Enantiodivergent Aldol Condensation in the Presence of Aziridine/Acid/Water Systems


Abstract
:
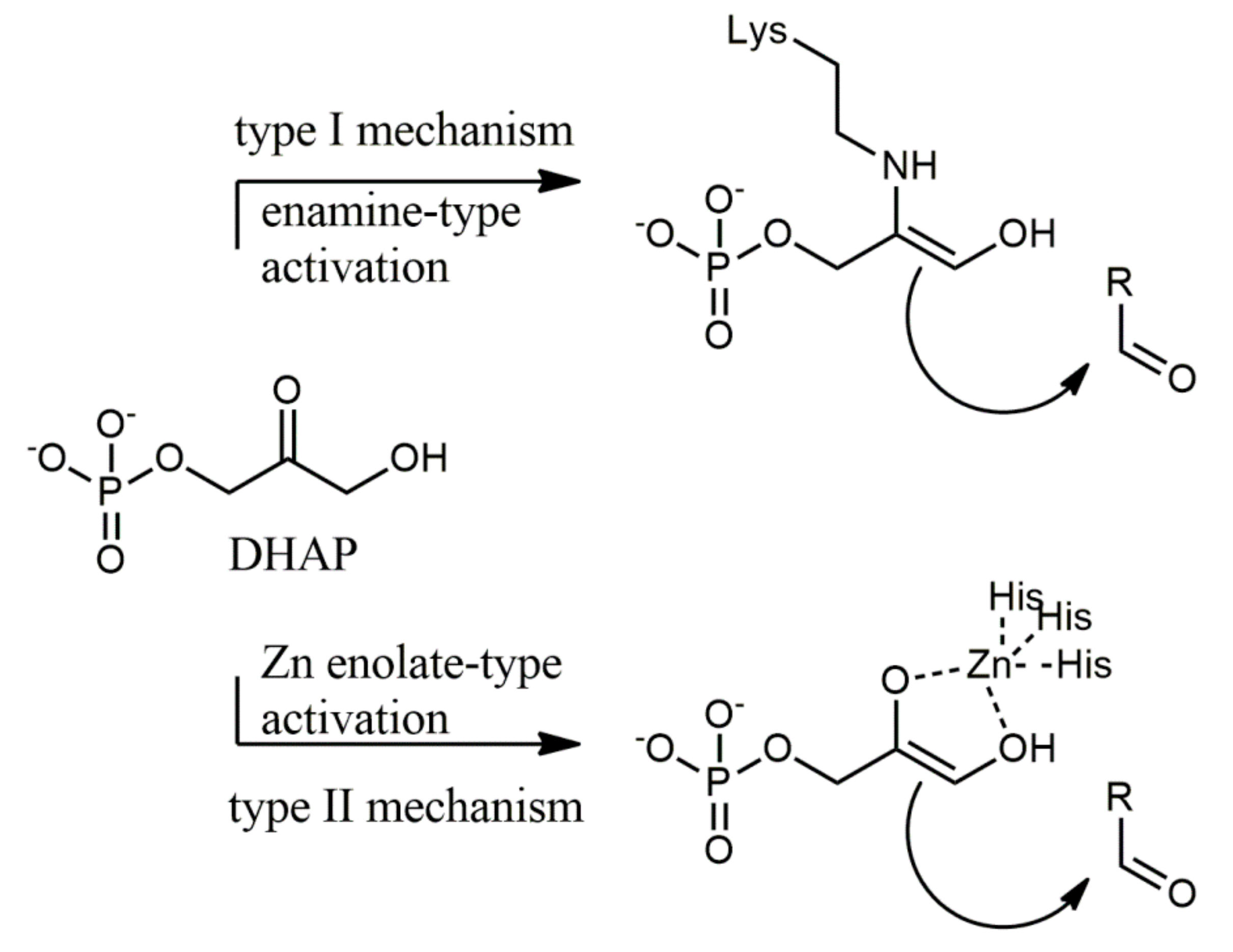
1. Introduction
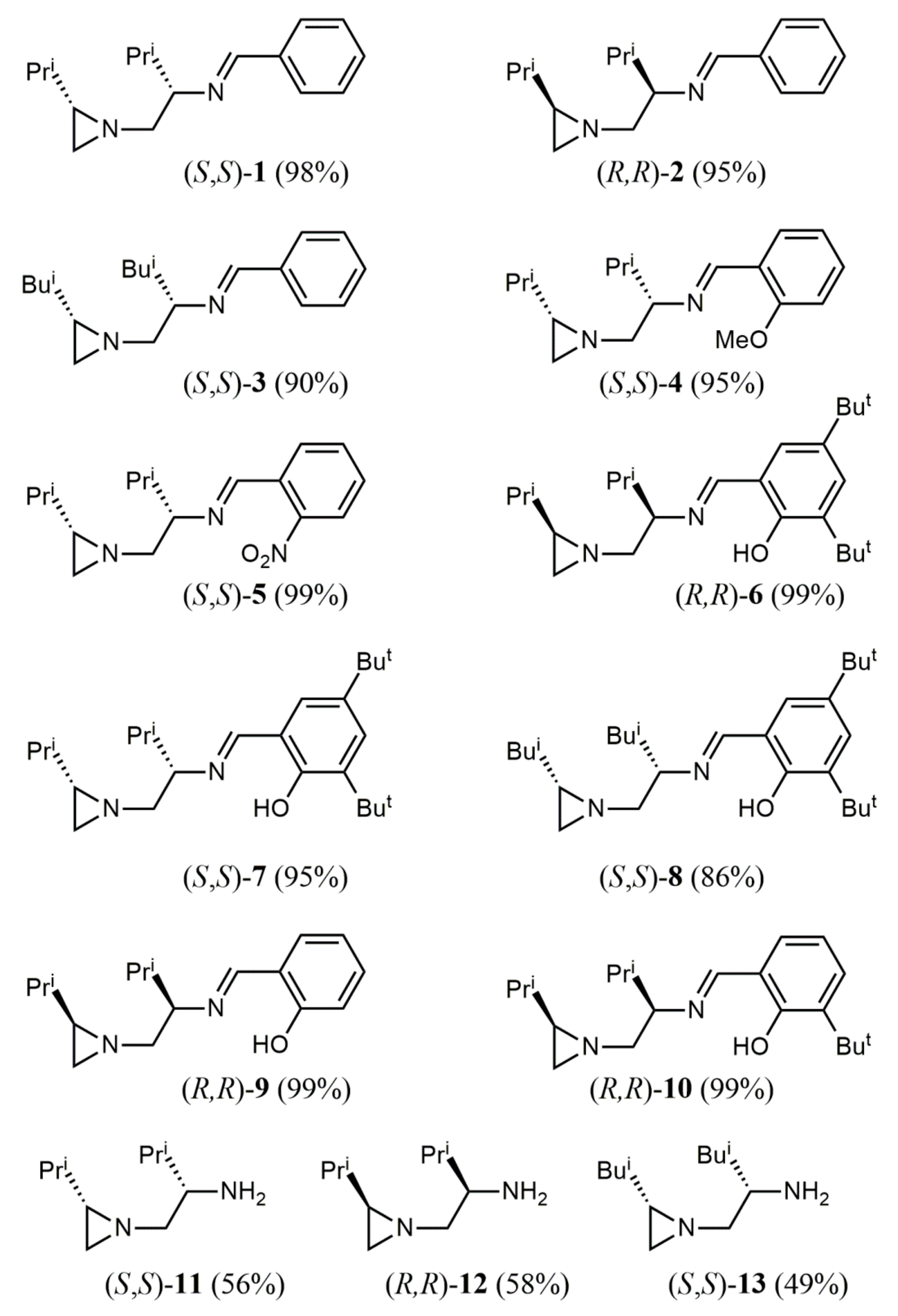
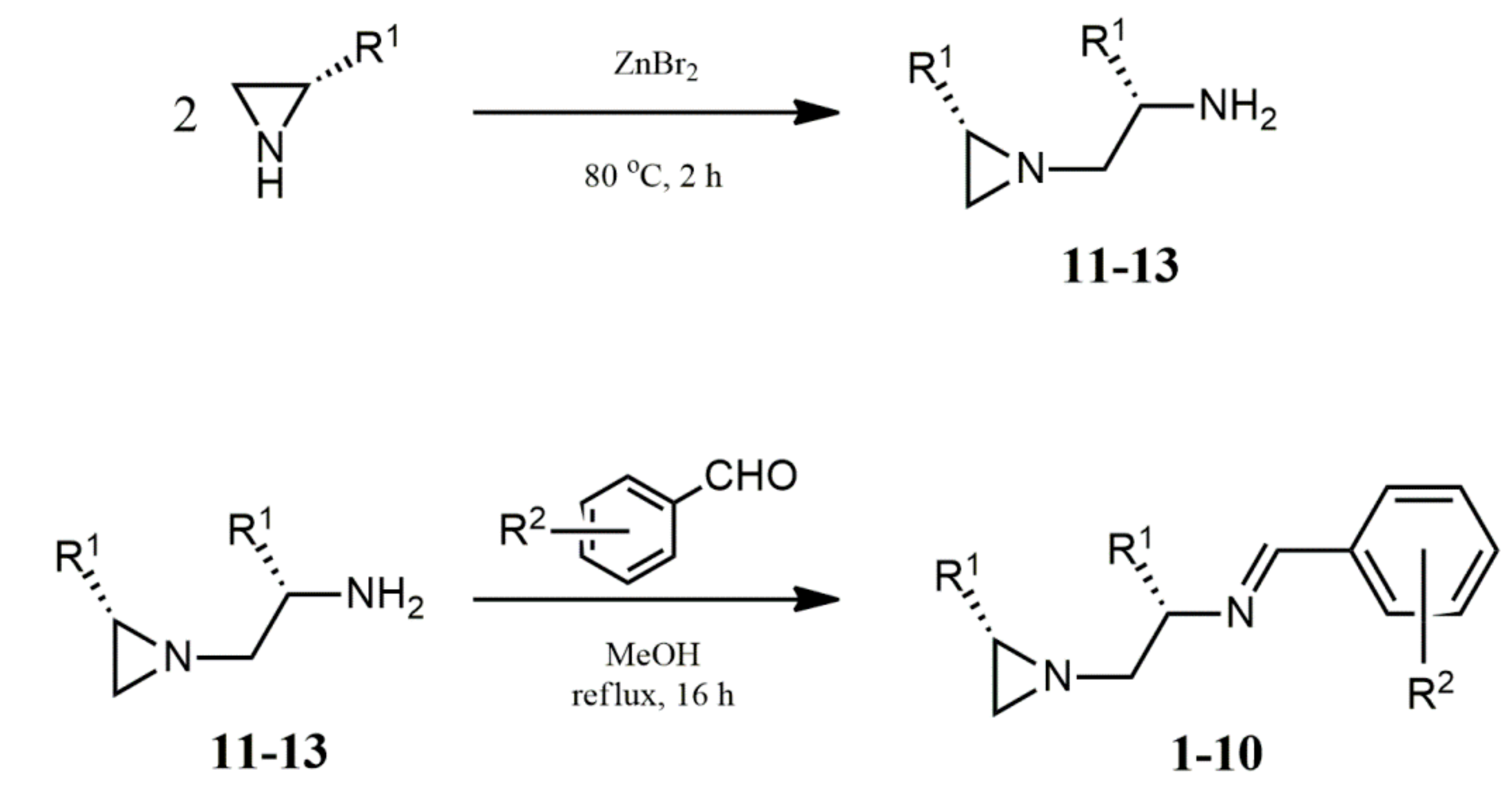
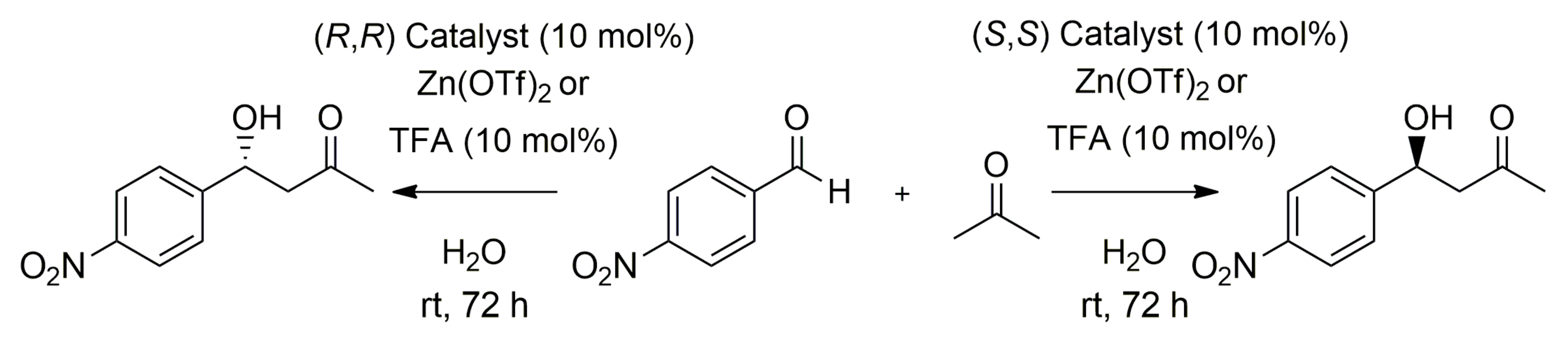
2. Results
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Appendix A
A.1. Materials and Methods
A.2. Synthesis of Chiral Imines 1–10—General Procedure [23]
A.3. Asymmetric Aldol Reaction—General Procedure
References
- Pellissier, H. Asymmetric organocatalysis. Tetrahedron 2007, 63, 9267–9331. [Google Scholar] [CrossRef]
- Trost, B.M.; Brindle, C.S. The direct catalytic asymmetric aldol reaction. Chem. Soc. Rev. 2010, 39, 1600–1632. [Google Scholar] [CrossRef] [Green Version]
- List, B.; Lerner, R.A.; Barbas, C.F. Proline-Catalyzed Direct Asymmetric Aldol Reactions. J. Am. Chem. Soc. 2000, 122, 2395–2396. [Google Scholar] [CrossRef]
- Guillena, G.; Nájera, C.; Ramón, D.J. Enantioselective direct aldol reaction: The blossoming of modern organocatalysis. Tetrahedron Asymmetry 2007, 18, 2249–2293. [Google Scholar] [CrossRef]
- Rosenberger, M.; McDougal, P.; Bahr, J. Reductive alkylation/arylation of arylcarbinols and ketones with organosilicon compounds. J. Org. Chem. 1982, 47, 2130–2134. [Google Scholar] [CrossRef]
- Akhtar, M.; Weedon, B.C.L. Carotenoids and related compounds. Part VIII. Novel syntheses of echinenone and canthaxanthin. J. Chem. Soc. 1959, 4058–4062. [Google Scholar] [CrossRef]
