A Hidden Side of the Conformational Mobility of the Quercetin Molecule Caused by the Rotations of the O3H, O5H and O7H Hydroxyl Groups: In Silico Scrupulous Study


Abstract
:
1. Introduction
2. Computational Methods
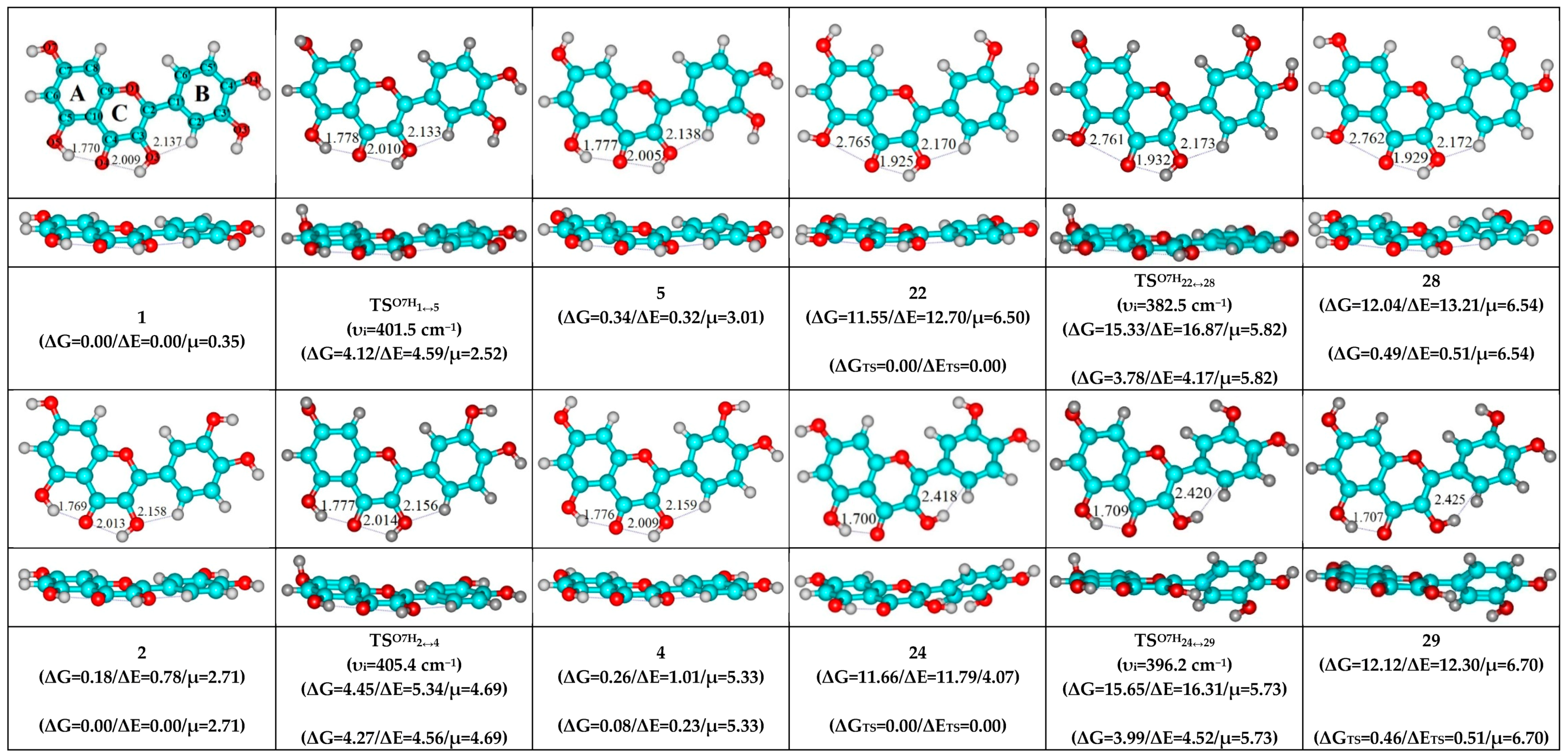
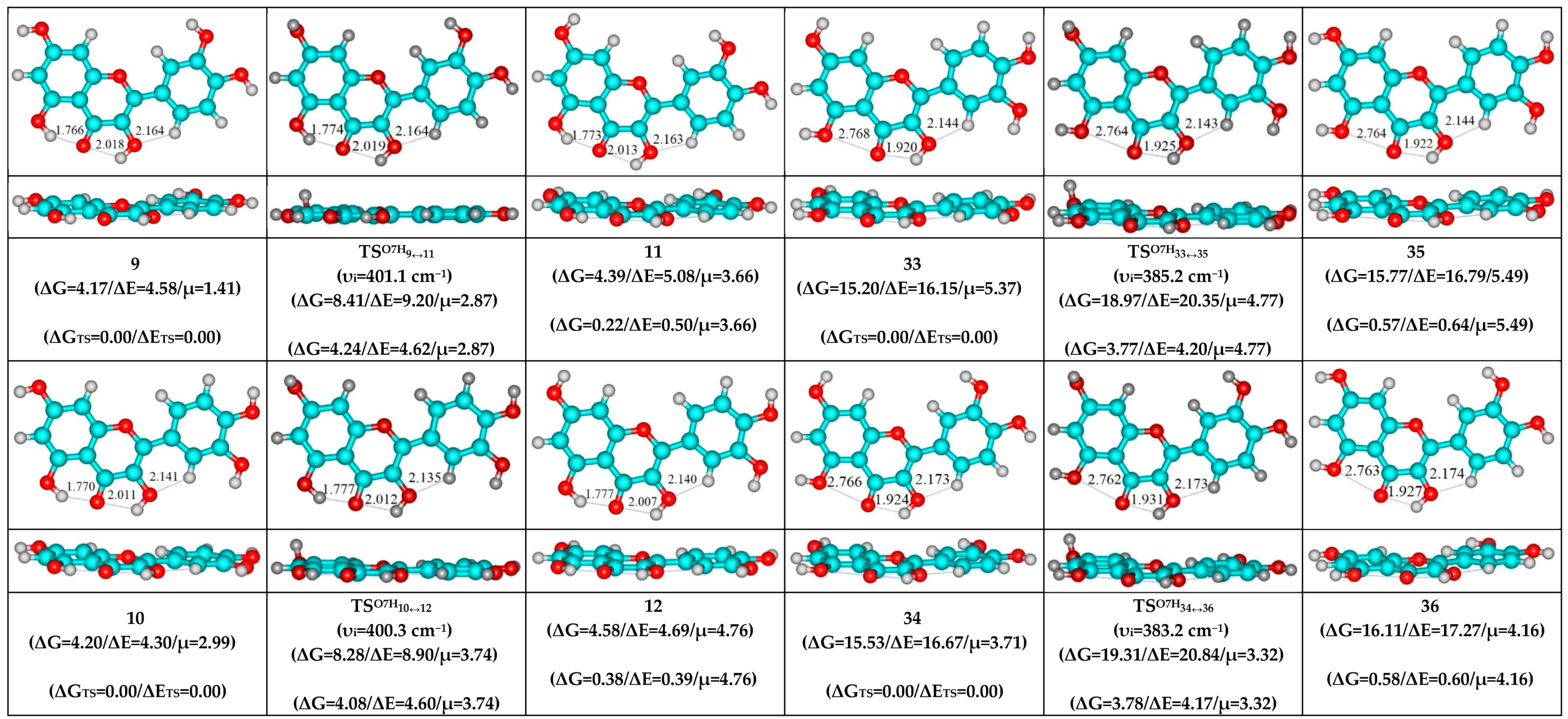
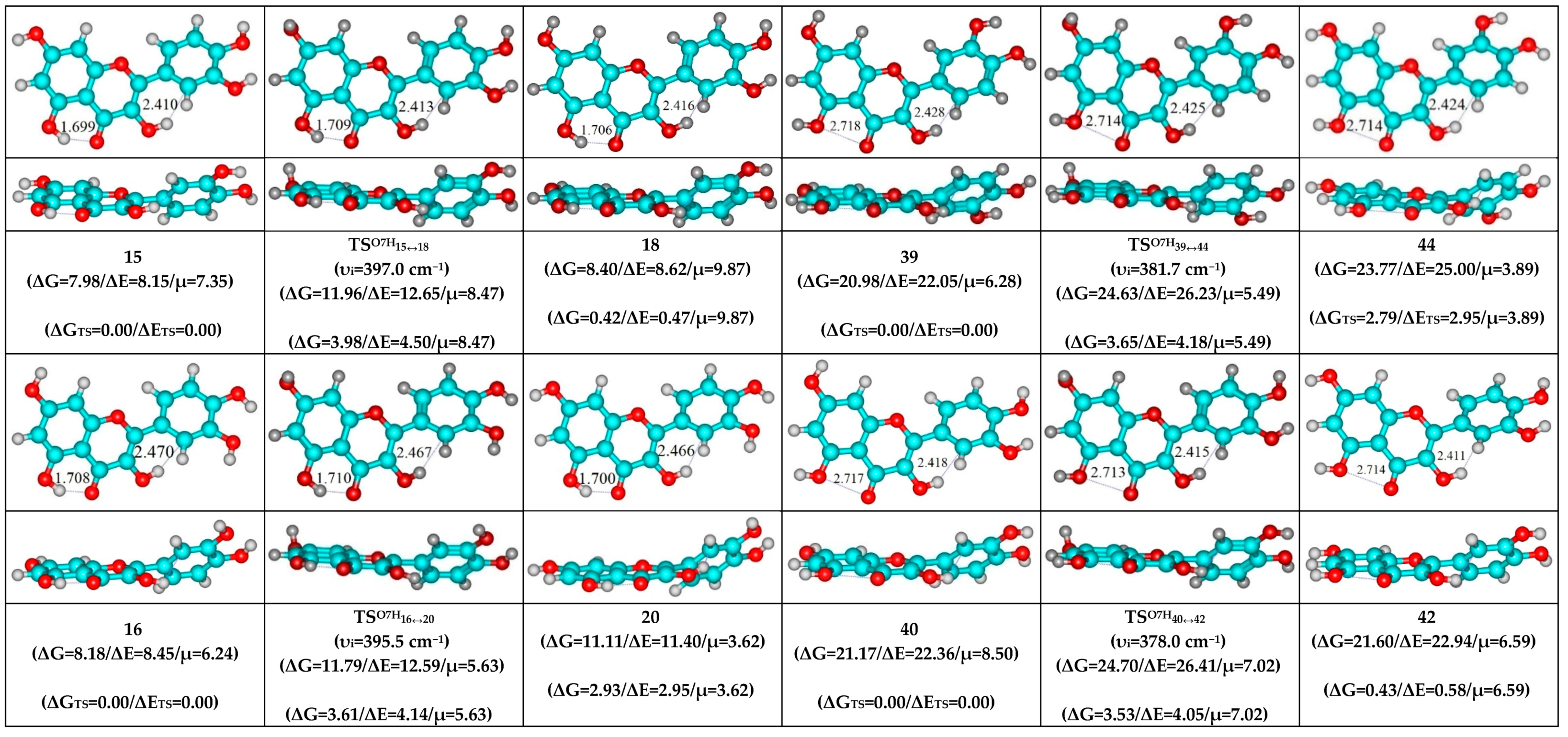
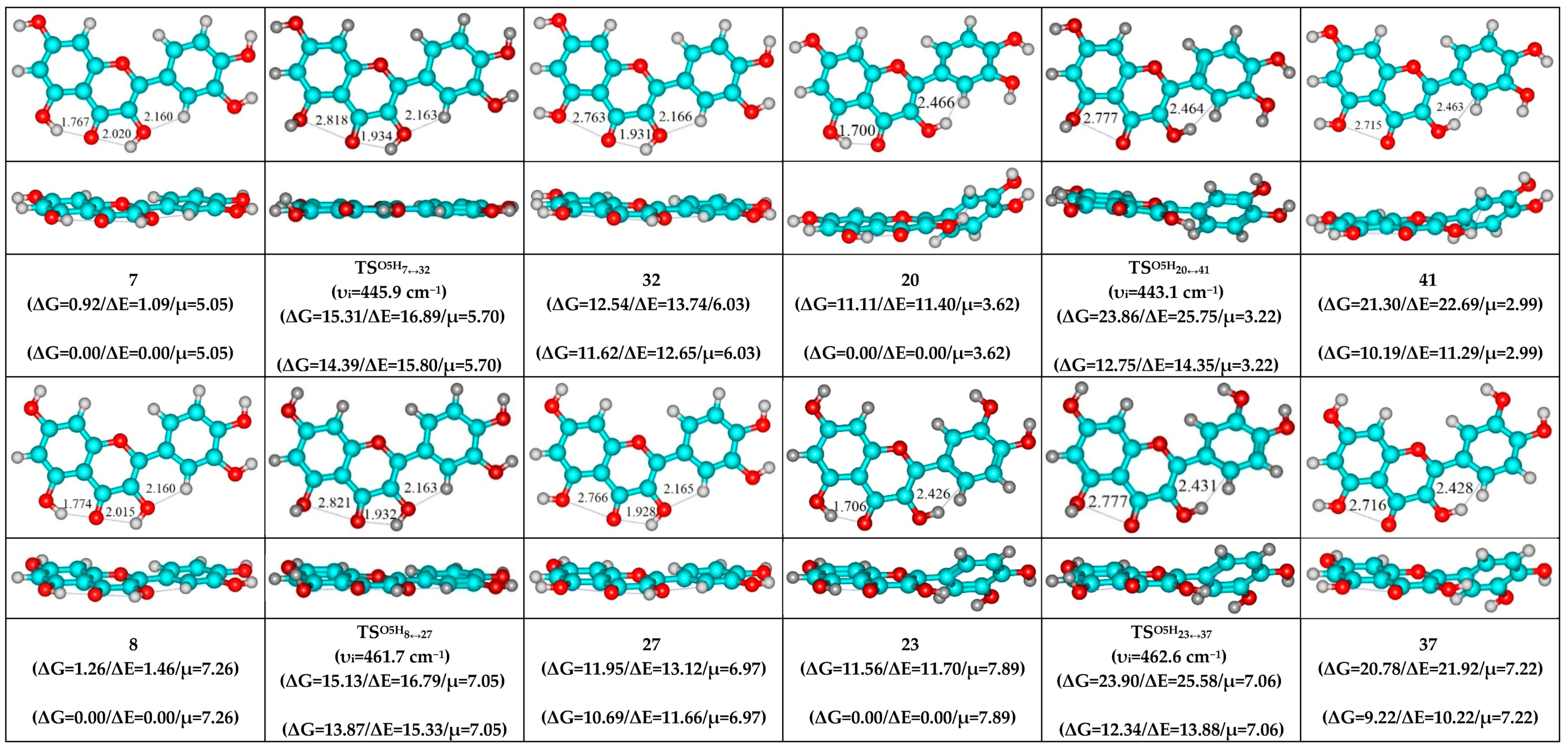
3. Results and Discussion
- -
- O5H…O4, O3H…O4 and C2′H…O3 H-bonds (initial conformer, TS, terminal conformer; 1↔5, 7↔8 and 10↔12);
- -
- O5H…O4, O3H…O4 and C6′H…O3 H-bonds (initial conformer, TS, terminal conformer; 2↔4, 3↔6 and 9↔11);
- -
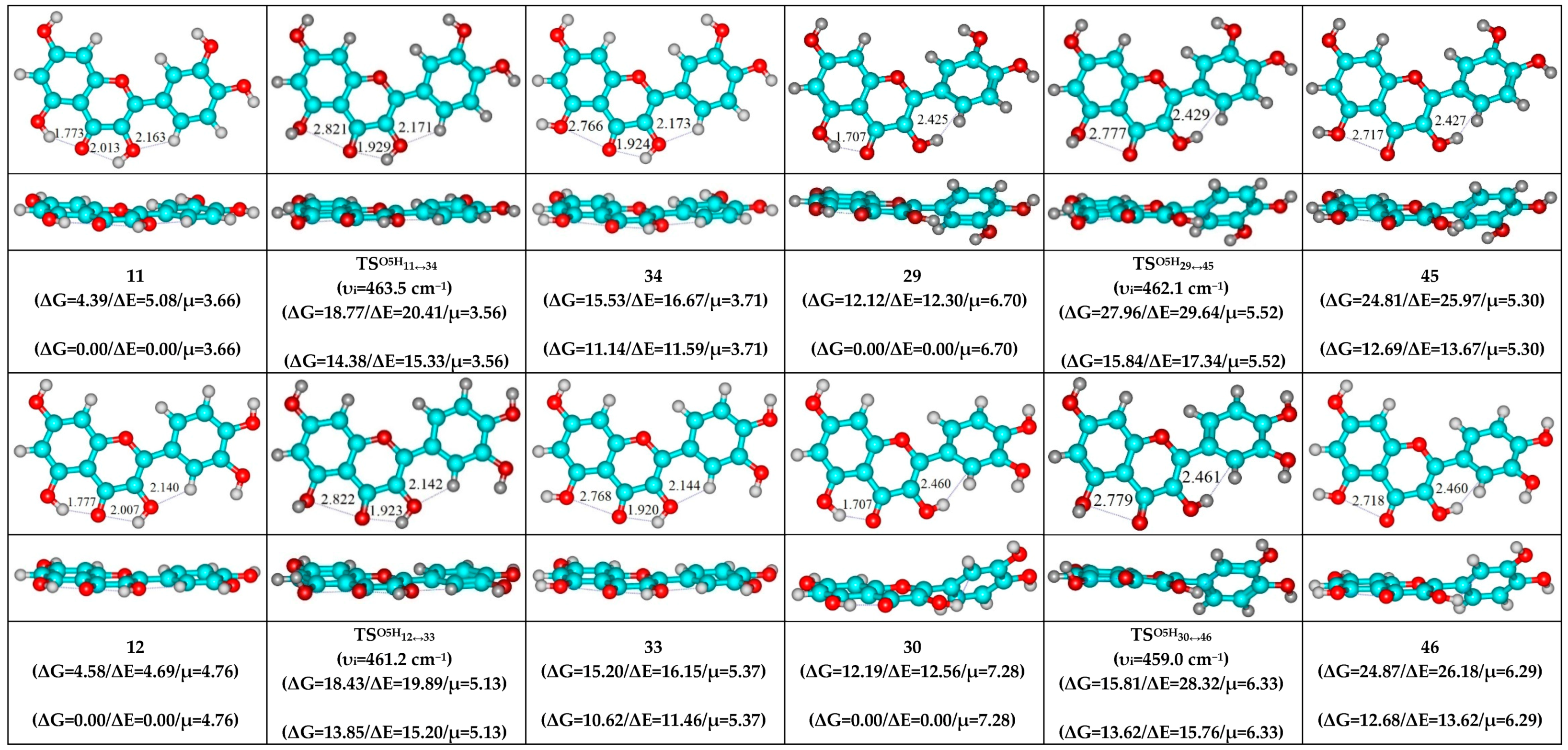
- O5…O4 vdW contact and O3H…O4, C2′H…O3 H-bonds (initial conformer, TS, terminal conformer; 19↔25, 27↔32 and 33↔35);
- -
- O5…O4 vdW contact and O3H…O4, C6′H…O3 H-bonds (initial conformer, TS, terminal conformer; 21↔31, 22↔28 and 34↔36).
- -
- O5H…O4 and O3H…C6′ H-bonds (initial conformer, TS, terminal conformer; 13↔23, 14↔17 and 24↔29);
- -
- O5H…O4 and O3H…C2′ H-bonds (initial conformer, TS, terminal conformer; 15↔18, 16↔20 and 26↔30);
- -
- O5…O4 vdW contact and O3H…C6′ H-bond (initial conformer, TS, terminal conformer; 37↔43, 39↔44 and 45↔47);
- -
- O5…O4 vdW contact and O3H…C2′ H-bond (initial conformer, TS, terminal conformer; 38↔41, 40↔42 and 46↔48).
- -
- O5H…O4, O3H…O4 and C2′H…O3 H-bonds (initial conformer) → O5…O4 vdW contact and O3H…O4, C2′H…O3 H-bonds (TS, terminal conformer; 1↔25, 3↔28, 5↔19, 7↔32, 8↔27, 10↔35 and 12↔33);
