Microsolvation of Histidine—A Theoretical Study of Intermolecular Interactions Based on AIM and SAPT Approaches


Abstract
:
1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. Metric Parameters Analysis
3.2. Atoms-In-Molecules (AIM) Analysis
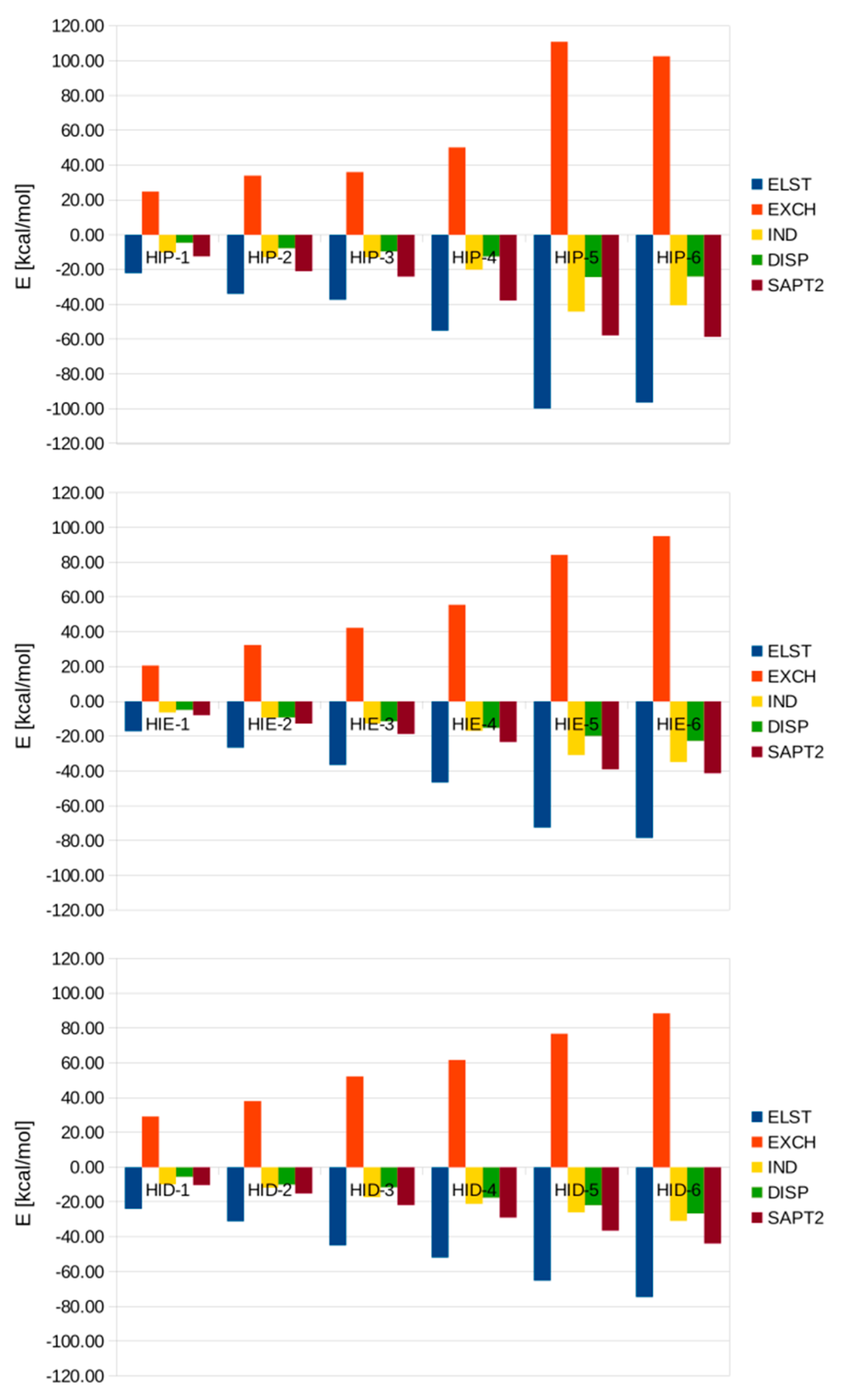
3.3. SAPT Analysis of Interaction Energies in the Microsolvated His-(H2O)n Complexes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Braga, D.; Grepioni, F.; Desiraju, G.R. Crystal Engineering and Organometallic Architecture. Chem. Rev. 1998, 98, 1375–1405. [Google Scholar] [CrossRef] [PubMed]
- Biedermann, F.; Schneider, H.-J. Experimental Binding Energies in Supramolecular Complexes. Chem. Rev. 2016, 116, 5216–5300. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Li, Y.; Xia, Y.-L.; Ai, S.-M.; Liang, J.; Sang, P.; Ji, X.-L.; Liu, S.-Q. Insights into Protein–Ligand Interactions: Mechanisms, Models, and Methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, G.A.; Saenger, W. Hydrogen Bonding in Biological Structures; Springer: New York, NY, USA, 1991. [Google Scholar]
- Wyttenbach, T.; Paizs, B.; Barran, P.; Breci, L.; Liu, D.; Suhai, S.; Wysocki, V.H.; Bowers, M.T. The Effect of the Initial Water of Hydration on the Energetics, Structures, and H/D Exchange Mechanism of a Family of Pentapeptides: An Experimental and Theoretical Study. J. Am. Chem. Soc. 2003, 125, 13768–13775. [Google Scholar] [CrossRef] [PubMed]
- Margenau, H.; Kestner, N. Theory of Inter-Molecular Forces, International Series of Monographs in Natural Philosophy; Pergamon Press: Oxford, UK, 1969. [Google Scholar]
- Chałasiński, G.; Gutowski, M. Weak interactions between small systems. Models for studying the nature of intermolecular forces and challenging problems for ab initio calculations. Chem. Rev. 1988, 88, 943–962. [Google Scholar] [CrossRef]
- Hobza, P.; Zahradnik, R. Intermolecular interactions between medium-sized systems. Nonempirical and empirical calculations of interaction energies. Successes and failures. Chem. Rev. 1988, 88, 871–897. [Google Scholar] [CrossRef]
- Kolář, M.H.; Hobza, P. Computer Modeling of Halogen Bonds and Other σ-Hole Interactions. Chem. Rev. 2016, 116, 5155–5187. [Google Scholar] [CrossRef] [Green Version]
- Grabowski, S.J. New type of halogen bond: Multivalent halogen interacting with π and σ-electrons. Molecules 2017, 22, 2150. [Google Scholar] [CrossRef] [Green Version]
- Jabłoński, M. Theoretical insight into the nature of the intermolecular charge-inverted hydrogen bond. Comput. Theor. Chem. 2012, 998, 39–45. [Google Scholar] [CrossRef]
- Jabłoński, M. Charge-inverted hydrogen bond vs. other interactions possessing a hydridic hydrogen atom. Chem. Phys. 2014, 433, 76–84. [Google Scholar] [CrossRef]
- Jensen, F. Introduction to Computational Chemistry, 2nd ed.; John Wiley & Sons Ltd.: Chichester, UK, 2007. [Google Scholar]
- Barone, V.; Cossi, M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Klamt, A.; Schüürmann, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 2 1993, 2, 799–805. [Google Scholar] [CrossRef]