- Warren, C.K.; Weedon, B.C.L. Carotenoids and related compounds. Part VII. Synthesis of canthaxanthin and echinenone. J. Chem. Soc. 1958, 3986–3993. [Google Scholar] [CrossRef]
- Matsumoto, H.; Ikoma, Y.; Kato, M.; Kuniga, T.; Nakajima, N.; Yoshida, T. Quantification of Carotenoids in Citrus Fruit by LC-MS and Comparison of Patterns of Seasonal Changes for Carotenoids among Citrus Varieties. J. Agric. Food Chem. 2007, 55, 2356–2368. [Google Scholar] [CrossRef]
- Młynarski, J.; Baś, S. Catalytic asymmetric aldol reactions in aqueous media – a 5 year update. Chem. Soc. Rev. 2014, 43, 577–587. [Google Scholar] [CrossRef]
- Beletskaya, I.P.; Nájera, C.; Yus, M. Stereodivergent Catalysis. Chem. Rev. 2018, 118, 5080–5200. [Google Scholar] [CrossRef]
- Krautwald, S.; Carreira, E.M. Stereodivergence in Asymmetric Catalysis. J. Am. Chem. Soc. 2017, 139, 5627–5639. [Google Scholar] [CrossRef] [PubMed]
- Bihani, M.; Zhao, J.C.G. Advances in Asymmetric Diastereodivergent Catalysis. Adv. Synth. Catal. 2017, 359, 534–575. [Google Scholar] [CrossRef]
- Martínez-Castañeda, A.; Rodríguez-Solla, H.; Concellón, C.; Amo, V. Switching Diastereoselectivity in Proline-Catalyzed Aldol Reactions. J. Org. Chem. 2012, 77, 10375–10381. [Google Scholar] [CrossRef]
- Pinaka, A.; Vougioukalakis, G.C.; Dimotikali, D.; Yannakopoulou, E.; Chankvetadze, B.; Papadopoulos, K. Green Asymmetric Synthesis: β -Amino Alcohol-Catalyzed Direct Asymmetric Aldol Reactions in Aqueous Micelles. Chirality 2013, 25, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Paradowska, J.; Pasternak, M.; Gut, M.; Gryzło, B.; Młynarski, J. Direct