- -
- O5H…O4, O3H…O4 and C6′H…O3 H-bonds (initial conformer) → O5…O4 vdW contact and O3H…O4, C6′H…O3 H-bonds (TS, terminal conformer; 2↔31, 4↔21, 6↔22, 9↔36 and 11↔34).
- -
- O5H…O4 and O3H…C6′ H-bonds (initial conformer) → O5…O4 vdW contact and O3H…C6′ H-bonds (TS, terminal conformer; 13↔43, 14↔44, 17↔39, 23↔37, 24↔47 and 29↔45);
- -
- O5H…O4 and O3H…C2′ H-bonds (initial conformer) → O5…O4 vdW contact and C6′H…O3 H-bonds (TS, terminal conformer; 15↔42, 16↔38, 18↔40, 20↔41, 26↔48 and 30↔46).
- -
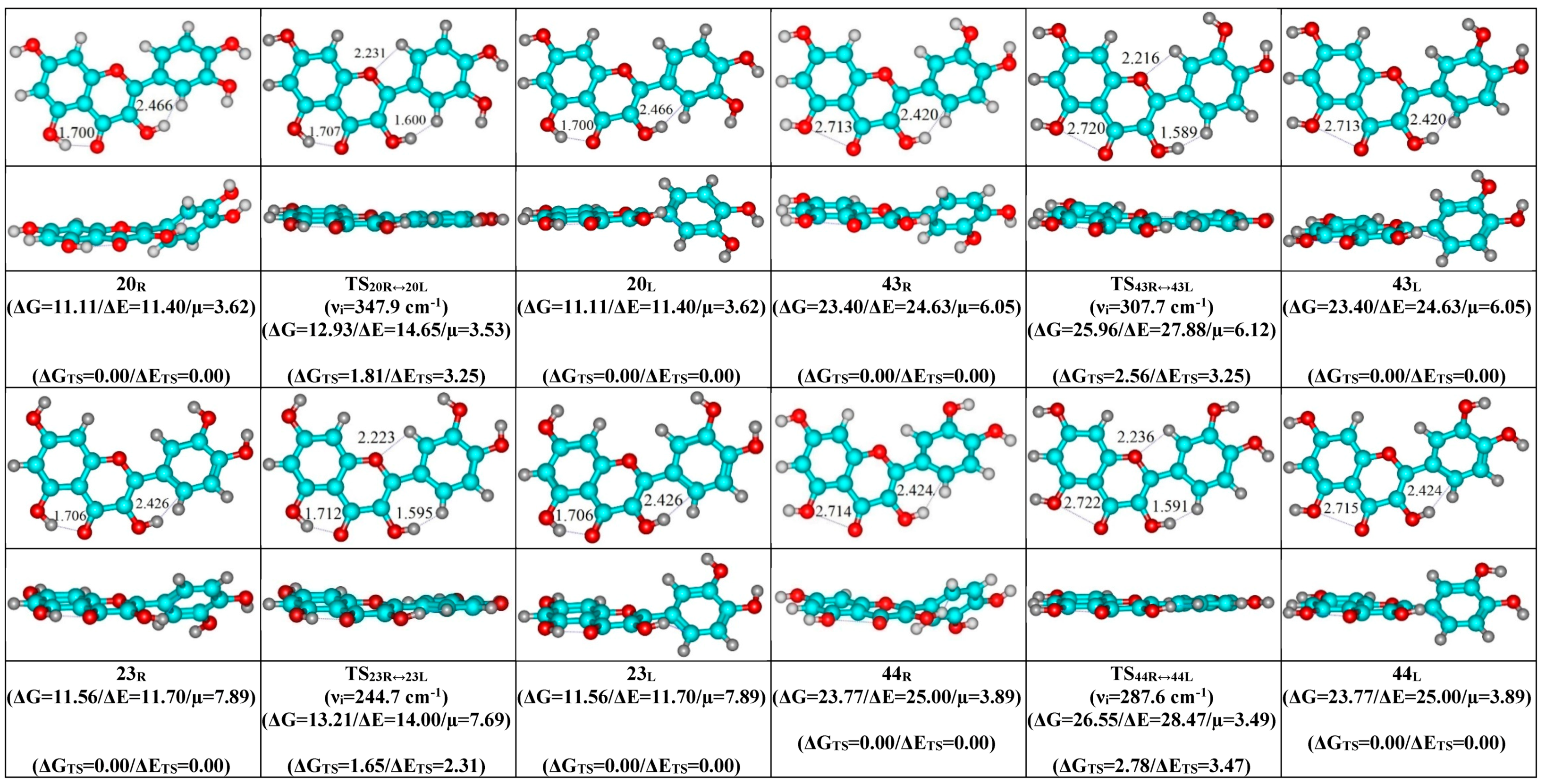
- O5H…O4, O3H…O4 and C2′H…O3 H-bonds (initial conformer) → O5H…O4, C2′H…O3H H-bonds (TS) → O5H…O4 and O3H…C2′ H-bonds (terminal conformer; 1↔20, 5↔16, 7↔15, 8↔18, 10↔26 and 12↔30);
- -
- O5H…O4, O3H…O4 and C6′H…O3 H-bonds (initial conformer) → O5H…O4, C6′H…O3H H-bonds (TS) → O5H…O4 and O3H…C6′ H-bonds (terminal conformer; 2↔14, 3↔13, 4↔17, 6↔23, 9↔24 and 11↔29);
- -
- O5…O4 vdW contact and O3H…O4, C2′H…O3 H-bonds (initial conformer) → O5…O4 vdW contact and C2′H…O3H H-bond (TS) → O5…O4 vdW contact and O3H…C2′ H-bond (terminal conformer; 19↔38, 25↔41, 27↔40, 32↔42, 33↔46 and 35↔48);
- -
- O5…O4 vdW contact and O3H…O4 and C6′H…O3 H-bonds (initial conformer) → O5…O4 vdW contact and C6′H…O3H H-bond (TS) → O5H…O4 and O3H…C6′ H-bonds (terminal conformer; 21↔39, 22↔37, 28↔43, 31↔44, 34↔45 and 36↔47).