- Skyner, R.E.; McDonagh, J.L.; Groom, C.R.; van Mourik, T.; Mitchell, J.B.O. A review of methods for the calculation of solution free energies and the modelling of systems in solution. Phys. Chem. Chem. Phys. 2015, 17, 6174–6191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López, J.C.; Sánchez, R.; Blanco, S.; Alonso, J.L. Microsolvation of 2-azetidinone: A model for the peptide group-water interactions. Phys. Chem. Chem. Phys. 2015, 17, 2054–2066. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, C.O.; Mennucci, B.; Vreven, T. Combining microsolvation and polarizable continuum studies: New insights in the rotation mechanism of amides in water. J. Phys. Chem. A 2003, 107, 6630–6637. [Google Scholar] [CrossRef]
- Li, S.; Hong, M. Protonation, Tautomerization, and Rotameric Structure of Histidine: A Comprehensive Study by Magic-Angle-Spinning Solid-State NMR. J. Am. Chem. Soc. 2011, 133, 1534–1544. [Google Scholar] [CrossRef] [Green Version]
- Day, R.M.; Thalhauser, C.J.; Sudmeier, J.L.; Vincent, M.P.; Torchilin, E.V.; Sanford, D.G.; Bachovchin, C.W.; Bachovchin, W.W. Tautomerism, acid-base equilibria, and H bonding of the six histidines in subtilisin BPN′ by NMR. Protein Sci. 2003, 12, 794–810. [Google Scholar] [CrossRef]
- Krishna Deepak, R.N.V.; Sankararamakrishnan, R. N-H…N Hydrogen Bonds Involving Histidine Imidazole Nitrogen Atoms: A New Structural Role for Histidine Residues in Proteins. Biochemistry 2016, 55, 3774–3783. [Google Scholar] [CrossRef]
- Olivieri, F.A.; Burastero, O.; Drusin, S.I.; Defelipe, L.A.; Wetzler, D.E.; Turjanski, A.; Marti, M. Conformational and Reaction Dynamic Coupling in Histidine Kinases: Insights from Hybrid QM/MM Simulations. J. Chem. Inf. Model. 2020, 60, 833–842. [Google Scholar] [CrossRef]
- Kawamura, K.; Yamada, T.; Kurihara, K.; Tamada, T.; Kuroki, R.; Tanaka, I.; Takahashi, H.; Niimura, N. X-ray and neutron protein crystallographic analysis of the trypsin-BPTI complex. Acta Cryst. D 2011, 67, 140–148. [Google Scholar] [CrossRef]
- Saxena, A.K.; Singh, T.P.; Peters, K.; Fittkau, S.; Visanji, M.; Wilson, K.S.; Betzel, C. Structure of a ternary complex of proteinase K, mercury, and a substrate-analogue hexa-peptide at 2.2 Å resolution. Proteins 1996, 25, 195–201. [Google Scholar] [CrossRef]
- Panek, J.J.; Mazzarello, R.; Novič, M.; Jezierska-Mazzarello, A. Impact of mercury(II) on proteinase K catalytic center: Investigations via classical and Born-Oppenheimer molecular dynamics. Mol. Divers. 2011, 15, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Kuczer, M.; Błaszak, M.; Czarniewska, E.; Rosiński, G.; Kowalik-Jankowska, T. Mono- and Polynuclear Copper(II) Complexes of Alloferons 1 with Point Mutations (H6A) and (H12A): Stability Structure and Cytotoxicity. Inorg. Chem. 2013, 52, 5951–5961. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.S.; Mathews, A.J.; Rohlfs, R.J.; Springer, B.A.; Egeberg, K.D.; Sligar, S.G.; Tame, J.; Renaud, J.P.; Nagai, K. The role of the distal histidine in myoglobin and haemoglobin. Nature 1988, 336, 265–266. [Google Scholar] [CrossRef] [PubMed]