Asymmetric Aldol Reactions Inspired by Two Types of Natural Aldolases: Water-Compatible Organocatalysts and ZnII Complexes. J. Org. Chem. 2012, 77, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Pieczonka, A.M.; Jarzyński, S.; Wujkowska, Z.; Leśniak, S.; Rachwalski, M. Zinc(II) mediated asymmetric aldol condensation catalyzed by chiral aziridine ligands. Tetrahedron Lett. 2015, 56, 6506–6507. [Google Scholar] [CrossRef]
- Pieczonka, A.M.; Leśniak, S.; Rachwalski, M. Direct asymmetric aldol condensation catalyzed by aziridine semicarbazide zinc(II) complexes. Tetrahedron Lett. 2014, 55, 2373–2375. [Google Scholar] [CrossRef]
- Wujkowska, Z.; Strojewska, A.; Pieczonka, A.M.; Leśniak, S.; Rachwalski, M. Highly Enantioselective Asymmetric Direct Aldol Reaction Promoted by Aziridine Amides Constructed on Chiral Terpene Scaffold. Chirality 2017, 29, 213–220. [Google Scholar] [CrossRef]
- Rachwalski, M.; Kaczmarczyk, S.; Leśniak, S.; Kiełbasiński, P. Highly Efficient Asymmetric Simmons–Smith Cyclopropanation Promoted by Chiral Heteroorganic Aziridinyl Ligands. ChemCatChem 2014, 6, 873–875. [Google Scholar] [CrossRef]
- Rachwalski, M. Limonene oxide derived aziridinyl alcohols as highly efficient catalysts for asymmetric additions of organozinc species to aldehydes. Tetrahedron Asymmetry 2014, 25, 219–223. [Google Scholar] [CrossRef]
- Jarzyński, S.; Leśniak, S.; Pieczonka, A.M.; Rachwalski, M. N-Trityl-aziridinyl alcohols as highly efficient chiral catalysts in asymmetric additions of organozinc species to aldehydes. Tetrahedron Asymmetry 2015, 26, 35–40. [Google Scholar] [CrossRef]