4. Conclusions
- It was established, that conformational mobility of the isolated quercetin molecule, which is connected with the mirror-symmetric torsional mobility of its O3H, O5H and O7H hydroxyl groups, were controlled by the 72 transitions states with the non-orthogonal geometry (C1 point symmetry). In the cases of the turnings of the O7H and O5H hydroxyl groups, TSs were stabilized by the participation of the specific intramolecular interactions—attractive O4…O5 vdW contacts and C2′/C6′H...O3 H-bonds, respectively. Activation barriers of the Gibbs free energies formed the following series under the standard conditions: ΔΔGTSO7H (3.51–4.24) < ΔΔGTSO3H (9.04–11.26) < ΔΔGTSO5H (12.34–16.17 kcal·mol−1).
- Conformational rearrangement of the O3H and O5H groups was partially controlled by the intramolecular specific interactions O3H…O4, C2′/C6′H…O3, O3H…C2′/C6′, O5H…O4 H-bonds and attractive O4…O5 vdW contacts, which were flexible and cooperative.
- Mutual transformation of the enantiomers of the non-planar conformers of the quercetin molecule realized via the 24 quasi-planar TSs with C1 point symmetry (ΔΔGTS = 1.65–5.59 kcal·mol−1), which were supported by the participation of the intramolecular O3H...HC2′/C6′ (~4.7/4.8) and C2′/C6′H…O1 (~4.7 kcal mol−1) H-bonds.
- All investigated conformational transitions were accompanied by the significant changes of the dipole moment of the molecule as by the absolute value, so by the spatial orientation.
- Investigated conformational transformations were quite quick processes—time, which is necessary to acquire thermal equilibrium, did not exceed 6.5 ns.
Author Contributions
Funding
Conflicts of Interest
References
- Grytsenko, O.M.; Degtyarev, L.S.; Pilipchuck, L.B. Physico-chemical properties and electronic structure of quercetin. Farmats. Zhurn. 1992, N2, 34–38. [Google Scholar]
- Burda, S.; Oleszek, W. Antioxidant and antiradical activities of flavonoids. J. Agric. Food Chem. 2001, 49, 2774–2779. [Google Scholar] [CrossRef] [PubMed]
- Grytsenko, O.M.; Pylypchuck, L.B.; Bogdan, T.V.; Trygubenko, S.A.; Hovorun, D.M.; Maksutina, N.P. Keto-enol prototropic tautomerism of quercetin molecule: Quantum-chemical calculations. Farmats. Zhurn. 2003, N5, 62–65. [Google Scholar]
- Olejniczak, S.; Potrzebowski, M.J. Solid state NMR studies and density functional theory (DFT) calculations of conformers of quercetin. Org. Biomol. Chem. 2004, 2, 2315–2322. [Google Scholar] [CrossRef] [PubMed]
- Bentz, A.B. A review of quercetin: Chemistry, antioxidant properties, and bioavailability. J. Young Investig. 2009, 19. [Google Scholar]
- Nathiya, S.; Durga, M.; Devasena, T. Quercetin, encapsulated quercetin and its application—A review. Int. J. Pharm. Pharm. Sci. 2014, 10, 20–26. [Google Scholar]
- David, A.V.A.; Arulmoli, R.; Parasuraman, S. Overviews of biological importance of quercetin: A bioactive flavonoid. Pharmacogn. Rev. 2016, 10, 84–89. [Google Scholar]
- van Acker, S.A.; de Groot, M.J.; van den Berg, D.J.; Tromp, M.N.; Donné-Op den Kelder, G.; van der Vijgh, W.J.; Bast, A.A. A quantum chemical explanation of the antioxidant activity of flavonoids. Chem. Res. Toxicol. 1996, 9, 1305–1312. [Google Scholar] [CrossRef]
- Bogdan, T.V.; Trygubenko, S.A.; Pylypchuck, L.B.; Potyahaylo, A.L.; Samijlenko, S.P.; Hovorun, D.M. Conformational analysis of the quercetin molecule. Sci. Notes NaUKMA 2001, 19, 456–460. [Google Scholar]
- Trouillas, P.; Marsal, P.; Siri, D.; Lazzaroni, R.; Duroux, J.-C. A DFT study of the reactivity of OH groups in quercetin and taxifolin antioxidants: The specificity of the 3-OH site. Food Chem. 2006, 97, 679–688. [Google Scholar] [CrossRef]
- Marković, Z.; Amić, D.; Milenković, D.; Dimitrić-Marković, J.M.; Marković, S. Examination of the chemical behavior of the quercetin radical cation towards some bases. Phys. Chem. Chem. Phys. 2013, 15, 7370–7378. [Google Scholar] [CrossRef] [PubMed]
- Protsenko, I.O.; Hovorun, D.M. Conformational properties of quercetin: Quantum chemistry investigation. Repts. Natl. Acad. Sci. Ukr. 2014, N3, 153–157. [Google Scholar] [CrossRef]
- Vinnarasi, S.; Radhika, R.; Vijayakumar, S.; Shankar, R. Structural insights into the anti-cancer activity of quercetin on G-tetrad, mixed G-tetrad, and G-quadruplex DNA using quantum chemical and molecular dynamics simulations. J. Biomol. Struct. Dyn. 2019. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, G. Quercetin: A flavonol with multifaceted therapeutic applications? Fitoterapia 2015, 106, 256–271. [Google Scholar] [CrossRef]
- Tu, B.; Liu, Z.-J.; Chen, Z.-F.; Ouyang, Y.; Hu, Y.-J. Understanding the structure–activity relationship between quercetin and naringenin: In vitro. RSC Adv. 2015, 5, 106171–106181. [Google Scholar] [CrossRef]
- Brovarets’, O.O.; Hovorun, D.M. Conformational diversity of the quercetin molecule: A quantum-chemical view. J. Biomol. Struct. Dyn. 2019. [Google Scholar] [CrossRef]
- Brovarets’, O.O.; Hovorun, D.M. Conformational transitions of the quercetin molecule via the rotations of its rings: A comprehensive theoretical study. J. Biomol. Struct. Dyn. 2019. [Google Scholar] [CrossRef]
- Brovarets’, O.O.; Protsenko, I.O.; Hovorun, D.M. Computational design of the conformational and tautomeric variability of the quercetin molecule. In Proceedings of the 6th Young Medicinal Chemist Symposium (EFMC-YMCS 2019), Athens, Greece, 5–6 September 2019; p. 50. [Google Scholar]
- Brovarets’, O.O.; Protsenko, I.O.; Hovorun, D.M. Comprehensive analysis of the potential energy surface of the quercetin molecule. In Proceedings of the Bioheterocycles 2019, XVIII International Conference on Heterocycles in Bioorganic Chemistry, Ghent, Belgium, 17–20 June 2019; p. 84. [Google Scholar]
- Brovarets’, O.O.; Protsenko, I.O.; Zaychenko, G. Computational modeling of the tautomeric interconversions of the quercetin molecule. In Proceedings of the International Symposium “EFMC-ACSMEDI Medicinal Chemistry Frontiers 2019” (MedChemFrontiers 2019), Krakow, Poland, 10–13 June 2019; p. 114. [Google Scholar]
- Jovanovic, S.V.; Steenken, S.; Hara, Y.; Simic, M.G. Reduction potentials of flavonoid and model phenoxyl radicals. Which ring in flavonoids is responsible for antioxidant activity? J. Chem. Soc. Trans. 1996, 2, 2497–2504. [Google Scholar] [CrossRef]
- Rice-Evans, C.A.; Miller, N.J.; Paganga, G. Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free Radical Med. 1996, 20, 933–956. [Google Scholar] [CrossRef]
- Rasulev, B.F.; Abdullaev, N.D.; Syrov, V.N.; Leszczynski, J. A Quantitative Structure-Activity Relationship (QSAR) study of the antioxidant activity of flavonoids. QSAR Comb. Sci. 2005, 24, 1056–1065. [Google Scholar] [CrossRef]
- Gao, S.; Sofic, E.; Prior, R.L. Antioxidant and prooxidant behavior of flavonoids: Structure-activity relationships. Free Radic. Biol. Med. 1997, 22, 749–760. [Google Scholar]
- Domagała, S.; Munshi, P.; Ahmed, M.; Guillot, B.; Jelsch, C. Structural analysis and multipole modelling of quercetin monohydrate—A quantitative and comparative study. Acta Cryst. Sect. B Struct. Sci. 2010, 67, 63–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhaouadi, Z.; Nsangou, M.; Garrab, N.; Anouar, E.H.; Marakchi, K.; Lahmar, S. DFT study of the reaction of quercetin with ·O2- and ·OH radicals. J. Mol. Struct. THEOCHEM 2009, 904, 35–42. [Google Scholar] [CrossRef]
- Yang, Y.; Zhao, J.; Li, Y. Theoretical study of the ESIPT process for a new natural product quercetin. Sci. Repts. 2016, 6, 32152. [Google Scholar] [CrossRef] [Green Version]
- Eliel, E.; Wilen, S.H.; Mander, L.N. Stereochemistry of Organic Compounds; Wiley: New York, NY, USA, 1994. [Google Scholar]
- Guijarro, A. The Origin of Chirality in the Molecules of Life: A Revision from Awareness to the Current Theories and Perspectives of this Unsolved Problem; Royal Society of Chemistry: Cambridge, UK, 2008. [Google Scholar]
- Palafox, M.A. Molecular structure differences between the antiviral nucleoside analogue 5-iodo-2′-deoxyuridine and the natural nucleoside 2′-deoxythymidine using MP2 and DFT methods: Conformational analysis, crystal simulations, DNA pairs and possible behavior. J. Biomol. Struct. Dyn. 2014, 32, 831–851. [Google Scholar] [CrossRef]
- Tirado-Rives, J.; Jorgensen, W.L. Performance of B3LYP Density Functional Methods for a large set of organic molecules. J. Chem. Theory Comput. 2008, 4, 297–306. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: Oxford, UK, 1989. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. GAUSSIAN 09; Revision B.01; Gaussian Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Brovarets’, O.O.; Hovorun, D.M. Atomistic understanding of the C·T mismatched DNA base pair tautomerization via the DPT: QM and QTAIM computational approaches. J. Comput. Chem. 2013, 34, 2577–2590. [Google Scholar] [CrossRef]
- Brovarets’, O.O.; Tsiupa, K.S.; Hovorun, D.M. Non-dissociative structural transitions of the Watson-Crick and reverse Watson-Crick A·T DNA base pairs into the Hoogsteen and reverse Hoogsteen forms. Sci. Repts. 2018, 8, 10371. [Google Scholar] [CrossRef] [Green Version]
- Hratchian, H.P.; Schlegel, H.B. Finding minima, transition states, and following reaction pathways on ab initio potential energy surfaces. In Theory and Applications of Computational Chemistry: The First 40 Years; Dykstra, C.E., Frenking, G., Kim, K.S., Scuseria, G., Eds.; Elsevier: Amsterdam, The Netherlands, 2005; pp. 195–249. [Google Scholar]
- Frisch, M.J.; Head-Gordon, M.; Pople, J.A. Semi-direct algorithms for the MP2 energy and gradient. Chem. Phys. Lett. 1990, 166, 281–289. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Atkins, P.W. Physical Chemistry; Oxford University Press: Oxford, UK, 1998. [Google Scholar]
- Wigner, E. Über das Überschreiten von Potentialschwellen bei chemischen Reaktionen [Crossing of potential thresholds in chemical reactions]. Zeits. Physik. Chem. 1932, B19, 203–216. [Google Scholar]
- Keith, T.A. AIMAll (Version 10.07.01). 2010. Available online: aim.tkgristmill.com (accessed on 18 January 2020).