- Vila, J.A.; Arnautova, Y.A.; Vorobjev, Y.; Scheraga, H.A. Assessing the fractions of tautomeric forms of the imidazole ring of histidine in proteins as a function of pH. Proc. Natl Acad. Sci. USA 2011, 108, 5602–5607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.O.; Nichols, S.E.; Wang, Y.; McCammon, A. Effects of histidine protonation and rotameric states on virtual screening of M. tuberculosis RmlC. J. Comput. Aided Mol. Des. 2013, 27, 235–246. [Google Scholar] [CrossRef] [Green Version]
- Cysewski, P.; Szefler, B. Environment influences on the aromatic character of nucleobases and amino acids. J. Mol. Model. 2010, 16, 1709–1720. [Google Scholar] [CrossRef] [Green Version]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Jeziorski, B.; Moszyński, R.; Szalewicz, K. Perturbation Theory Approach to Intermolecular Potential Energy Surfaces of van der Waals Complexes. Chem. Rev. 1994, 7, 1887–1930. [Google Scholar] [CrossRef]
- Jezierska, A.; Panek, J.J. Cooperativity of hydrogen bonding network in microsolvated biotin, the ligand of avidin class proteins. J. Mol. Model. 2019, 25, 361. [Google Scholar] [CrossRef] [PubMed]
- Rai, A.K.; Fei, W.; Lu, Z.; Lin, Z. Effects of microsolvation and aqueous solvation on the tautomers of histidine: A computational study on energy, structure and IR spectrum. Theor. Chem. Acc. 2009, 124, 37–47. [Google Scholar] [CrossRef]
- Lee, S.-S.; Kim, J.-Y.; Han, Y.; Shim, H.-J.; Lee, S. Thermodynamic and kinetic stability of zwitterionic histidine: Effects of gas phase hydration. Chem. Phys. Lett. 2015, 637, 42–50. [Google Scholar] [CrossRef]
- Cambridge Crystallographic Data Centre. Available online: https://www.ccdc.cam.ac.uk/ (accessed on 1 July 2020).
- Skelton, B.W.; Piggott, M.J.; Walkey, M.C. CCDC 1840707: Experimental Crystal Structure Determination; The Cambridge Crystallographic Data Centre (CCDC): Cambridge, UK, 2018. [Google Scholar]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [Green Version]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Schaftenaar, G.; Noordik, J.H. Molden: A pre- and post-processing program for molecular and electronic structures. J. Comput.-Aided Mol. Design 2000, 14, 123–134. Available online: http://cheminf.cmbi.ru.nl/molden/ (accessed on 4 June 2020). [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parameterization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Hohenstein, E.G.; Sherrill, C.D. Density fitting of intramonomer correlation effects in symmetry-adapted perturbation theory. J. Chem. Phys. 2010, 133, 014101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Møller, C.; Plesset, M.S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef] [Green Version]
- Kendall, R.A.; Dunning, T.H., Jr.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef] [Green Version]
- PSI4 Open-Source Quantum Chemistry. Available online: http://www.psicode.org/psi4manual/master/dfmp2.html# (accessed on 4 June 2020).
