Abstract
Consensus string is a significant feature of a deoxyribonucleic acid (DNA) sequence. The median string is one of the most popular exact algorithms to find DNA consensus. A DNA sequence is represented using the alphabet Σ= {a, c, g, t}. The algorithm generates a set of all the 4l possible motifs or l-mers from the alphabet to search a motif of length l. Out of all possible l-mers, it finds the consensus. This algorithm guarantees to return the consensus but this is NP-complete and runtime increases with the increase in l-mer size. Using transitional probability from the Markov chain, the proposed algorithm symmetrically generates four subsets of l-mers. Each of the subsets contains a few l-mers starting with a particular letter. We used these reduced sets of l-mers instead of using 4l l-mers. The experimental result shows that the proposed algorithm produces a much lower number of l-mers and takes less time to execute. In the case of l-mer of length 7, the proposed system is 48 times faster than the median string algorithm. For l-mer of size 7, the proposed algorithm produces only 2.5% l-mer in comparison with the median string algorithm. While compared with the recently proposed voting algorithm, our proposed algorithm is found to be 4.4 times faster for a longer l-mer size like 9.
1. Introduction
Within the gene, there are short DNA sequences that control the organic phenomenon. These short DNA sequences are known as gene regulatory binding motifs. It is very difficult to identify those short motifs. Those are solely 6–15 nucleotide pairs long. Regulatory motifs don’t have any specific point to begin and stop. These motifs can start from and stop anywhere within the sequence. A restrictive motif is indistinguishable from an equivalent random sequence. It is a specific pattern that happens more often than expected beneath the background sequence. The location of such a pattern may differ from one sample to another. In addition, the frequency of a particular pattern may differ from one sequence to another. Discovery of such a regulatory motif from a DNA sequence therefore involves finding such a continuation of the pattern [1]; this can be illustrated in Figure 1.
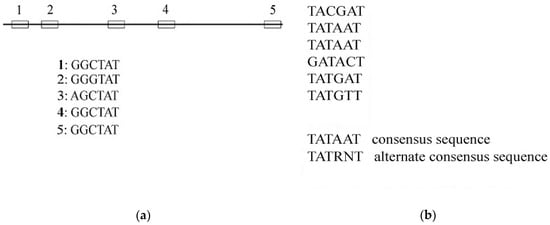
Figure 1.
Illustration of consensus string: (a) Aligning the DNA sequences; (b) The most common and most frequent sequence is the consensus string.
DNA sequences are composed of the nitrogen bases adenine, cytosine, thiamin, and guanine. These four bases are represented by four symbols a, c, g, and t. Finding a specific motif from a DNA sequence incurs a search for a short pattern over the alphabet Σ = {a, c, g, t}. The motif search process emphasizes two points: (a) a way to figure out the sequence motif by the application of a proper model; (b) an efficient way to formulate an algorithm for motif finding. The foremost unremarkably used models for motif representation are position weight matrices (PWM) [2] and consensus sequences [3]. The position weight metrics system generally takes a shorter time to report. These metrics use statistical methods but there is no guarantee about finding a global optimum [4,5,6]. The exact algorithms, which use consensus sequences to represent motifs, are sure to report all (l, d) motifs by traversing the full search area. Most of the exact algorithms are driven by patterns. In the case of pattern-driven algorithms, the search space is O(|Σ|l). It rises dramatically with the increment of |Σ|. As a consequence, most of the existing exact algorithms have been designed only for looking for motifs in deoxyribonucleic acid sequences wherever |Σ|=4, and that they cannot search low-conserved motifs within a reasonable time within the data sets over larger alphabets, like the protein data sets wherever |Σ| = 20.
To increase the capability of the exact algorithms for larger alphabets, Ref. [7] proposed the idea of the motif stem within the field of motif search. The consensus is a representative string of a given set S of strings. This is sometimes called the closest string or center string. In the case of multiple string comparison, the consensus is one of the major issues. It has been explored elaborately [8,9,10,11,12,13,14,15] to address several questions arising from computational biology. Though finding the consensus sequence is an NP-complete problem, researchers even have developed fixed-parameter algorithms [10,16,17,18], approximation algorithms [13,17,19,20,21,22,23] and algorithms for a short range of strings [10,24,25]. All the fixed-parameter algorithm shows how to solve an NP-complete problem in linear time in the case of consensus search. Given k sequences of equal length and a positive integer d, find a “closest string” s such that none of the given sequences has a hamming distance longer than d from s. The closest string is one of the prime obstacles in consensus analysis. Solving the closest string is an NP-complete problem. The authors of [10] and [16] proposed an algorithm to solve this problem in polynomial time. In addition, the authors of [17] proposed an exact algorithm to solve the closest string problem with time complexity O(n|Σ| O(d)), where Σ is the alphabet. The 1-mismatch problem is the determination of all the maximal sections from an alignment where there exists a “center” string. The inside of the section alignment rows are within Hamming distance 1 from the center. Solving the center string is also NP-complete. The authors of [18] introduced a polynomial-time algorithm to solve the center string problem. In [13], an application of a genetic algorithm to solve the closest string problem was proposed. A polynomial-time approximation algorithm was proposed in [19]. An approximation algorithm for solving center string problem was introduced in [20]. An approximation scheme for the hamming clustering problem was proposed in [21]. A comprehensive study of all recent research in the field of motif search is summarized in [25]. Some new algorithms have been proposed. Among them, oligonucleotide analysis [26], SLI-REST [7], MCES [27], Pruner [28] and Voting [29] algorithms are the most recent.