- Matta, C.F.; Hernández-Trujillo, J. Bonding in polycyclic aromatic hydrocarbons in terms of the electron density and of electron delocalization. J. Phys. Chem A 2003, 107, 7496–7504. [Google Scholar] [CrossRef]
- Brovarets’, O.O.; Yurenko, Y.P.; Hovorun, D.M. The significant role of the intermolecular CH···O/N hydrogen bonds in governing the biologically important pairs of the DNA and RNA modified bases: A comprehensive theoretical investigation. J. Biomol. Struct. Dyn. 2015, 33, 1624–1652. [Google Scholar] [CrossRef]
- Lecomte, C.; Espinosa, E.; Matta, C.F. On atom–atom ‘short contact’ bonding interactions in crystals. IUCrJ 2015, 2, 161–163. [Google Scholar] [CrossRef] [Green Version]
- Matta, C.F.; Castillo, N.; Boyd, R.J. Extended weak bonding interactions in DNA: π-stacking (base-base), base-backbone, and backbone-backbone interactions. J. Phys. Chem. B 2006, 110, 563–578. [Google Scholar] [CrossRef]
- Nikolaienko, T.Y.; Bulavin, L.A.; Hovorun, D.M. Bridging QTAIM with vibrational spectroscopy: The energy of intramolecular hydrogen bonds in DNA-related biomolecules. Phys. Chem. Chem. Phys. 2012, 14, 7441–7447. [Google Scholar] [CrossRef]
- Raczyńska, E.; Kosińska, W.; Ośmiałowski, B.; Gawinecki, R. Tautomeric equilibria in relation to π-electron delocalization. Chem. Rev. 2005, 105, 3561–3612. [Google Scholar] [CrossRef]
- Brovarets’, O.O.; Hovorun, D.M. A new era in the prototropic tautomerism of the quercetin molecule: A QM/QTAIM computational advances. J. Biomol. Struct. Dyn. 2019. [Google Scholar] [CrossRef]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conformational Transition | µTS a | νi b | ∆G c | ∆E d | ∆∆GTS e | ∆∆ETS f | ∆∆G g | ∆∆E h | kf i | kr j | τ99.9% k | τ l | HO7C7C8/HO7C7C6 m |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1↔5 | 2.52 | 401.5 | 0.34 | 0.32 | 4.12 | 4.59 | 3.78 | 4.27 | 6.72 × 109 | 1.20 × 1010 | 3.70 × 10−10 | 8.36 × 10−11 | ±90.6 |
| 2↔4 | 4.69 | 405.4 | 0.08 | 0.23 | 4.27 | 4.56 | 4.19 | 4.32 | 5.23 × 109 | 5.99 × 109 | 6.16 × 10−10 | 1.67 × 10−10 | ±90.8 |
| 3↔6 | 4.57 | 400.9 | 0.28 | 0.53 | 4.11 | 4.63 | 3.83 | 4.10 | 6.87 × 109 | 1.10 × 1010 | 3.86 × 10−10 | 9.07 × 10−11 | ±89.9 |
| 7↔8 | 6.11 | 401.0 | 0.34 | 0.37 | 3.88 | 4.59 | 3.54 | 4.22 | 1.01 × 1010 | 1.79 × 1010 | 2.46 × 10−10 | 5.59 × 10−11 | ±90.3 |
| 9↔11 | 2.87 | 401.1 | 0.22 | 0.50 | 4.24 | 4.62 | 4.02 | 4.12 | 5.48 × 109 | 8.00 × 109 | 5.12 × 10−10 | 1.25 × 10−10 | ±90.1 |
| 10↔12 | 3.74 | 400.3 | 0.38 | 0.39 | 4.08 | 4.60 | 3.71 | 4.22 | 7.17 × 109 | 1.34 × 1010 | 3.35 × 10−10 | 7.45 × 10−11 | ±90.3 |
| 13↔23 | 6.44 | 395.1 | 3.97 | 4.04 | 3.98 | 4.52 | 0.01 | 0.48 | 8.50 × 109 | 6.94 × 1012 | 9.94 × 10−13 | 1.44 × 10−13 | ±89.9 |
| 14↔17 | 7.92 | 399.3 | 0.35 | 0.36 | 3.98 | 4.52 | 3.63 | 4.16 | 8.57 × 109 | 1.54 × 1010 | 2.88 × 10−10 | 6.49 × 10−11 | ±90.4 |
| 15↔18 | 8.47 | 397.0 | 0.42 | 0.47 | 3.98 | 4.50 | 3.56 | 4.03 | 8.61 × 109 | 1.75 × 1010 | 2.65 × 10−10 | 5.72 × 10−11 | ±90.1 |
| 16↔20 | 5.63 | 395.5 | 2.93 | 2.95 | 3.61 | 4.14 | 0.68 | 1.19 | 1.59 × 1010 | 2.24 × 1012 | 3.06 × 10−12 | 4.47 × 10−13 | ±90.3 |
| 19↔25 | 2.54 | 385.9 | 0.60 | 0.79 | 3.78 | 4.40 | 3.18 | 3.61 | 1.19 × 1010 | 3.28 × 1010 | 1.55 × 10−10 | 3.05 × 10−11 | ±93.0 |
| 21↔31 | 2.26 | 385.4 | 0.74 | 0.88 | 3.91 | 4.38 | 3.17 | 3.50 | 9.62 × 109 | 3.36 × 1010 | 1.60 × 10−10 | 2.98 × 10−11 | ±93.3 |
| 22↔28 | 5.82 | 382.5 | 0.49 | 0.51 | 3.78 | 4.17 | 3.24 | 3.66 | 1.18 × 1010 | 2.95 × 1010 | 1.67 × 10−10 | 3.39 × 10−11 | ±92.3 |
| 24↔29 | 5.73 | 396.2 | 0.46 | 0.51 | 3.99 | 4.52 | 3.53 | 4.01 | 8.46 × 109 | 1.84 × 1010 | 2.57 × 10−10 | 5.43 × 10−11 | ±93.3 |
| 26↔30 | 4.82 | 395.0 | 0.45 | 0.44 | 4.02 | 4.53 | 3.57 | 4.08 | 7.93 × 109 | 1.70 × 1010 | 2.77 × 10−10 | 5.88 × 10−11 | ±90.2 |
| 27↔32 | 5.97 | 385.0 | 0.59 | 0.62 | 3.79 | 4.16 | 3.20 | 3.55 | 1.16 × 1010 | 3.14 × 1010 | 1.61 × 10−10 | 3.18 × 10−11 | ±92.7 |
| 33↔35 | 4.77 | 385.2 | 0.57 | 0.64 | 3.77 | 4.20 | 3.20 | 3.56 | 1.20 × 1010 | 3.16 × 1010 | 1.58 × 10−10 | 3.16 × 10−11 | ±92.8 |
| 34↔36 | 3.32 | 383.2 | 0.58 | 0.60 | 3.78 | 4.17 | 3.20 | 3.57 | 1.18 × 1010 | 3.16 × 1010 | 1.59 × 10−10 | 3.17 × 10−11 | ±92.5 |
| 37↔43 | 5.89 | 378.4 | 2.62 | 2.71 | 3.51 | 3.97 | 0.89 | 1.26 | 1.87 × 1010 | 1.56 × 1012 | 4.37 × 10−12 | 6.40 × 10−13 | ±92.3 |
| 38↔41 | 3.94 | 379.3 | 0.46 | 0.66 | 3.63 | 4.15 | 3.17 | 3.49 | 1.53 × 1010 | 3.33 × 1010 | 1.42 × 10−10 | 3.00 × 10−11 | ±92.8 |
| 39↔44 | 5.49 | 381.7 | 2.79 | 2.95 | 3.65 | 4.18 | 0.86 | 1.23 | 1.47 × 1010 | 1.63 × 1012 | 4.20 × 10−12 | 6.13 × 10−13 | ±92.8 |
| 40↔42 | 7.02 | 378.0 | 0.43 | 0.58 | 3.53 | 4.05 | 3.10 | 3.46 | 1.80 × 1010 | 3.72 × 1010 | 1.25 × 10−10 | 2.69 × 10−11 | ±92.6 |
| 45↔47 | 4.33 | 379.3 | 0.42 | 0.54 | 3.54 | 4.03 | 3.12 | 3.49 | 1.78 × 1010 | 3.63 × 1010 | 1.28 × 10−10 | 2.76 × 10−11 | ±92.5 |
| 46↔48 | 5.03 | 445.0 | 0.43 | 0.61 | 3.57 | 4.10 | 3.14 | 3.49 | 1.68 × 1010 | 3.48 × 1010 | 1.34 × 10−10 | 2.88 × 10−11 | ±92.7 |
| Conformational Transition | µTS | νi | ∆G | ∆E | ∆∆GTS | ∆∆ETS | ∆∆G | ∆∆E | kf | kr | τ99.9% | τ | HO5 C5C10 a |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1↔25 | 2.71 | 445.0 | 11.68 | 12.45 | 14.41 | 15.69 | 2.00 | 2.48 | 1.96 × 102 | 2.49 × 1011 | 2.77 × 10−11 | 4.01 × 10−12 | ±110.9 |
| 2↔31 | 1.69 | 443.5 | 12.08 | 12.63 | 14.85 | 15.78 | 2.77 | 3.15 | 9.37 × 101 | 6.80 × 1010 | 1.02 × 10−10 | 1.47 × 10−11 | ±111.0 |
| 3↔28 | 5.85 | 445.1 | 11.79 | 12.66 | 14.57 | 15.83 | 2.78 | 3.18 | 1.51 × 102 | 6.71 × 1010 | 1.03 × 10−10 | 1.49 × 10−11 | ±111.1 |
| 4↔21 | 3.79 | 462.2 | 11.26 | 11.52 | 14.51 | 15.24 | 3.25 | 3.72 | 1.69 × 102 | 3.07 × 1010 | 2.25 × 10−10 | 3.26 × 10−11 | ±109.0 |
| 5↔19 | 2.54 | 461.0 | 10.74 | 11.34 | 13.86 | 15.16 | 3.12 | 3.82 | 5.04 × 102 | 3.81 × 1010 | 1.81 × 10−10 | 2.62 × 10−11 | ±108.9 |
| 6↔22 | 6.20 | 463.9 | 11.02 | 11.62 | 14.24 | 15.43 | 3.28 | 3.81 | 2.64 × 102 | 2.89 × 1010 | 2.39 × 10−10 | 3.46 × 10−11 | ±109.1 |
| 7↔32 | 5.70 | 445.9 | 11.62 | 12.65 | 14.39 | 15.80 | 2.77 | 3.15 | 2.08 × 102 | 6.88 × 1010 | 1.00 × 10−10 | 1.45 × 10−11 | ±111.1 |
| 8↔27 | 7.05 | 461.7 | 10.69 | 11.66 | 13.87 | 15.33 | 3.18 | 3.66 | 4.97 × 102 | 3.44 × 1010 | 2.01 × 10−10 | 2.90 × 10−11 | ±109.1 |
| 9↔36 | 3.38 | 444.9 | 11.94 | 12.69 | 14.72 | 15.86 | 2.78 | 3.17 | 1.17 × 102 | 6.68 × 1010 | 1.03 × 10−10 | 1.50 × 10−11 | ±111.0 |
| 10↔35 | 4.78 | 445.0 | 11.57 | 12.49 | 14.33 | 15.67 | 2.76 | 3.17 | 1.92 × 102 | 5.87 × 1010 | 1.18 × 10−10 | 1.70 × 10−11 | ±111.0 |
| 11↔34 | 3.56 | 463.5 | 11.14 | 11.59 | 14.38 | 15.33 | 3.24 | 3.74 | 2.09 × 102 | 3.12 × 1010 | 2.22 × 10−10 | 3.21 × 10−11 | ±109.0 |