- Kowalczyk, A.; Pieczonka, A.M.; Rachwalski, M.; Lesniak, S.; Staczek, P. Synthesis and Evaluation of Biological Activities of Aziridine Derivatives of Urea and Thiourea. Molecules 2018, 23, 45. [Google Scholar] [CrossRef] [Green Version]
- Pieczonka, A.M.; Misztal, E.; Rachwalski, M.; Leśniak, S. Synthesis of chiral 1-(2-aminoalkyl)aziridines via the self-opening reaction of aziridine. Arkivoc 2017, 2, 223–234. [Google Scholar]
- Pieczonka, A.M.; Leśniak, S.; Rachwalski, M. Chiral imines prepared from 1-(2-aminoalkyl) aziridines as novel chiral shifts reagents for efficient recognition of acids. Tetrahedron 2018, 74, 1571–1579. [Google Scholar] [CrossRef]
- Chen, X.; Lin, C.; Du, H.; Xu, J. Efficient Direct Synthesis of Aziridine-Containing Chiral Tridentate Ligands by the Iminium-Mediated Self-Ring Opening Reaction of Enantiopure Aziridines and Salicylaldehydes. Adv. Synth. Catal. 2019, 361, 1647–1661. [Google Scholar] [CrossRef]
- Shah, E.; Soni, H.P. Inducing chirality on ZnS nanoparticles for asymmetric aldol condensation reactions. RSC Adv. 2013, 3, 17453–17461. [Google Scholar] [CrossRef]
- Bayat, S.; Tejo, B.A.; Salleh, A.B.; Abdmalek, E.; Normi, Y.M.; Rahman, B.A. Various Polar Tripeptides as Asymmetric Organocatalyst in Direct Aldol Reactions in Aqueous Media. Chirality 2013, 25, 726–734. [Google Scholar] [CrossRef]
- Simon, A.; Lam, Y.; Houk, K.N. Transition States of Vicinal Diamine-Catalyzed Aldol Reactions. J. Am. Chem. Soc. 2016, 138, 503–506. [Google Scholar] [CrossRef]
