.G.; Bannwarth, C. Dispersion-Corrected Mean-Field Electronic Structure Methods. Chem. Rev. 2016, 116, 5105–5154. [Google Scholar] [CrossRef]
- Herschlag, D.; Pinney, M.M. Hydrogen Bonds: Simple after All? Biochemistry 2018, 57, 3338–3352. [Google Scholar] [CrossRef] [PubMed]
- Sigala, P.A.; Ruben, E.A.; Liu, C.W.; Piccoli, P.M.B.; Hohenstein, E.G.; Martínez, T.J.; Schultz, A.J.; Herschlag, D. Determination of hydrogen bond structure in water versus aprotic environments to test the relationship between length and stability. J. Am. Chem. Soc. 2015, 137, 5730–5740. [Google Scholar] [CrossRef] [PubMed]
- Ashkar, R.; Bilheux, H.Z.; Bordallo, H.; Briber, R.; Callaway, D.J.E.; Cheng, X.; Chu, X.-Q.; Curtis, J.E.; Dadmun, M.; Fenimore, P.; et al. Neutron scattering in the biological sciences: Progress and prospects. Acta Cryst. 2018, D74, 1129–1168. [Google Scholar] [CrossRef] [PubMed]
- Hudson, B.S. Inelastic Neutron Scattering: A Tool in Molecular Vibrational Spectroscopy and a Test of ab Initio Methods. Phys. Chem. A 2001, 105, 3949–3960. [Google Scholar] [CrossRef]
- Hudson, B.S. Vibrational spectroscopy using inelastic neutron scattering: Overview and outlook. Vibr. Spectr. 2006, 42, 25–32. [Google Scholar] [CrossRef]
- Pusztai, L. Neutron Scattering Methods in Chemistry. In Handbook of Nuclear Chemistry; Vértes, A., Nagy, S., Klencsár, Z., Lovas, R.G., Rösch, F., Eds.; Springer: Boston, MA, USA, 2011. [Google Scholar]
- Albers, P.W.; Parker, S.W. IINS Inelastic Incoherent Neutron Scattering in Catalysis Research. Adv. Catal. 2007, 51, 99–132. [Google Scholar]
- Tsapatsaris, N.; Kolesov, B.A.; Fischer, J.; Boldyreva, E.V.; Daemen, L.; Eckert, J.; Bordallo, H.N. Polymorphism of Paracetamol: A New Understanding of Molecular Flexibility through Local Methyl Dynamics. Mol. Pharm. 2014, 11, 1032–1041. [Google Scholar] [CrossRef] [PubMed]
- Pawlukojć, A.; Prager, M.; Sawka-Dobrowolska, W.; Bator, G.; Sobczyk, L.; Ivanov, A.; Rols, S.; Grech, E.; Nowicka-Scheibe, J.; Unruh, T. The structure, methyl rotation reflected in elestic and quasielastic neutron scattering and vibrational spectra of 1,2,3,5-tetramethoxybenzene and its 2:1 complex with 1,2,4,5-tetracyanobenzene. J. Chem. Phys. 2008, 129, 154506–154512. [Google Scholar] [CrossRef] [PubMed]
- Bordallo, H.N.; Zakharov, B.A.; Boldyreva, E.V.; Johnson, M.R.; Koza, M.M.; Seydel, T.; Fischer, J. Application of Incoherent Inelastic Neutron Scattering in Pharmaceutical Analysis: Relaxation Dynamics in Phenacetin. Mol. Pharm. 2012, 9, 2434–2441. [Google Scholar] [CrossRef] [PubMed]
- Bordallo, H.N.; Barthes, M.; Eckert, J. Vibrational dynamics of crystalline L-alanine. Phys. B 1998, 241-243, 1138–1140. [Google Scholar] [CrossRef]
- Pawlukojć, A.; Natkaniec, I.; Bator, G.; Sobczyk, L.; Grech, E.; Nowicka-Scheibe, J. Low frequency internal modes of 1,2,4,5-tetramethylbenzene, tetramethylpyrazine and tetramethyl-1,4-benzoquinone. INS, Raman, IR and theoretical DFT studies. Spectrochim. Acta A 2006, 63, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Prager, M.; Pawlukojc, A.; Wischnewski, A.; Wuttke, J. Inelastic neutron scattering study of methyl groups rotation in some methylxanthines. J. Chem. Phys. 2007, 127, 214509–214519. [Google Scholar] [CrossRef] [PubMed]
- Bator, G.; Sawka-Dobrowolska, W.; Sobczyk, L.; Grech, E.; Nowicka-Scheibe, J.; Pawlukojc, A.; Wuttke, J.; Baran, J.; Owczarek, M. 4,4’-, 5,5’-, and 6,6’-dimethyl-2,2’-bipyridyls: The structures, phase transitions, vibrations, and methyl group tunneling of their complexes with chloranilic acid. J. Chem. Phys. 2011, 135, 044509. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Gangopadhyay, D.; Nandi, R.; Sharma, P.; Singh, R.K. Raman signatures of strong and weak hydrogen bonds in binary mixtures of phenol with acetonitrile, benzene and orthodichlorobenzene. Raman Spectr. 2016, 47, 712–719. [Google Scholar] [CrossRef]
- Abe, N.; Ito, M. Effects of hydrogen bonding on the Raman intensities of methanol, ethanol and water. Raman Spectr. 1978, 7, 161–167. [Google Scholar] [CrossRef]
- Zundel, G. The Hydrogen Bond. Recent Developments in Theory and Experiments; Schuster, P., Zundel, G., Sandorfy, C., Eds.; North-Holland: Amsterdam, The Netherlands, 1976; Volume 2, pp. 683–766. [Google Scholar]
- Zundel, G. Hydrogen bonds with large proton polarizability and proton transfer processes in electrochemistry and biology. Adv. Chem. Phys. 2000, 111, 1–217. [Google Scholar]
- Kwocz, A.; Panek, J.J.; Jezierska, A.; Hetmańczyk, Ł.; Pawlukojć, A.; Kochel, A.; Lipkowski, P.; Filarowski, A. A molecular roundabout: Triple cycle-arranged hydrogen bonds in light of experiment and theory. New J. Chem. 2018, 42, 19467–19477. [Google Scholar] [CrossRef]
- Jóźwiak, K.; Jezierska, A.; Panek, J.J.; Goremychkin, E.A.; Tolstoy, P.M.; Shenderovich, I.G.; Filarowski, A. Inter- vs. intra-molecular hydrogen bond patterns and proton dynamics in phthalic acid associates. Molecules 2020, 25, 4720. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).