| 12↔33 | 5.13 | 461.2 | 10.62 | 11.46 | 13.85 | 15.20 | 3.23 | 3.74 | 5.14 × 102 | 3.18 × 1010 | 2.18 × 10−10 | 3.15 × 10−11 | ±108.9 |
| 13↔43 | 5.57 | 446.8 | 15.81 | 16.97 | 16.17 | 17.79 | 0.36 | 0.82 | 1.01 × 101 | 3.97 × 1012 | 1.74 × 10−12 | 2.52 × 10−13 | ±114.2 |
| 14↔44 | 4.58 | 445.9 | 15.81 | 16.97 | 16.16 | 17.76 | 0.35 | 0.79 | 1.03 × 101 | 4.06 × 1012 | 1.70 × 10−12 | 2.46 × 10−13 | ±114.3 |
| 15↔42 | 6.47 | 443.3 | 13.62 | 14.79 | 16.16 | 17.82 | 2.54 | 3.02 | 1.02 × 101 | 9.94 × 1010 | 6.95 × 10−11 | 1.01 × 10−11 | ±114.4 |
| 16↔38 | 5.12 | 458.6 | 12.66 | 13.58 | 15.78 | 17.26 | 3.12 | 3.68 | 1.96 × 101 | 3.80 × 1010 | 1.82 × 10−10 | 2.63 × 10−11 | ±112.3 |
| 17↔39 | 7.06 | 461.6 | 12.67 | 13.66 | 15.83 | 17.32 | 3.16 | 3.67 | 1.83 × 101 | 3.59 × 1010 | 1.92 × 10−10 | 2.79 × 10−11 | ±112.3 |
| 18↔40 | 8.61 | 458.5 | 12.77 | 13.74 | 15.85 | 17.38 | 3.07 | 3.64 | 1.75 × 101 | 4.11 × 1010 | 1.68 × 10−10 | 2.44 × 10−11 | ±112.4 |
| 20↔41 | 3.22 | 443.1 | 10.19 | 11.29 | 12.75 | 14.35 | 2.56 | 3.06 | 3.24 × 103 | 9.70 × 1010 | 7.12 × 10−11 | 1.03 × 10−11 | ±114.3 |
| 23↔37 | 7.06 | 462.6 | 9.22 | 10.22 | 12.34 | 13.88 | 3.12 | 3.66 | 6.61 × 103 | 3.78 × 1010 | 1.83 × 10−10 | 2.64 × 10−11 | ±112.3 |
| 24↔47 | 3.71 | 446.8 | 13.57 | 14.72 | 16.17 | 17.78 | 2.60 | 3.06 | 1.01 × 101 | 9.10 × 1010 | 7.59 × 10−11 | 1.10 × 10−11 | ±114.2 |
| 26↔48 | 4.82 | 440.5 | 13.56 | 14.67 | 16.11 | 17.73 | 2.55 | 3.06 | 1.12 × 101 | 9.87 × 1010 | 7.00 × 10−11 | 1.01 × 10−11 | ±114.3 |
| 29↔45 | 5.52 | 462.1 | 12.69 | 13.67 | 15.84 | 17.34 | 3.15 | 3.67 | 1.78 × 101 | 3.62 × 1010 | 1.91 × 10−10 | 2.76 × 10−11 | ±112.3 |
| 30↔46 | 6.33 | 459.0 | 12.68 | 13.62 | 15.76 | 17.29 | 3.08 | 3.67 | 2.04 × 101 | 4.09 × 1010 | 1.69 × 10−10 | 2.44 × 10−11 | ±112.3 |
| Conformational Transition | µTS | νi | ∆G | ∆E | ∆∆GTS | ∆∆ETS | ∆∆G | ∆∆E | kf | kr | τ99.9% | τ | HO3 C3C4 a | C3C2 C1′C2′/C3C2 C1′C6′ b |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1↔20 | 2.77 | 274.2 | 11.11 | 11.40 | 9.16 | 9.61 | −1.95 | −1.80 | 1.26 × 106 | 1.78 × 1014 | 3.88 × 10−14 | 5.61 × 10−15 | ±118.6 | ±32.0 |
| 2↔14 | 5.04 | 326.5 | 7.78 | 7.25 | 9.53 | 9.47 | 1.75 | 2.21 | 6.96 × 105 | 3.54 × 1011 | 1.95 × 10−11 | 2.83 × 10−12 | ±114.1 | ±31.9 |
| 3↔13 | 5.40 | 330.4 | 7.34 | 7.11 | 9.20 | 9.46 | 1.86 | 2.35 | 1.21 × 106 | 2.95 × 1011 | 2.34 × 10−11 | 3.39 × 10−12 | ±113.8 | ±29.4 |
| 4↔17 | 7.71 | 329.3 | 8.05 | 7.38 | 9.85 | 9.64 | 1.80 | 2.26 | 4.10 × 105 | 3.29 × 1011 | 2.10 × 10−11 | 3.04 × 10−12 | ±114.3 | ±32.2 |
| 5↔16 | 5.45 | 278.6 | 7.84 | 8.13 | 9.19 | 9.70 | 1.36 | 1.57 | 1.20 × 106 | 6.70 × 1011 | 1.03 × 10−11 | 1.49 × 10−12 | ±118.6 | ±32.5 |
| 6↔23 | 7.46 | 335.1 | 11.03 | 10.62 | 9.38 | 9.62 | −1.65 | −0.99 | 8.99 × 105 | 1.11 × 1014 | 6.22 × 10−14 | 9.00 × 10−15 | ±113.7 | ±30.8 |
| 7↔15 | 6.95 | 324.7 | 7.06 | 7.06 | 9.04 | 9.32 | 2.01 | 2.26 | 1.59 × 106 | 2.27 × 1011 | 3.04 × 10−11 | 4.40 × 10−12 | ±113.6 | ±30.5 |
| 8↔18 | 9.37 | 328.3 | 7.14 | 7.16 | 9.21 | 9.53 | 2.07 | 2.36 | 1.19 × 106 | 2.07 × 1011 | 3.34 × 10−11 | 4.84 × 10−12 | ±113.7 | ±31.3 |
| 9↔24 | 3.36 | 328.8 | 7.49 | 7.21 | 9.27 | 9.49 | 1.77 | 2.29 | 1.08 × 106 | 3.42 × 1011 | 2.02 × 10−11 | 2.93 × 10−12 | ±114.2 | ±29.9 |
| 10↔26 | 4.47 | 284.1 | 7.54 | 7.82 | 9.12 | 9.51 | 1.59 | 1.69 | 1.36 × 106 | 4.57 × 1011 | 1.51 × 10−11 | 2.19 × 10−12 | ±117.4 | ±31.5 |
| 11↔29 | 5.93 | 333.3 | 7.73 | 7.22 | 9.49 | 9.54 | 1.76 | 2.33 | 7.49 × 105 | 3.49 × 1011 | 1.98 × 10−11 | 2.86 × 10−12 | ±114.1 | ±31.2 |
| 12↔30 | 2.71 | 288.7 | 7.61 | 7.87 | 9.23 | 9.66 | 1.61 | 1.78 | 1.14 × 106 | 4.39 × 1011 | 1.57 × 10−11 | 2.28 × 10−12 | ±117.3 | ±32.1 |
| 19↔38 | 3.75 | 301.8 | 9.76 | 10.37 | 11.08 | 11.98 | 1.33 | 1.62 | 5.00 × 104 | 7.17 × 1011 | 9.64 × 10−12 | 1.39 × 10−12 | ±118.8 | ±32.2 |
| 21↔39 | 5.33 | 351.6 | 9.46 | 9.52 | 11.12 | 11.74 | 1.66 | 2.23 | 4.80 × 104 | 4.19 × 1011 | 1.65 × 10−11 | 2.38 × 10−12 | ±114.8 | ±32.1 |
| 22↔37 | 7.07 | 359.0 | 9.23 | 9.22 | 11.08 | 11.74 | 1.85 | 2.52 | 5.19 × 104 | 3.05 × 1011 | 2.26 × 10−11 | 3.27 × 10−12 | ±114.1 | ±31.3 |
| 25↔41 | 2.53 | 298.0 | 9.62 | 10.24 | 11.03 | 11.85 | 1.41 | 1.61 | 5.41 × 104 | 6.15 × 1011 | 1.12 × 10−11 | 1.62 × 10−12 | ±118.8 | ±31.7 |
| 27↔40 | 8.24 | 346.3 | 9.22 | 9.24 | 11.26 | 11.63 | 2.04 | 2.39 | 3.80 × 104 | 2.20 × 1011 | 3.14 × 10−11 | 4.55 × 10−12 | ±114.4 | ±31.2 |
| 28↔43 | 6.21 | 354.7 | 11.36 | 11.42 | 10.99 | 11.68 | −0.37 | 0.26 | 6.04 × 104 | 1.30 × 1013 | 5.32 × 10−13 | 7.70 × 10−14 | ±114.2 | ±29.7 |
| 31↔44 | 1.73 | 351.6 | 11.51 | 11.59 | 10.39 | 10.86 | −1.12 | −0.72 | 1.64 × 105 | 4.55 × 1013 | 1.52 × 10−13 | 2.20 × 10−14 | ±114.8 | ±32.1 |
| 32↔42 | 6.03 | 343.1 | 9.06 | 9.20 | 11.13 | 11.58 | 2.07 | 2.38 | 4.69 × 104 | 2.08 × 1011 | 3.33 × 10−11 | 4.82 × 10−12 | ±114.3 | ±30.4 |
| 33↔46 | 6.07 | 310.8 | 9.67 | 10.03 | 11.20 | 11.79 | 1.53 | 1.76 | 4.10 × 104 | 5.13 × 1011 | 1.35 × 10−11 | 1.95 × 10−12 | ±117.7 | ±31.9 |
| 34↔45 | 4.65 | 356.8 | 9.28 | 9.30 | 10.98 | 11.65 | 1.70 | 2.35 | 6.15 × 104 | 3.91 × 1011 | 1.77 × 10−11 | 2.56 × 10−12 | ±114.5 | ±31.5 |
| 35↔48 | 5.03 | 307.1 | 9.53 | 10.00 | 11.08 | 11.74 | 1.55 | 1.74 | 4.99 × 104 | 4.88 × 1011 | 1.42 × 10−11 | 2.05 × 10−12 | ±117.7 | ±31.2 |
| 36↔47 | 3.52 | 352.8 | 9.12 | 9.24 | 10.83 | 11.56 | 1.71 | 2.33 | 7.88 × 104 | 3.86 × 1011 | 1.79 × 10−11 | 2.59 × 10−12 | ±113.9 | ±30.1 |
| Interconversion of Enantiomers | µTS | νi | ∆∆GTS | ∆∆ETS | kf,r | τ99.9% | τ | HO3C3C2C1′ a | HC2′/C6′C1′C2 b | C3C2C1′C2′/C3C2C1′C6′ c |
|---|---|---|---|---|---|---|---|---|---|---|
| 13R/L↔13L/R | 5.47 | 266.3 | 4.98 | 5.56 | 1.48 × 109 | 2.34 × 10−9 | 6.78 × 10−10 | ±14.6 | ±0.7 | ±7.6 |
| 14R/L↔14L/R | 5.92 | 256.6 | 4.01 | 5.92 | 7.51 × 109 | 4.60 × 10−10 | 1.33 × 10−10 | ±15.7 | ±0.6 | ±7.7 |
| 15R/L↔15L/R | 7.31 | 227.0 | 5.27 | 5.68 | 8.87 × 108 | 3.89 × 10−9 | 1.13 × 10−9 | ±18.1 | ±0.7 | ±8.4 |
| 16R/L↔16L/R | 6.20 | 344.0 | 5.17 | 6.72 | 1.11 × 109 | 3.10 × 10−9 | 8.99 × 10−10 | ±16.4 | ±2.9 | ±7.7 |
| 17R/L↔17L/R | 8.57 | 250.1 | 5.21 | 5.92 | 9.82 × 108 | 3.52 × 10−9 | 1.02 × 10−9 | ±18.0 | ±0.6 | ±9.1 |
| 18R/L↔18L/R | 9.79 | 221.3 | 5.33 | 5.82 | 8.00 × 108 | 4.32 × 10−9 | 1.25 × 10−9 | ±19.1 | ±0.7 | ±8.7 |
| 20R/L↔20L/R | 3.53 | 347.9 | 1.81 | 3.25 | 3.22 × 1011 | 1.07 × 10−11 | 3.11 × 10−12 | ±16.2 | ±2.8 | ±7.7 |
| 23R/L↔23L/R | 7.69 | 244.7 | 1.65 | 2.31 | 4.06 × 1011 | 8.52 × 10−12 | 2.47 × 10−12 | ±16.1 | ±0.6 | ±8.1 |
| 24R/L↔24L/R | 3.89 | 268.7 | 4.96 | 5.64 | 1.53 × 109 | 2.25 × 10−9 | 6.52 × 10−10 | ±16.0 | ±0.7 | ±8.2 |