- Zhou, Y.; Shan, Z. (R)- or (S)-Bi-2-naphthol assisted, l-proline catalyzed direct aldol reaction. Tetrahedron Asymmetry 2006, 17, 1671–1677. [Google Scholar] [CrossRef]
- Reddy, B.V.S.; Bhavani, K.; Raju, B.V.; Yadav, J.S. A novel trifunctional organocatalyst for the asymmetric aldol reaction: A facile enantioselective synthesis of β-hydroxyketones. Tetrahedron Asymmetry 2011, 22, 881–886. [Google Scholar] [CrossRef]
- Qin, L.; Zhang, L.; Jin, Q.; Zhang, J.; Han, B.; Liu, M. Supramolecular Assemblies of Amphiphilic L -Proline Regulated by Compressed CO2 as a Recyclable Organocatalyst for the Asymmetric Aldol Reaction. Angew. Chem. Int. Ed. 2013, 52, 7761–7765. [Google Scholar] [CrossRef]
- Ying, A.; Liu, S.; Li, Z.; Chen, G.; Yang, J.; Yan, H.; Xu, S. Magnetic Nanoparticles-Supported Chiral Catalyst with an Imidazolium Ionic Moiety: An Efficient and Recyclable Catalyst for Asymmetric Michael and Aldol Reactions. Adv. Synth. Catal. 2016, 358, 2116–2125. [Google Scholar] [CrossRef]
- Li, L.; Gou, S.; Liu, F. Highly stereoselective anti-aldol reactions catalyzed by simple chiral diamines and their unique application in configuration switch of aldol products. Tetrahedron Lett. 2013, 54, 6358–6362. [Google Scholar] [CrossRef]