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; rev. B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- AIMAll (Version 19.10.12) Software by T. A. Keith. Available online: aim.tkgristmill.com (accessed on 4 June 2020).
- Parrish, R.M.; Burns, L.A.; Smith, D.G.A.; Simmonett, A.C.; DePrince, A.E.; Hohenstein, E.G.; Bozkaya, U.; Sokolov, A.Y.; Di Remigio, R.; Richard, R.M.; et al. Psi4 1.1: An Open-Source Electronic Structure Program Emphasizing Automation, Advanced Libraries, and Interoperability. J. Chem. Theory Comput. 2017, 13, 3185–3197. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. [Google Scholar] [CrossRef]
- The GIMP Development Team, GIMP. 2019. Available online: https://www.gimp.org (accessed on 1 July 2020).
- Koch, U.; Popelier, P.L.A. Characterization of C-H-O Hydrogen Bonds on the Basis of the Charge Density. J. Phys. Chem. 1995, 99, 9747–9754. [Google Scholar] [CrossRef]
- Grimme, S.; Mück-Lichtenfeld, C.; Erker, G.; Kehr, G.; Wang, H.; Beckers, H.; Willner, H. When Do Interacting Atoms Form a Chemical Bond? Spectroscopic Measurements and Theoretical Analyses of Dideuteriophenanthrene. Angew. Chem. Int. Ed. 2009, 48, 2592–2595. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomete, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]




{kind=link}
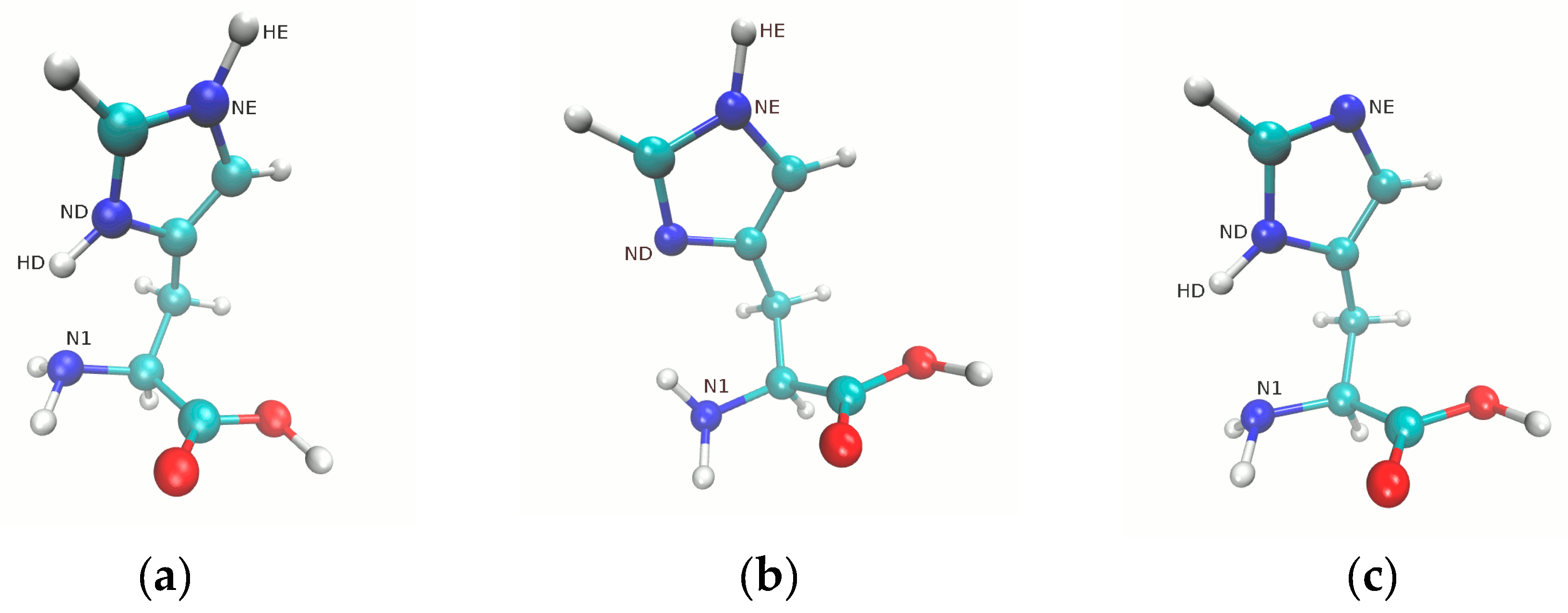{kind=link}
{kind=link}
{kind=link}
| Metric Parameters | Gas Phase | PCM |
|---|---|---|
| HIP | ||
| ND…N1 | 2.713 | 2.769 |
| ND-HD | 1.036 | 1.026 |
| HD…N1 | 1.917 | 2.030 |
| ∠NHN [°] | 131.2 | 126.9 |
| HIE | ||
| ND…N1 | 3.219 | 3.160 |
| N1-H | 1.012 | 1.017 |
| H…ND | 2.816 | 2.411 |
| ∠NHN [°] | 104.2 | 129.8 |
| HID | ||
| ND…N1 | 2.905 | 2.872 |
| ND-HD | 1.011 | 1.014 |
| HD…N1 | 2.247 | 2.197 |
| ∠NHN [°] | 121.4 | 122.5 |
| Metric Parameters | Histidine HIP Form | |
|---|---|---|
| HIP with 1–6 Water Molecules | HIP with 1–6 Water Molecules and with PCM | |
| One water molecule (HIP-1) | ||
| Intramolecular HB | ||
| ND...N1 | 2.706 | 2.763 |
| ND-HD | 1.038 | 1.027 |
| HD...N1 | 1.901 | 2.015 |
| ∠NHN [°] | 131.9 | 127.5 |
| Intermolecular HB | ||
| O1...O | 2.644 | 2.671 |
| O1-H1 | 0.992 | 0.992 |
| H1...O | 1.671 | 1.679 |
| ∠O1H1O [°] | 165.7 | 178.0 |
| Two water molecules (HIP-2) | ||
| Intramolecular HB | ||
| ND...N1 [A] | 2.668 | 2.737 |
| ND-HD | 1.048 | 1.030 |
| HD...N1 | 1.824 | 1.966 |
| ∠NHN [°] | 134.7 | 129.3 |
| Intermolecular HB | ||