| 26R/L↔26L/R | 4.73 | 326.0 | 5.49 | 6.50 | 6.37 × 108 | 5.43 × 10−9 | 1.57 × 10−9 | ±15.9 | ±2.3 | ±7.7 |
| 29R/L↔29L/R | 6.52 | 263.7 | 5.09 | 5.85 | 1.22 × 109 | 2.83 × 10−9 | 8.19 × 10−10 | ±17.5 | ±0.7 | ±8.7 |
| 30R/L↔30L/R | 7.19 | 322.1 | 5.54 | 6.65 | 5.88 × 108 | 5.88 × 10−9 | 1.70 × 10−9 | ±16.1 | ±2.4 | ±7.7 |
| 37R/L↔37L/R | 7.17 | 270.5 | 5.02 | 5.65 | 1.38 × 109 | 2.51 × 10−9 | 7.26 × 10−10 | ±12.6 | ±0.6 | ±7.0 |
| 38R/L↔38L/R | 4.02 | 328.0 | 5.53 | 6.50 | 5.98 × 108 | 5.78 × 10−9 | 1.67 × 10−9 | ±15.6 | ±2.3 | ±8.0 |
| 39R/L↔39L/R | 6.02 | 282.3 | 5.04 | 5.73 | 1.33 × 109 | 2.60 × 10−9 | 7.53 × 10−10 | ±13.4 | ±0.7 | ±7.6 |
| 40R/L↔40L/R | 8.61 | 241.6 | 5.20 | 5.67 | 1.01 × 109 | 3.43 × 10−9 | 9.93 × 10−10 | ±14.0 | ±0.6 | ±7.2 |
| 41R/L↔41L/R | 2.21 | 339.2 | 5.51 | 6.44 | 6.27 × 108 | 5.51 × 10−9 | 1.60 × 10−9 | ±14.8 | ±0.7 | ±7.7 |
| 42R/L↔42L/R | 6.82 | 256.9 | 5.13 | 5.58 | 1.13 × 109 | 3.05 × 10−9 | 8.82 × 10−10 | ±13.1 | ±0.7 | ±6.8 |
| 43R/L↔43L/R | 6.12 | 307.7 | 2.56 | 3.25 | 8.97 × 1010 | 3.85 × 10−11 | 1.11 × 10−11 | ±11.0 | ±0.7 | ±6.3 |
| 44R/L↔44L/R | 3.49 | 287.6 | 2.78 | 3.47 | 6.14 × 1010 | 5.62 × 10−11 | 1.63 × 10−11 | ±13.3 | ±0.7 | ±7.6 |
| 45R/L↔45L/R | 4.90 | 277.7 | 4.96 | 5.70 | 1.53 × 109 | 2.25 × 10−9 | 6.52 × 10−10 | ±13.4 | ±0.7 | ±7.4 |
| 46R/L↔46L/R | 6.21 | 312.2 | 5.59 | 6.44 | 5.35 × 108 | 6.46 × 10−9 | 1.87 × 10−9 | ±14.8 | ±1.8 | ±7.7 |
| 47R/L↔47L/R | 3.40 | 308.0 | 4.75 | 5.56 | 2.22 × 109 | 1.56 × 10−9 | 4.51 × 10−10 | ±12.1 | ±0.7 | ±6.9 |
| 48R/L↔48L/R | 4.93 | 315.5 | 5.59 | 6.35 | 5.35 × 108 | 6.46 × 10−9 | 1.87 × 10−9 | ±14.6 | ±1.7 | ±1.7 |
| Transition State | AH···B H-Bond/AH···HB Dihydrogen H-Bond/A···B vdW Contact | EAH···B/EAH···HB/EA··B a | ρ b | Δρ c | dA···B d | dH···B/dH···H e | AH···B f | C3C2C1′C6′ g |
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
| TSO7H1↔5 | O5H...O4 | 6.42 | 0.040 | 0.124 | 2.659 | 1.778 | 147.0 | 179.5 |
| O3H...O4 | 3.24 | 0.026 | 0.103 | 2.625 | 2.010 | 118.8 | ||
| C2′H...O3H * | 4.07 | 0.018 | 0.077 | 2.879 | 2.133 | 123.9 | ||
| TSO7H2↔4 | O5H...O4 | 6.45 | 0.040 | 0.124 | 2.658 | 1.777 | 147.0 | −0.5 |
| O3H...O4 | 3.20 | 0.026 | 0.103 | 2.626 | 2.014 | 118.7 | ||
| C6′H...O3H * | 3.86 | 0.018 | 0.073 | 2.892 | 2.156 | 123.3 | ||
| TSO7H3↔6 | O5H...O4 | 6.52 | 0.040 | 0.124 | 2.655 | 1.773 | 147.0 | −3.2 |
| O3H...O4 | 3.13 | 0.026 | 0.102 | 2.630 | 2.020 | 118.4 | ||
| C6′H...O3H * | 3.80 | 0.017 | 0.072 | 2.898 | 2.163 | 123.2 | ||
| TSO7H7↔8 | O5H...O4 | 6.50 | 0.040 | 0.124 | 2.656 | 1.775 | 147.0 | 177.8 |
| O3H...O4 | 3.12 | 0.026 | 0.102 | 2.629 | 2.021 | 118.4 | ||
| C2′H...O3H * | 3.87 | 0.018 | 0.074 | 2.882 | 2.156 | 122.4 | ||
| TSO7H9↔11 | O5H...O4 | 6.50 | 0.040 | 0.124 | 2.656 | 1.774 | 147.0 | −2.4 |
| O3H...O4 | 3.14 | 0.026 | 0.102 | 2.629 | 2.019 | 118.5 | ||
| C6′H...O3H * | 3.80 | 0.017 | 0.072 | 2.895 | 2.164 | 122.9 | ||
| TSO7H10↔12 | O5H...O4 | 6.44 | 0.040 | 0.124 | 2.658 | 1.777 | 147.0 | 179.1 |
| O3H...O4 | 3.21 | 0.026 | 0.103 | 2.626 | 2.012 | 118.7 | ||
| C2′H...O3H * | 4.03 | 0.018 | 0.076 | 2.885 | 2.135 | 124.1 | ||
| TSO7H13↔23 | O5H...O4 | 8.24 | 0.047 | 0.135 | 2.604 | 1.709 | 148.2 | −42.3 |
| O3H…C6′ * | 2.47 | 0.013 | 0.046 | 3.068 | 2.422 | 124.0 | ||
| TSO7H14↔17 | O5H...O4 | 8.20 | 0.047 | 0.135 | 2.605 | 1.710 | 148.2 | −43.1 |
| O3H…C6′ * | 2.45 | 0.013 | 0.046 | 3.064 | 2.426 | 123.3 | ||
| TSO7H15↔18 | O5H...O4 | 8.25 | 0.047 | 0.135 | 2.604 | 1.709 | 148.2 | 137.2 |
| O3H…C2′ * | 2.48 | 0.013 | 0.046 | 3.063 | 2.413 | 124.4 | ||
| TSO7H16↔20 | O5H...O4 | 8.20 | 0.047 | 0.135 | 2.605 | 1.710 | 148.2 | 137.7 |
| O3H…C2′ * | 2.35 | 0.012 | 0.045 | 3.052 | 2.467 | 118.8 | ||
| TSO7H19↔25 | O5...O4 * | 2.91 | 0.012 | 0.049 | 2.765 | - | - | 179.7 |
| O3H...O4 | 4.60 | 0.032 | 0.117 | 2.571 | 1.923 | 121.1 | ||
| C2′H...O3H * | 3.98 | 0.018 | 0.075 | 2.886 | 2.141 | 123.8 | ||
| TSO7H21↔31 | O5...O4 * | 2.92 | 0.012 | 0.049 | 2.764 | - | - | −0.1 |
| O3H...O4 | 4.55 | 0.032 | 0.116 | 2.572 | 1.926 | 120.9 | ||
| C6′H...O3H * | 3.76 | 0.017 | 0.071 | 2.901 | 2.166 | 123.2 | ||
| TSO7H22↔28 | O5...O4 * | 2.94 | 0.012 | 0.049 | 2.761 | - | - | −2.4 |
| O3H...O4 | 4.46 | 0.032 | 0.116 | 2.575 | 1.932 | 120.7 | ||
| C6′H...O3H * | 3.71 | 0.017 | 0.070 | 2.907 | 2.173 | 123.2 | ||
| TSO7H24↔29 | O5H...O4 | 8.23 | 0.047 | 0.135 | 2.604 | 1.709 | 148.2 | −42.4 |
| O3H…C6′ * | 2.47 | 0.013 | 0.046 | 3.065 | 2.420 | 123.9 | ||
| TSO7H26↔30 | O5H...O4 | 8.22 | 0.047 | 0.135 | 2.605 | 1.710 | 148.2 | 137.4 |
| O3H…C2′ * | 2.37 | 0.012 | 0.045 | 3.057 | 2.457 | 120.1 | ||
| TSO7H27↔32 | O5...O4 * | 2.93 | 0.012 | 0.049 | 2.762 | - | - | 178.7 |
| O3H...O4 | 4.43 | 0.031 | 0.115 | 2.575 | 1.934 | 120.6 | ||
| C2′H...O3H * | 3.79 | 0.017 | 0.072 | 2.891 | 2.165 | 122.4 | ||
| TSO7H33↔35 | O5...O4 * | 2.92 | 0.012 | 0.049 | 2.764 | - | - | 179.4 |
| O3H...O4 | 4.57 | 0.032 | 0.117 | 2.572 | 1.925 | 121.0 | ||
| C2′H...O3H * | 3.95 | 0.018 | 0.074 | 2.892 | 2.143 | 124.1 | ||
| TSO7H34↔36 | O5...O4 * | 2.93 | 0.012 | 0.049 | 2.762 | - | - | −1.7 |
| O3H...O4 | 4.48 | 0.032 | 0.116 | 2.574 | 1.931 | 120.7 | ||
| C6′H...O3H * | 3.71 | 0.017 | 0.070 | 2.903 | 2.173 | 122.9 | ||
| TSO7H37↔43 | O5...O4 * | 3.26 | 0.013 | 0.055 | 2.712 | - | - | −42.7 |
| O3H…C6′ * | 2.44 | 0.013 | 0.046 | 3.079 | 2.423 | 124.8 | ||
| TSO7H38↔41 | O5...O4 * | 3.25 | 0.013 | 0.055 | 2.714 | - | - | 137.6 |
| O3H…C2′ * | 2.35 | 0.012 | 0.045 | 3.061 | 2.465 | 119.7 | ||
| TSO7H39↔44 | O5...O4 * | 3.25 | 0.013 | 0.055 | 2.714 | - | - | −43.1 |
| O3H…C6′ * | 2.44 | 0.013 | 0.046 | 3.072 | 2.425 | 124.0 | ||
| TSO7H40↔42 | O5...O4 * | 3.26 | 0.013 | 0.055 | 2.713 | - | - | 137.2 |
| O3H…C2′ * | 2.46 | 0.013 | 0.046 | 3.070 | 2.415 | 124.9 | ||
| TSO7H45↔47 | O5...O4 * | 3.26 | 0.013 | 0.055 | 2.713 | - | - | −42.7 |
| O3H…C6′ * | 2.45 | 0.013 | 0.046 | 3.075 | 2.422 | 124.6 | ||
| TSO7H46↔48 | O5...O4 * | 3.25 | 0.013 | 0.055 | 2.714 | - | - | 137.3 |
| O3H…C2′ * | 2.36 | 0.012 | 0.045 | 3.065 | 2.456 | 120.8 | ||
| TSO5H1↔25 | O5...O4 * | 2.98 | 0.012 | 0.045 | 2.821 | - | - | −178.7 |
| O3H...O4 | 4.60 | 0.032 | 0.117 | 2.571 | 1.923 | 121.1 | ||
| C2′H...O3H * | 4.00 | 0.018 | 0.075 | 2.884 | 2.139 | 123.8 | ||
| TSO5H2↔31 | O5...O4 * | 2.98 | 0.012 | 0.045 | 2.820 | - | - | 1.9 |
| O3H...O4 | 4.55 | 0.032 | 0.116 | 2.572 | 1.926 | 120.9 | ||
| C2′H...O3H * | 3.78 | 0.017 | 0.072 | 2.899 | 2.165 | 123.2 | ||
| TSO5H3↔28 | O5...O4 * | 2.99 | 0.012 | 0.045 | 2.818 | - | - | 1.3 |
| O3H...O4 | 4.45 | 0.032 | 0.116 | 2.575 | 1.933 | 120.7 | ||
| C2′H...O3H * | 3.73 | 0.017 | 0.071 | 2.905 | 2.171 | 123.2 | ||