- Qu, C.; Zhao, W.; Zhang, L.; Cui, Y. Preparation of Immobilized L-Prolinamide Via Enzymatic Polymerization of Phenolic L-Prolinamide and Evaluation of Its Catalytic Performance for Direct Asymmetric Aldol Reaction. Chirality 2014, 26, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Fu, X.; Wu, C.; Li, C. A new chiral primary–tertiary diamine-Brønsted acid salt organocatalyst for the highly enantioselective direct anti-aldol and syn-Mannich reactions. Res. Chem. Intermed. 2013, 39, 1069–1087. [Google Scholar] [CrossRef]
- Martínez, R.; Berbegal, L.; Guillena, G.; Ramón, D.J. Bio-renewable enantioselective aldol reaction in natural deep eutectic solvents. Green Chem. 2016, 18, 1724–1730. [Google Scholar] [CrossRef] [Green Version]

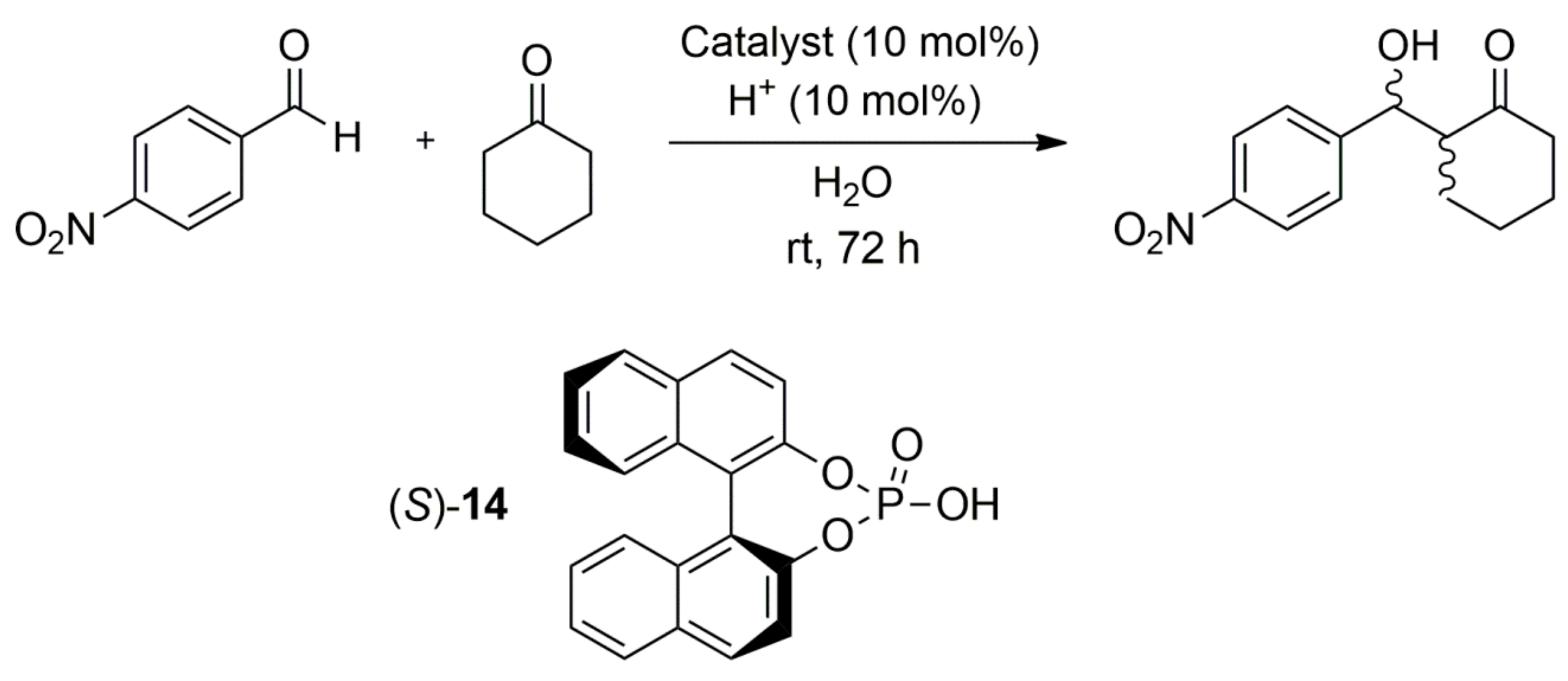

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst | Additive | Yield (%) | eea (%) | Abs. Conf. |
|---|---|---|---|---|---|
| 1 | 1 | Zn(OTf)2 | 96 | 99 | (S) |
| 2 | 1 | TFA | 98 | 99 | (S) |
| 3 | 2 | Zn(OTf)2 | 86 | 78 | (R) |
| 4 | 2 | TFA | 96 | 99 | (R) |
| 5 | 3 | Zn(OTf)2 | 85 | 65 | (S) |
| 6 | 3 | TFA | 91 | 20 | (S) |
| 7 | 4 | Zn(OTf)2 | 87 | 69 | (S) |
| 8 | 4 | TFA | 87 | 42 | (S) |
| 9 | 5 | Zn(OTf)2 | 99 | 54 | (S) |
| 10 | 5 | TFA | 99 | 53 | (S) |
| 11 | 6 | Zn(OTf)2 | 98 | 86 | (R) |
| 12 | 6 | TFA | 84 | 8 | (R) |
| 13 | 7 | Zn(OTf)2 | 96 | 64 | (S) |
| 14 | 7 | TFA | 91 | 30 | (S) |
| 15 | 8 | Zn(OTf)2 | 99 | 54 | (S) |
| 16 | 8 | TFA | 98 | 50 | (S) |
| 17 | 9 | Zn(OTf)2 | 94 | 65 | (R) |
| 18 | 9 | TFA | 98 | 45 | (R) |
| 19 | 10 | Zn(OTf)2 | 91 | 36 | (R) |
| 20 | 10 | TFA | 99 | 50 | (R) |
| 21 | 11 | TFA | 99 | 51 | (S) |
| 22 | 12 | TFA | 98 | 50 | (R) |
| 23 | 13 | TFA | 93 | 23 | (S) |
| 24 b | 1 | Zn(OTf)2 | 96 | 87 | (S) |