| O1...O | 2.651 | 2.676 |
| O1-H1 | 0.991 | 0.991 |
| H1...O | 1.685 | 1.686 |
| ∠O1H1O [°] | 163.6 | 178.1 |
| N1...O | 2.996 | 3.005 |
| N1-H3 | 1.018 | 1.018 |
| H3...O | 2.067 | 2.052 |
| ∠N1H3O [°] | 150.5 | 154.9 |
| Three water molecules (HIP-3) | ||
| Intramolecular HB | ||
| ND...N1 | 2.644 | 2.739 |
| ND-HD | 1.055 | 1.030 |
| HD...N1 | 1.780 | 1.968 |
| ∠NDHDN [°] | 136.2 | 129.3 |
| Intermolecular HB | ||
| O1...O | 2.661 | 2.677 |
| O1-H1 | 0.989 | 0.991 |
| H1...O | 1.703 | 1.686 |
| ∠O1H1O [°] | 161.7 | 178.2 |
| N1...O | 3.225 | 3.081 |
| N1-H3 | 1.017 | 1.018 |
| H3...O | 2.300 | 2.091 |
| ∠N1H3O [°] | 150.6 | 163.6 |
| Four water molecules (HIP-4) | ||
| Intramolecular HB | ||
| ND...N1 [A] | 2.699 | 2.750 |
| ND-HD | 1.039 | 1.028 |
| HD...N1 | 1.881 | 1.990 |
| ∠NDHDN1 [°] | 133.0 | 128.5 |
| Intermolecular HB | ||
| O1...O | 2.664 | 2.676 |
| O1-H1 | 0.989 | 0.991 |
| H1...O | 1.707 | 1.686 |
| ∠O1H1O [°] | 161.6 | 177.3 |
| N1...O | 3.281 | 3.092 |
| N1-H3 | 1.016 | 1.018 |
| H3...O | 2.291 | 2.074 |
| ∠N1H3O [°] | 164.6 | 180.0 |
| NE...O | 2.781 | 2.773 |
| NE-HE | 1.029 | 1.033 |
| HE...O | 1.753 | 1.741 |
| ∠NEHEO [°] | 178.2 | 177.8 |
| Five water molecules (HIP-5) | ||
| No intramolecular HB | ||
| Intermolecular HB | ||
| O1...O | 2.664 | 2.655 |
| O1-H1 | 0.990 | 0.995 |
| H1...O | 1.687 | 1.661 |
| ∠O1H1O [°] | 168.1 | 177.7 |
| N1...O | 2.751 | 2.764 |
| O-H | 0.988 | 0.991 |
| H...N1 | 1.789 | 1.785 |
| ∠N1HO [°] | 163.5 | 169.0 |
| NE...O | 2.785 | 2.769 |
| NE-HE | 1.029 | 1.033 |
| HE...O | 1.756 | 1.736 |
| ∠NEHEO [°] | 178.1 | 178.1 |
| ND...O | 2.676 | 2.742 |
| ND-HD | 1.051 | 1.039 |
| HD...O | 1.640 | 1.721 |
| ∠NDHDO [°] | 167.6 | 166.7 |
| O...O2 | 2.869 | 2.816 |
| O-H | 0.970 | 0.971 |
| H...O2 | 1.910 | 1.851 |
| ∠OHO2 [°] | 169.5 | 171.7 |
| Six water molecules (HIP-6) | ||
| No intramolecular HB | ||
| Intermolecular HB | ||
| O1...O | 2.696 | 2.643 |
| O1-H1 | 0.986 | 0.998 |
| H1...O | 1.741 | 1.659 |
| ∠O1H1O [°] | 161.7 | 168.1 |
| N1...O | 2.754 | 2.780 |
| O-H | 0.990 | 0.990 |
| H...N1 | 1.789 | 1.801 |
| ∠N1HO [°] | 163.8 | 169.3 |
| NE...O | 2.811 | 2.773 |
| NE-HE | 1.023 | 1.022 |
| HE...O | 1.884 | 1.910 |
| ∠NEHEO [°] | 149.2 | 140.1 |
| ND...O | 2.673 | 2.743 |
| ND-HD | 1.050 | 1.037 |
| HD...O | 1.652 | 1.746 |
| ∠NDHDO [°] | 162.6 | 160.0 |
| O...O2 | 2.837 | 2.819 |
| O-H | 0.971 | 0.971 |
| H...O2 | 1.880 | 1.855 |