| TSO5H4↔21 | O5...O4 * | 2.97 | 0.012 | 0.044 | 2.823 | - | - | 1.9 |
| O3H...O4 | 4.57 | 0.032 | 0.116 | 2.572 | 1.925 | 120.9 | ||
| C2′H...O3H * | 3.76 | 0.017 | 0.071 | 2.900 | 2.167 | 123.1 | ||
| TSO5H5↔19 | O5...O4 * | 2.97 | 0.012 | 0.044 | 2.823 | - | - | −178.7 |
| O3H...O4 | 4.63 | 0.032 | 0.117 | 2.570 | 1.921 | 121.1 | ||
| C2′H...O3H * | 3.99 | 0.018 | 0.075 | 2.885 | 2.139 | 123.8 | ||
| TSO5H6↔22 | O5...O4 * | 2.99 | 0.012 | 0.045 | 2.820 | - | - | 1.4 |
| O3H...O4 | 4.49 | 0.032 | 0.116 | 2.574 | 1.930 | 120.7 | ||
| C6′H...O3H * | 3.74 | 0.017 | 0.071 | 2.904 | 2.169 | 123.2 | ||
| TSO5H7↔32 | O5...O4 * | 2.99 | 0.012 | 0.045 | 2.818 | - | - | −178.4 |
| O3H...O4 | 4.43 | 0.031 | 0.115 | 2.576 | 1.934 | 120.6 | ||
| C2′H...O3H * | 3.80 | 0.017 | 0.072 | 2.889 | 2.163 | 122.4 | ||
| TSO5H8↔27 | O5...O4 * | 2.99 | 0.012 | 0.045 | 2.821 | - | - | −178.3 |
| O3H...O4 | 4.46 | 0.032 | 0.116 | 2.574 | 1.932 | 120.6 | ||
| C2′H...O3H * | 3.80 | 0.017 | 0.072 | 2.889 | 2.163 | 122.4 | ||
| TSO5H9↔36 | O5...O4 * | 2.99 | 0.012 | 0.045 | 2.819 | - | - | 1.6 |
| O3H...O4 | 4.48 | 0.032 | 0.116 | 2.575 | 1.931 | 120.7 | ||
| C6′H...O3H * | 3.73 | 0.017 | 0.071 | 2.901 | 2.171 | 122.8 | ||
| Transition State | AH···B H-Bond/AH···HB Dihydrogen H-Bond/A···B vdW Contact | EAH···B/EAH···HB/EA··B a | ρ b | Δρ c | dA···B d | dH···B/dH···H e | AH···Bf | C3C2C1′C6′ g |
| TSO5H10↔35 | O5...O4 * | 2.98 | 0.012 | 0.045 | 2.820 | - | - | −178.8 |
| O3H...O4 | 4.57 | 0.032 | 0.116 | 2.572 | 1.925 | 121.0 | ||
| C2′H...O3H * | 3.97 | 0.018 | 0.075 | 2.890 | 2.142 | 124.1 | ||
| TSO5H11↔34 | O5...O4 * | 2.98 | 0.012 | 0.045 | 2.821 | - | - | 1.7 |
| O3H...O4 | 4.51 | 0.032 | 0.116 | 2.573 | 1.929 | 120.8 | ||
| C6′H...O3H * | 3.73 | 0.017 | 0.071 | 2.901 | 2.171 | 122.9 | ||
| TSO5H12↔33 | O5...O4 * | 2.98 | 0.012 | 0.045 | 2.822 | - | - | −178.8 |
| O3H...O4 | 4.60 | 0.032 | 0.117 | 2.571 | 1.923 | 121.0 | ||
| C2′H...O3H * | 3.97 | 0.018 | 0.075 | 2.890 | 2.142 | 124.1 | ||
| TSO5H13↔43 | O5...O4 * | 3.27 | 0.014 | 0.050 | 2.774 | - | - | −42.4 |
| O3H…C6′ * | 2.45 | 0.013 | 0.046 | 3.075 | 2.422 | 124.6 | ||
| TSO5H14↔44 | O5...O4 * | 3.26 | 0.014 | 0.049 | 2.776 | - | - | −43.1 |
| O3H…C6′ * | 2.44 | 0.013 | 0.046 | 3.071 | 2.426 | 123.9 | ||
| TSO5H15↔42 | O5...O4 * | 3.26 | 0.014 | 0.049 | 2.774 | - | - | 137.6 |
| O3H…C2′ * | 2.48 | 0.013 | 0.046 | 3.067 | 2.412 | 124.7 | ||
| TSO5H16↔38 | O5...O4 * | 3.24 | 0.014 | 0.049 | 2.780 | - | - | 137.1 |
| O3H…C2′ * | 2.33 | 0.012 | 0.045 | 3.064 | 2.470 | 119.6 | ||
| TSO5H17↔39 | O5...O4 * | 3.24 | 0.014 | 0.049 | 2.779 | - | - | −43.2 |
| O3H…C6′ * | 2.42 | 0.013 | 0.045 | 3.074 | 2.430 | 123.8 | ||
| TSO5H18↔40 | O5...O4 * | 3.25 | 0.014 | 0.049 | 2.777 | - | - | 136.6 |
| O3H…C2′ * | 2.43 | 0.013 | 0.045 | 3.073 | 2.420 | 124.7 | ||
| TSO5H20↔41 | O5...O4 * | 3.25 | 0.014 | 0.049 | 2.777 | - | - | 137.7 |
| O3H…C2′ * | 2.36 | 0.012 | 0.045 | 3.058 | 2.464 | 119.6 | ||
| TSO5H23↔37 | O5...O4 * | 3.24 | 0.014 | 0.049 | 2.777 | - | - | −43.7 |
| O3H…C6′ * | 2.42 | 0.013 | 0.045 | 3.084 | 2.431 | 124.7 | ||
| TSO5H24↔47 | O5...O4 * | 3.25 | 0.014 | 0.049 | 2.775 | - | - | −42.5 |
| O3H…C6′ * | 2.48 | 0.013 | 0.046 | 3.072 | 2.421 | 124.5 | ||
| TSO5H26↔48 | O5...O4 * | 3.25 | 0.014 | 0.049 | 2.776 | - | - | 137.6 |
| O3H…C2′ * | 2.38 | 0.012 | 0.045 | 3.062 | 2.454 | 120.7 | ||
| TSO5H29↔45 | O5...O4 * | 3.25 | 0.014 | 0.049 | 2.777 | - | - | −43.6 |
| O3H…C6′ * | 2.40 | 0.013 | 0.045 | 3.080 | 2.429 | 124.4 | ||
| TSO5H30↔46 | O5...O4 * | 3.24 | 0.014 | 0.049 | 2.779 | - | - | 136.5 |
| O3H…C2′ * | 2.34 | 0.012 | 0.045 | 3.069 | 2.461 | 120.8 | ||
| TSO3H1↔20 | O5H...O4 | 8.54 | 0.049 | 0.136 | 2.600 | 1.699 | 148.7 | 149.8 |
| C2′H...O3H * | 2.92 | 0.014 | 0.050 | 2.941 | 2.369 | 111.3 | ||
| TSO3H2↔14 | O5H...O4 | 8.54 | 0.049 | 0.136 | 2.600 | 1.699 | 148.7 | −31.9 |
| C2′H...O3H * | 2.89 | 0.014 | 0.050 | 2.950 | 2.376 | 111.6 | ||
| TSO3H3↔13 | O5H...O4 | 8.58 | 0.049 | 0.136 | 2.598 | 1.697 | 148.7 | −29.4 |
| C2′H...O3H * | 2.99 | 0.014 | 0.051 | 2.948 | 2.353 | 113.0 | ||
| TSO3H4↔17 | O5H...O4 | 8.31 | 0.048 | 0.135 | 2.605 | 2.382 | 111.4 | −32.2 |
| C2′H...O3H * | 2.86 | 0.014 | 0.049 | 2.953 | 1.925 | 120.9 | ||
| TSO3H5↔16 | O5H...O4 | 8.31 | 0.048 | 0.135 | 2.605 | 1.707 | 148.6 | 149.3 |
| C2′H...O3H * | 2.88 | 0.014 | 0.050 | 2.943 | 2.376 | 111.0 | ||
| TSO3H6↔23 | O5H...O4 | 8.37 | 0.048 | 0.135 | 2.603 | 1.705 | 148.6 | −30.8 |
| C6′H...O3H * | 2.92 | 0.014 | 0.050 | 2.952 | 2.368 | 112.2 | ||
| TSO3H7↔15 | O5H...O4 | 8.37 | 0.048 | 0.135 | 148.747 | 1.698 | 148.7 | 150.8 |
| C2′H...O3H * | 2.92 | 0.014 | 0.050 | 2.937 | 2.365 | 111.4 | ||
| TSO3H8↔18 | O5H...O4 | 8.35 | 0.048 | 0.135 | 2.604 | 1.705 | 148.6 | 150.0 |
| C2′H...O3H * | 2.90 | 0.014 | 0.050 | 2.940 | 2.374 | 111.0 | ||
| TSO3H9↔24 | O5H...O4 | 8.57 | 0.049 | 0.136 | 2.599 | 1.698 | 148.7 | −29.9 |
| C6′H...O3H * | 2.95 | 0.014 | 0.051 | 2.947 | 2.361 | 112.4 | ||
| TSO3H10↔26 | O5H...O4 | 8.54 | 0.049 | 0.136 | 2.599 | 1.699 | 148.7 | 150.3 |
| C2′H...O3H * | 2.94 | 0.014 | 0.051 | 2.943 | 2.363 | 111.8 | ||
| TSO3H11↔29 | O5H...O4 | 8.35 | 0.048 | 0.135 | 2.604 | 1.705 | 148.6 | −31.2 |
| C6′H...O3H * | 2.89 | 0.014 | 0.049 | 2.951 | 2.376 | 111.7 | ||
| TSO3H12↔30 | O5H...O4 | 8.32 | 0.048 | 0.135 | 2.605 | 1.706 | 148.6 | 149.5 |
| C2′H...O3H * | 2.91 | 0.014 | 0.050 | 2.945 | 2.371 | 111.5 | ||
| TSO3H19↔38 | O5...O4 * | 3.19 | 0.013 | 0.054 | 2.723 | - | - | 149.5 |
| C2′H...O3H * | 2.87 | 0.014 | 0.049 | 2.946 | 2.377 | 111.1 | ||
| TSO3H21↔39 | O5...O4 * | 3.18 | 0.013 | 0.054 | 2.724 | - | - | −32.1 |
| C6′H...O3H * | 2.83 | 0.014 | 0.048 | 2.957 | 2.388 | 111.3 | ||
| TSO3H22↔37 | O5...O4 * | 3.19 | 0.013 | 0.054 | 2.723 | - | - | −31.3 |
| C6′H...O3H * | 2.85 | 0.014 | 0.049 | 2.959 | 2.382 | 111.8 | ||
| TSO3H25↔41 | O5...O4 * | 3.22 | 0.013 | 0.054 | 2.719 | - | - | 150.0 |
| C2′H...O3H * | 2.91 | 0.014 | 0.050 | 2.943 | 2.370 | 111.4 | ||
| TSO3H27↔40 | O5...O4 * | 3.19 | 0.013 | 0.054 | 2.723 | - | - | 150.0 |
| C2′H...O3H * | 2.87 | 0.014 | 0.049 | 2.944 | 2.380 | 110.9 | ||
| TSO3H28↔43 | O5...O4 * | 3.21 | 0.013 | 0.054 | 2.719 | - | - | −29.7 |
| C6′H...O3H * | 2.93 | 0.014 | 0.050 | 2.954 | 2.364 | 112.7 | ||
| TSO3H31↔44 | O5...O4 * | 3.18 | 0.013 | 0.054 | 2.724 | - | - | −32.1 |
| C6′H...O3H * | 2.83 | 0.014 | 0.048 | 2.957 | 2.388 | 111.3 | ||
| TSO3H32↔42 | O5...O4 * | 3.21 | 0.013 | 0.054 | 2.720 | - | - | 150.9 |
| C2′H...O3H * | 2.91 | 0.014 | 0.050 | 2.942 | 2.371 | 111.3 | ||
| TSO3H33↔46 | O5...O4 * | 3.19 | 0.013 | 0.054 | 2.723 | - | - | 149.7 |