| Entry | R | Yield (%) | eea (%) | Abs. Conf. |
|---|---|---|---|---|
| 1 | 2-NO2 | 86 | 82 | (S) |
| 2 | 2,4-diNO2 | >99 | >99 | (S) |
| 3 | 4-CN | 98 | 42 | (S) |
| 4 | 4-CF3 | 62 | 38 | (S) |
| 5 | H | 24 | 11 | (S) |
| Entry | Catalyst | Additive | Yield (%) | dra (Syn/Anti) | eebSyn (%) | eebAnti (%) |
|---|---|---|---|---|---|---|
| 1 | 9 (S,S) | TFA | 86 | 25:75 | <2 | 77 (S,R) |
| 2 | 9 (R,R) | TFA | 83 | 27:73 | <2 | 80 (R,S) |
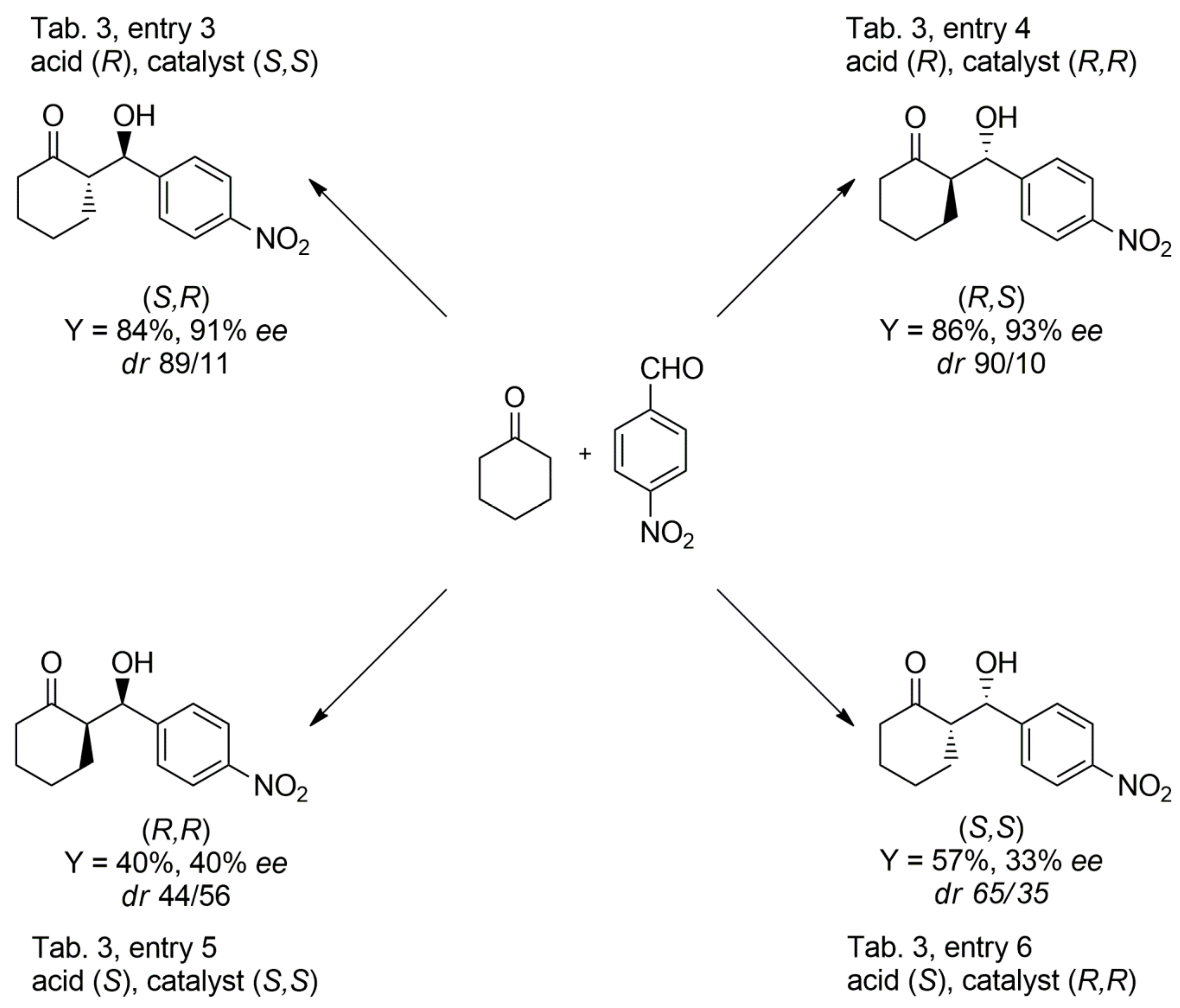
| 3 | 9 (S,S) | 14 (R) | 93 | 11:89 | 11 (R,R) | 91 (S,R) |
| 4 | 9 (R,R) | 14 (R) | 95 | 10:90 | 8 (R,R) | 93 (R,S) |
| 5 | 9 (S,S) | 14 (S) | 90 | 44:56 | 40 (R,R) | 70 (S,R) |
| 6 | 9 (R,R) | 14 (S) | 88 | 65:35 | 33 (S,S) | 45 (R,S) |
| Entry | Catalyst | Additive | Yield (%) | dra (Syn/Anti) | eebSyn (%) | eebAnti (%) |
|---|---|---|---|---|---|---|
| 1 | 9 (R,R) | 14 (R) | 95 | 10:90 | 8 (R,R) | 93 (R,S) |
| 2 | 9 (R,R) | 14 (S) | 88 | 65:35 | 33 (S,S) | 45 (R,S) |
| 3 | 2 (R,R) | 14 (R) | 35 | 15:85 | 10 (R,R) | 94 (R,S) |
| 4 | 2 (R,R) | 14 (S) | 99 | 19:81 | 14 (S,S) | 95 (R,S) |
| 5 | 6 (R,R) | 14 (R) | 99 | 17:83 | 24 (S,S) | 94 (R,S) |
| 6 | 6 (R,R) | 14 (S) | 99 | 24:76 | 30 (S,S) | 92 (R,S) |
| Entry | Substrate 1 | Substrate 2 | Catalyst | Additive | Yield (%) | dra (Syn/Anti) | eeb Syn (%) | eeb Anti (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | 2-NO2 | Cyclohexanone | 9 (S,S) | 14 (R) | 47 | 10:90 | 20 (R,R) | 94 (S,R) |
| 2 | 2-NO2 | Cyclohexanone | 9 (R,R) | 14 (R) | 46 | 28:72 | 52 (R,R) | 92 (R,S) |