| ∠OHO2 [°] | 168.0 | 171.7 |
| HIP-(H2O)n | Simulation Environment | ND | HD | NE | HE | O2 | Mol. Charge |
|---|---|---|---|---|---|---|---|
| HIP-1 n = 1 | Gas phase | −1.246 | 0.537 | −1.206 | 0.476 | −1.179 | 0.958 |
| PCM H2O | −1.240 | 0.528 | −1.209 | 0.496 | −1.203 | 0.952 | |
| Δ(PCM-gas) | 0.006 | −0.009 | −0.003 | 0.020 | −0.024 | ||
| HIP-2 n = 2 | Gas phase | −1.251 | 0.544 | −1.207 | 0.473 | −1.182 | 0.946 |
| PCM H2O | −1.244 | 0.531 | −1.209 | 0.495 | −1.205 | 0.939 | |
| Δ(PCM-gas) | 0.007 | −0.013 | −0.002 | 0.022 | −0.023 | ||
| HIP-3 n = 3 | Gas phase | −1.252 | 0.549 | −1.207 | 0.472 | −1.184 | 0.953 |
| PCM H2O | −1.244 | 0.531 | −1.209 | 0.495 | −1.206 | 0.933 | |
| Δ(PCM-gas) | 0.008 | −0.018 | −0.002 | 0.024 | −0.022 | ||
| HIP-4 n = 4 | Gas phase | −1.254 | 0.533 | −1.249 | 0.539 | −1.190 | 0.916 |
| PCM H2O | −1.246 | 0.526 | −1.249 | 0.545 | −1.206 | 0.893 | |
| Δ(PCM-gas) | 0.008 | −0.008 | 0.000 | 0.006 | −0.016 | ||
| HIP-5 n = 5 | Gas phase | −1.253 | 0.553 | −1.256 | 0.538 | −1.207 | 0.927 |
| PCM H2O | −1.252 | 0.541 | −1.255 | 0.547 | −1.212 | 0.943 | |
| Δ(PCM-gas) | 0.001 | −0.012 | 0.001 | 0.008 | −0.004 | ||
| HIP-6 n = 6 | Gas phase | −1.251 | 0.552 | −1.246 | 0.517 | −1.201 | 0.938 |
| PCM H2O | −1.248 | 0.540 | −1.239 | 0.529 | −1.211 | 0.956 | |
| Δ(PCM-gas) | 0.003 | −0.012 | 0.007 | 0.012 | −0.010 |
| HIE-(H2O)n | Simulation Environment | ND | NE | HE | O2 | Mol. Charge |
|---|---|---|---|---|---|---|
| HIE-1 n = 1 | Gas phase | −1.150 | −1.227 | 0.424 | −1.183 | 0.047 |
| PCM H2O | −1.185 | −1.231 | 0.459 | −1.207 | 0.059 | |
| Δ(PCM-gas) | −0.036 | −0.004 | 0.035 | −0.024 | ||
| HIE-2 n = 2 | Gas phase | −1.156 | −1.228 | 0.424 | −1.174 | 0.058 |
| PCM H2O | −1.188 | −1.232 | 0.458 | −1.198 | 0.067 | |
| Δ(PCM-gas) | −0.031 | −0.003 | 0.035 | −0.024 | ||
| HIE-3 n = 3 | Gas phase | −1.164 | −1.263 | 0.486 | −1.176 | 0.041 |
| PCM H2O | −1.194 | −1.269 | 0.508 | −1.199 | 0.039 | |
| Δ(PCM-gas) | −0.030 | −0.005 | 0.023 | −0.023 | ||
| HIE-4 n = 4 | Gas phase | −1.188 | −1.263 | 0.492 | −1.176 | 0.062 |
| PCM H2O | −1.203 | −1.267 | 0.513 | −1.199 | 0.089 | |
| Δ(PCM-gas) | −0.015 | −0.003 | 0.021 | −0.023 | ||
| HIE-5 n = 5 | Gas phase | −1.180 | −1.255 | 0.493 | −1.222 | 0.051 |
| PCM H2O | −1.198 | −1.259 | 0.505 | −1.225 | 0.058 | |
| Δ(PCM-gas) | −0.017 | −0.004 | 0.012 | −0.003 | ||
| HIE-6 n = 6 | Gas phase | −1.176 | −1.253 | 0.471 | −1.232 | 0.021 |