| C2′H...O3H * | 2.89 | 0.014 | 0.050 | 2.948 | 2.373 | 111.5 | ||
| TSO3H34↔45 | O5...O4 * | 3.19 | 0.013 | 0.054 | 2.723 | - | - | −31.5 |
| C6′H...O3H * | 2.83 | 0.014 | 0.048 | 2.957 | 2.387 | 111.3 | ||
| TSO3H35↔48 | O5...O4 * | 3.22 | 0.013 | 0.054 | 2.719 | - | - | 150.4 |
| C2′H...O3H * | 2.93 | 0.014 | 0.050 | 2.946 | 2.365 | 111.9 | ||
| TSO3H36↔47 | O5...O4 * | 3.21 | 0.013 | 0.054 | 2.720 | - | - | −30.1 |
| C6′H...O3H * | 2.90 | 0.014 | 0.050 | 2.952 | 2.371 | 112.1 | ||
| TS13R↔13L | O5H...O4 | 8.37 | 0.048 | 0.135 | 2.603 | 1.705 | 148.4 | −7.6 |
| O3H...HC6′ * | 4.81 | 0.023 | 0.075 | 2.496 | 1.592 | 155.4 | ||
| C2′H...O1 * | 4.74 | 0.019 | 0.090 | 2.619 | 2.218 | 99.3 | ||
| TS14R↔14L | O5H...O4 | 8.33 | 0.048 | 0.135 | 2.605 | 1.707 | 148.4 | −7.7 |
| O3H...HC6′ * | 4.77 | 0.023 | 0.075 | 2.490 | 1.590 | 154.6 | ||
| C2′H...O1 * | 4.72 | 0.019 | 0.090 | 2.617 | 2.234 | 98.3 | ||
| TS15R↔15L | O5H...O4 | 8.37 | 0.048 | 0.135 | 2.603 | 1.705 | 148.4 | 171.9 |
| O3H...HC2′ * | 4.67 | 0.022 | 0.074 | 2.500 | 1.603 | 153.9 | ||
| C6′H...O1 * | 4.69 | 0.019 | 0.089 | 2.627 | 2.233 | 98.9 | ||
| TS16R↔16L | O5H...O4 | 8.09 | 0.047 | 0.134 | 2.610 | 1.715 | 148.3 | 171.2 |
| O3H...HC2′ * | 4.67 | 0.023 | 0.074 | 2.502 | 1.601 | 155.4 | ||
| C6′H...O1 * | 4.66 | 0.019 | 0.089 | 2.631 | 2.234 | 99.2 | ||
| TS17R↔17L | O5H...O4 | 8.10 | 0.047 | 0.134 | 2.610 | 1.714 | 148.3 | −9.1 |
| O3H...HC6′ * | 4.73 | 0.023 | 0.074 | 2.493 | 1.600 | 153.2 | ||
| C2′H...O1 * | 4.71 | 0.019 | 0.089 | 2.621 | 2.241 | 98.1 | ||
| TS18R↔18L | O5H...O4 | 8.16 | 0.047 | 0.134 | 2.608 | 1.712 | 148.3 | 170.8 |
| O3H...HC2′ * | 4.58 | 0.022 | 0.073 | 2.496 | 1.612 | 151.5 | ||
| C6′H...O1 * | 4.64 | 0.019 | 0.088 | 2.631 | 2.239 | 98.9 | ||
| TS20R↔20L | O5H...O4 | 8.30 | 0.048 | 0.135 | 2.605 | 1.707 | 148.4 | 171.3 |
| O3H...HC2′ * | 4.68 | 0.023 | 0.074 | 2.502 | 1.600 | 155.5 | ||
| C6′H...O1 * | 4.69 | 0.019 | 0.089 | 2.629 | 2.231 | 99.2 | ||
| TS23R↔23L | O5H...O4 | 8.18 | 0.047 | 0.134 | 2.608 | 1.712 | 148.3 | −8.1 |
| O3H...HC6′ * | 4.78 | 0.023 | 0.075 | 2.493 | 1.595 | 154.2 | ||
| C2′H...O1 * | 4.69 | 0.019 | 0.089 | 2.624 | 2.223 | 99.2 | ||
| TS24R↔24L | O5H...O4 | 8.36 | 0.048 | 0.135 | 2.603 | 1.705 | 148.4 | 8.2 |
| O3H...HC6′ * | 4.77 | 0.023 | 0.075 | 2.497 | 1.597 | 154.8 | ||
| C2′H...O1 * | 4.71 | 0.019 | 0.089 | 2.624 | 2.219 | 99.5 | ||
| TS26R↔26L | O5H...O4 | 8.32 | 0.048 | 0.135 | 2.605 | 1.707 | 148.4 | 171.4 |
| O3H...HC2′ * | 4.70 | 0.023 | 0.074 | 2.499 | 1.597 | 155.3 | ||
| C6′H...O1 * | 4.70 | 0.019 | 0.089 | 2.626 | 2.235 | 98.8 | ||
| TS29R↔29L | O5H...O4 | 8.16 | 0.047 | 0.134 | 2.608 | 1.712 | 148.3 | −8.2 |
| O3H...HC6′ * | 4.76 | 0.023 | 0.075 | 2.497 | 1.597 | 154.6 | ||
| C2′H...O1 * | 4.67 | 0.019 | 0.089 | 2.627 | 2.223 | 99.5 | ||
| TS30R↔30L | O5H...O4 | 8.11 | 0.047 | 0.134 | 2.610 | 1.714 | 148.3 | 171.3 |
| O3H...HC2′ * | 4.69 | 0.023 | 0.074 | 2.499 | 1.598 | 155.1 | ||
| C6′H...O1 * | 4.66 | 0.019 | 0.089 | 2.629 | 2.238 | 98.8 | ||
| TS37R↔37L | O5...O4 * | 3.20 | 0.013 | 0.054 | 2.722 | - | - | −7.0 |
| O3H...HC6′ * | 4.82 | 0.023 | 0.075 | 2.501 | 1.591 | 156.8 | ||
| C2′H...O1 * | 4.68 | 0.019 | 0.089 | 2.624 | 2.221 | 99.4 | ||
| TS38R↔38L | O5...O4 * | 13.75 | 0.047 | 0.134 | 2.725 | - | - | 171.2 |
| O3H...HC2′ * | 4.67 | 0.023 | 0.074 | 2.507 | 1.602 | 156.0 | ||
| C6′H...O1 * | 4.66 | 0.019 | 0.089 | 2.633 | 2.235 | 99.3 | ||
| TS39R↔39L | O5...O4 * | 3.17 | 0.013 | 0.054 | 2.726 | - | - | −7.6 |
| O3H...HC6′ * | 4.80 | 0.023 | 0.075 | 2.502 | 1.593 | 156.6 | ||
| C2′H...O1 * | 4.70 | 0.019 | 0.089 | 2.621 | 2.236 | 98.4 | ||
| TS40R↔40L | O5...O4 * | 3.19 | 0.013 | 0.054 | 2.723 | - | - | −172.5 |
| O3H...HC2′ * | 4.69 | 0.023 | 0.074 | 2.505 | 1.601 | 155.6 | ||
| C6′H...O1 * | 4.63 | 0.019 | 0.088 | 2.631 | 2.235 | 99.1 | ||
| TS41R↔41L | O5...O4 * | 3.20 | 0.013 | 0.054 | 2.722 | - | - | 171.6 |
| O3H...HC2′ * | 4.68 | 0.023 | 0.074 | 2.507 | 1.599 | 156.6 | ||
| C6′H...O1 * | 4.66 | 0.019 | 0.088 | 2.631 | 2.232 | 99.3 | ||
| TS42R↔42L | O5...O4 * | 3.21 | 0.013 | 0.054 | 2.720 | - | - | −172.9 |
| O3H...HC2′ * | 4.71 | 0.023 | 0.074 | 2.506 | 1.600 | 156.3 | ||
| C6′H...O1 * | 4.67 | 0.019 | 0.089 | 2.628 | 2.232 | 99.1 | ||
| TS43R↔43L | O5...O4 * | 3.21 | 0.013 | 0.054 | 2.720 | - | - | 6.3 |
| O3H...HC6′ * | 4.84 | 0.023 | 0.075 | 2.503 | 1.589 | 157.8 | ||
| C2′H...O1 * | 4.73 | 0.019 | 0.090 | 2.620 | 2.216 | 99.4 | ||
| TS44R↔44L | O5...O4 * | 3.20 | 0.013 | 0.054 | 2.722 | - | - | 7.6 |
| O3H...HC6′ * | 4.84 | 0.023 | 0.075 | 2.500 | 1.591 | 156.8 | ||
| C2′H...O1 * | 4.71 | 0.019 | 0.090 | 2.620 | 2.236 | 98.3 | ||
| TS45R↔45L | O5...O4 * | 3.19 | 0.013 | 0.054 | 2.723 | - | - | 7.5 |
| O3H...HC6′ * | 4.79 | 0.023 | 0.075 | 2.504 | 1.595 | 156.7 | ||
| C2′H...O1 * | 4.66 | 0.019 | 0.088 | 2.628 | 2.221 | 99.6 | ||
| TS46R↔46L | O5...O4 * | 3.18 | 0.013 | 0.054 | 2.724 | - | - | 171.6 |
| O3H...HC2′ * | 4.69 | 0.023 | 0.074 | 2.503 | 1.598 | 156.0 | ||
| C6′H...O1 * | 4.63 | 0.019 | 0.088 | 2.631 | 2.238 | 98.9 | ||
| TS47R↔47L | O5...O4 * | 3.21 | 0.013 | 0.054 | 2.720 | - | - | 6.9 |
| O3H...HC6′ * | 4.81 | 0.023 | 0.075 | 2.505 | 1.592 | 157.6 | ||
| C2′H...O1 * | 4.70 | 0.019 | 0.089 | 2.625 | 2.216 | 99.7 | ||
| TS48R↔48L | O5...O4 * | 3.20 | 0.013 | 0.054 | 2.721 | - | - | 171.7 |
| O3H...HC2′ * | 4.70 | 0.023 | 0.074 | 2.503 | 1.598 | 156.2 | ||
| C6′H...O1 * | 4.66 | 0.019 | 0.089 | 2.628 | 2.235 | 98.9 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brovarets’, O.O.; Hovorun, D.M. A Hidden Side of the Conformational Mobility of the Quercetin Molecule Caused by the Rotations of the O3H, O5H and O7H Hydroxyl Groups: In Silico Scrupulous Study. Symmetry 2020, 12, 230. https://doi.org/10.3390/sym12020230
Brovarets’ OO, Hovorun DM. A Hidden Side of the Conformational Mobility of the Quercetin Molecule Caused by the Rotations of the O3H, O5H and O7H Hydroxyl Groups: In Silico Scrupulous Study. Symmetry. 2020; 12(2):230. https://doi.org/10.3390/sym12020230
Chicago/Turabian StyleBrovarets’, Ol’ha O., and Dmytro M. Hovorun. 2020. "A Hidden Side of the Conformational Mobility of the Quercetin Molecule Caused by the Rotations of the O3H, O5H and O7H Hydroxyl Groups: In Silico Scrupulous Study" Symmetry 12, no. 2: 230. https://doi.org/10.3390/sym12020230
APA StyleBrovarets’, O. O., & Hovorun, D. M. (2020). A Hidden Side of the Conformational Mobility of the Quercetin Molecule Caused by the Rotations of the O3H, O5H and O7H Hydroxyl Groups: In Silico Scrupulous Study. Symmetry, 12(2), 230. https://doi.org/10.3390/sym12020230