| 3 | 2-NO2 | Cyclohexanone | 9 (S,S) | 14 (S) | 99 | 17:83 | 72 (R,R) | 96 (S,R) |
| 4 | 2-NO2 | Cyclohexanone | 9 (R,R) | 14 (S) | 99 | 18:82 | 48 (R,R) | 92 (R,S) |
| 5 | 2,4-diNO2 | Cyclohexanone | 9 (S,S) | 14 (R) | 62 | 1:99 | 64 (S,S) | 94 (R,S) |
| 6 | 2,4-diNO2 | Cyclohexanone | 9 (R,R) | 14 (R) | 99 | 12:88 | 41 (R,R) | 96 (S,R) |
| 7 | 2,4-diNO2 | Cyclohexanone | 9 (S,S) | 14 (S) | 65 | 2:98 | 32 (S,S) | 92(R,S) |
| 8 | 2,4-diNO2 | Cyclohexanone | 9 (R,R) | 14 (S) | 99 | 33:67 | 85 (R,R) | 96 (S,R) |
| 9 | 4-NO2 | OH-acetone | 9 (S,S) | 14 (R) | 99 | 87:13 | 92 (R,S) | 26 (R,R) |
| 10 | 4-NO2 | OH-acetone | 9 (R,R) | 14 (R) | 99 | 89:11 | 92 (S,R) | 34 (S,S) |
| 11 | 4-NO2 | OH-acetone | 9 (S,S) | 14 (S) | 99 | 89:11 | 90 (R,S) | 32 (R,R) |
| 12 | 4-NO2 | OH-acetone | 9 (R,R) | 14 (S) | 99 | 91:9 | 96 (S,R) | 36 (S,S) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pieczonka, A.M.; Marciniak, L.; Rachwalski, M.; Leśniak, S. Enantiodivergent Aldol Condensation in the Presence of Aziridine/Acid/Water Systems. Symmetry 2020, 12, 930. https://doi.org/10.3390/sym12060930
Pieczonka AM, Marciniak L, Rachwalski M, Leśniak S. Enantiodivergent Aldol Condensation in the Presence of Aziridine/Acid/Water Systems. Symmetry. 2020; 12(6):930. https://doi.org/10.3390/sym12060930
Chicago/Turabian StylePieczonka, Adam Marek, Lena Marciniak, Michał Rachwalski, and Stanisław Leśniak. 2020. "Enantiodivergent Aldol Condensation in the Presence of Aziridine/Acid/Water Systems" Symmetry 12, no. 6: 930. https://doi.org/10.3390/sym12060930