| PCM H2O | −1.196 | −1.256 | 0.498 | −1.229 | 0.052 | |
| Δ(PCM-gas) | −0.020 | −0.003 | 0.027 | 0.003 |
| HID-(H2O)n | Simulation Environment | ND | HD | NE | O2 | Mol. Charge |
|---|---|---|---|---|---|---|
| HID-1 n = 1 | Gas phase | −1.246 | 0.459 | −1.134 | −1.194 | −0.025 |
| PCM H2O | −1.252 | 0.480 | −1.192 | −1.208 | −0.043 | |
| Δ(PCM-gas) | −0.006 | 0.021 | −0.058 | −0.014 | ||
| HID-2 n = 2 | Gas phase | −1.266 | 0.478 | −1.133 | −1.193 | −0.014 |
| PCM H2O | −1.254 | 0.482 | −1.193 | −1.210 | −0.051 | |
| Δ(PCM-gas) | 0.012 | 0.004 | −0.060 | −0.017 | ||
| HID-3 n = 3 | Gas phase | −1.254 | 0.475 | −1.165 | −1.195 | 0.015 |
| PCM H2O | −1.250 | 0.488 | −1.210 | −1.207 | 0.017 | |
| Δ(PCM-gas) | 0.004 | 0.013 | −0.045 | −0.012 | ||
| HID-4 n = 4 | Gas phase | −1.260 | 0.495 | −1.166 | −1.193 | 0.032 |
| PCM H2O | −1.258 | 0.495 | −1.192 | −1.209 | 0.010 | |
| Δ(PCM-gas) | 0.002 | 0.001 | −0.026 | −0.016 | ||
| HID-5 n = 5 | Gas phase | −1.260 | 0.497 | −1.168 | −1.209 | 0.035 |
| PCM H2O | −1.251 | 0.488 | −1.194 | −1.212 | 0.023 | |
| Δ(PCM-gas) | 0.008 | −0.009 | −0.026 | −0.002 | ||
| HID-6 n = 6 | Gas phase | −1.262 | 0.507 | −1.174 | −1.206 | 0.022 |
| PCM H2O | −1.249 | 0.490 | −1.192 | −1.213 | 0.028 | |
| Δ(PCM-gas) | 0.013 | −0.017 | −0.018 | −0.008 |
| MP2/aug-cc-pVDZ | MP2/aug-cc-pVTZ | SAPT2/aug-cc-pVDZ | SAPT2/aug-cc-pVTZ | |||||
|---|---|---|---|---|---|---|---|---|
| Eint | per 1 H2O | Eint | per 1 H2O | Eint | per 1 H2O | Eint | per 1 H2O | |
| HIP-1 | −12.50 | −12.50 | −13.49 | −13.49 | −12.57 | −12.57 | −13.79 | −13.79 |
| HIP-2 | −21.03 | −10.52 | −22.47 | −11.23 | −21.02 | −10.51 | −22.76 | −11.38 |
| HIP-3 | −23.87 | −7.96 | −25.43 | −8.48 | −24.11 | −8.04 | −25.99 | −8.66 |
| HIP-4 | −37.99 | −9.50 | −40.23 | −10.06 | −37.88 | −9.47 | −40.54 | −10.13 |
| HIP-5 | −57.64 | −11.53 | −61.90 | −12.38 | −57.87 | −11.57 | ||
| HIP-6 | −58.74 | −9.79 | −62.72 | −10.45 | −58.78 | −9.80 | ||
| HIE-1 | −8.02 | −8.02 | −8.74 | −8.74 | −7.98 | −7.98 | −8.89 | −8.89 |
| HIE-2 | −12.84 | −6.42 | −14.16 | −7.08 | −12.78 | −6.39 | −14.44 | −7.22 |
| HIE-3 | −18.87 | −6.29 | −20.58 | −6.86 | −18.70 | −6.23 | −20.85 | −6.95 |
| HIE-4 | −23.38 | −5.85 | −25.63 | −6.41 | −23.43 | −5.86 | −26.26 | −6.57 |
| HIE-5 | −38.97 | −7.79 | −42.25 | −8.45 | −39.14 | −7.83 | ||
| HIE-6 | −40.74 | −6.79 | −44.39 | −7.40 | −41.33 | −6.89 | ||
| HID-1 | −10.09 | −10.09 | −11.19 | −11.19 | −10.31 | −10.31 | −11.70 | −11.70 |
| HID-2 | −14.93 | −7.46 | −16.56 | −8.28 | −15.16 | −7.58 | −17.15 | −8.58 |
| HID-3 | −21.62 | −7.21 | −23.64 | −7.88 | −21.87 | −7.29 | −24.41 | −8.14 |
| HID-4 | −29.12 | −7.28 | −31.80 | −7.95 | −29.05 | −7.26 | −32.29 | −8.07 |
| HID-5 | −36.72 | −7.34 | −40.10 | −8.02 | −36.53 | −7.31 | ||
| HID-6 | −43.71 | −7.28 | −47.70 | −7.95 | −43.87 | −7.31 | ||
| ELST | EXCH | IND | DISP | Total SAPT2 | |
|---|---|---|---|---|---|
| HIP-1 | −22.19 | 24.75 | −10.39 | −4.74 | −12.57 |
| HIP-2 | −34.12 | 33.95 | −12.95 | −7.89 | −21.02 |
| HIP-3 | −37.44 | 36.04 | −13.01 | −9.70 | −24.11 |
| HIP-4 | -55.27 | 50.20 | −20.16 | −12.66 | −37.88 |
| HIP−5 | −100.08 | 110.86 | −44.28 | −24.38 | −57.87 |
| HIP-6 | −96.58 | 102.61 | −40.69 | −24.12 | −58.78 |
| HIE-1 | −17.19 | 20.65 | −6.48 | −4.96 | −7.98 |
| HIE-2 | −26.68 | 32.45 | −9.27 | −9.28 | −12.78 |
| HIE-3 | −36.61 | 42.29 | −12.79 | −11.59 | −18.70 |
| HIE-4 | −46.72 | 55.55 | −16.98 | −15.28 | −23.43 |
| HIE-5 | −72.60 | 84.21 | −30.90 | −19.85 | −39.14 |
| HIE-6 | −78.58 | 94.95 | −34.93 | −22.78 | −41.33 |
| HID-1 | −23.95 | 29.14 | −9.87 | −5.63 | −10.31 |
| HID-2 | −31.15 | 37.99 | −11.90 | −10.10 | −15.16 |
| HID-3 | −45.01 | 52.18 | −17.34 | −11.69 | −21.87 |
| HID-4 | −52.01 | 61.66 | −21.06 | −17.65 | −29.05 |
| HID-5 | −65.31 | 76.76 | −26.04 | −21.94 | −36.53 |
| HID-6 | −74.77 | 88.46 | −30.94 | −26.63 | −43.87 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kizior, B.; Panek, J.J.; Jezierska, A. Microsolvation of Histidine—A Theoretical Study of Intermolecular Interactions Based on AIM and SAPT Approaches. Symmetry 2020, 12, 1153. https://doi.org/10.3390/sym12071153
Kizior B, Panek JJ, Jezierska A. Microsolvation of Histidine—A Theoretical Study of Intermolecular Interactions Based on AIM and SAPT Approaches. Symmetry. 2020; 12(7):1153. https://doi.org/10.3390/sym12071153
Chicago/Turabian StyleKizior, Beata, Jarosław J. Panek, and Aneta Jezierska. 2020. "Microsolvation of Histidine—A Theoretical Study of Intermolecular Interactions Based on AIM and SAPT Approaches" Symmetry 12, no. 7: 1153. https://doi.org/10.3390/sym12071153
APA StyleKizior, B., Panek, J. J., & Jezierska, A. (2020). Microsolvation of Histidine—A Theoretical Study of Intermolecular Interactions Based on AIM and SAPT Approaches. Symmetry, 12(7), 1153. https://doi.org/10.3390/sym12071153