Among all the algorithms, the median string algorithm provides the highest level of guarantee that it must find the consensus. That is because the median string is a brute force algorithm. The only thing it lacks is that it takes a long time to execute. The median string algorithm searches all the |Σ|l l-mers. As the l-mer size grows, the execution time grows exponentially. As the size of the alphabet and l-mer size both grow, the execution time makes the median sting an inferior algorithm. In this paper, we used a Markov chain to reduce the search space. As a result, the execution time reduces dramatically. We combined the statistical method to reduce the search space of an exact algorithm. We got the same output within a shorter period. Experiments indicate our proposed algorithm executes faster than the median string algorithm. Furthermore, the paper includes mathematical proof about why this method will produce the same output as that of the brute force algorithm. Finally, we compared the execution time of our proposed algorithm with the execution time of the voting algorithm for motif search. We found our proposed algorithm is faster than the voting algorithm for longer l-mer size.
2. Background Information
2.1. Median String Algorithm for Consensus Sequence
The input parameters of the median string algorithm are a set of DNA sequences, number of sequences, number of nucleotides in each sequence, and the length of the motif to be searched. A set of t number of DNA sequences of length n and the length of the motif to be searched l are included. The algorithm produces all possible motifs of length l. Since DNA sequences are composed of four types of nucleotides, the number of all possible l-mers is 4l. It then takes one l-mer, compares the nucleotides of the l-mer with the l nucleotide starting from the first nucleotide of the first sequence, and calculates the distance. After that, it shifts one nucleotide right, places the l-mer, and calculates the distance. The process continues up to the end of the sequence. The minimum distance is the score for the l-mer with the sequence. In this way, scores for the l-mer with all the sequences are calculated. The sum of scores is the score for the l-mer with the set of DNA sequences. The scores for all the l-mers are calculated using the same process. The l-mer with the lowest score is the consensus string. The algorithm is listed below:
| Algorithm 1: Median String Search |
| { inputs: DNA,t,n,l output: bestFit_Motif procedure: MedianStringSearch (DNA, t, n, l) bestFit_Motif ← AAA…A bestFit_Score ← ∞ for each l-mer s from AAA…A to TTT…T if TotalFit_Score(s, DNA) < bestFit_Score bestFit_Score ← TotalFit_Score(s, DNA) bestFit_Motif ← s return bestFit_Motif } |
The median string algorithm examines all the 4l combinations of l-mers. Consequently, the number of candidate l-mers, as well as execution time, grows exponentially with the rise of l; however, a significant amount of the generated 4l l-mers, will not be the consensus. Even some of them can be absent in a particular sequence. Those motifs were generated as we were interested in generating all possible combinations. For those motifs too, the median string still performs the calculation and contributes to the time complexity. In this paper, we introduced the Markov chain to exclude infrequent candidate motifs. The exclusion of those unnecessary candidate motifs leads to a smaller search space and reduces the time complexity of the median string algorithm.
2.2. Markov Chain
In the case of a series of chance experiments, all the outcomes of the previous experiment could influence the result of the next experiment. The Markov chain can answer questions regarding the probability of these types of chance experiments. Let S be a set of states where S = {s1, ss,…, sr}. The process can start from any one of the states and can proceed successively from one position to another. Markov defined these moves as steps. If the current position of the chain is in position si and it proceeds to position sj in the next move, then the probability of this move is expressed by Pij. Pij is called transition probability. The chain can reside in the same position as probability Pii. If we plot the probabilities for all possible moves in a matrix, that matrix is called a transition matrix.
In bioinformatics, DNA sequences can be modeled using Markov chains. A DNA sequence is a series of four-letter alphabet ∑ = {a, c, g, t}. In the case of DNA sequences, a Markov chain is defined by ∑ and a probability for transition Pij. The transition probabilities are stored in a transition matrix. A sequence defined by the trajectory of the process through the state space. The scenario is depicted in Figure 2. The diagram exhibits that the sequence can start from any nucleotide and the probability of the next nucleotide is dependent on the current nucleotide. If we start from the state a, the next nucleotide can be any one of a, c, g, t, and the probability is given by Paa, Pac, Pag, and Pat. Starting from another nucleotide will incur another set of transition probabilities. If we start from state g, then the probabilities would be Pga, Pgc, Pgg, and Pgt. DNA sequences are defined by Markov Chain in this manner. All the transition probabilities are listed in the transition matrix T and the start state probabilities are given by a vector Π = (Πa, Πc, Πg,Πt).
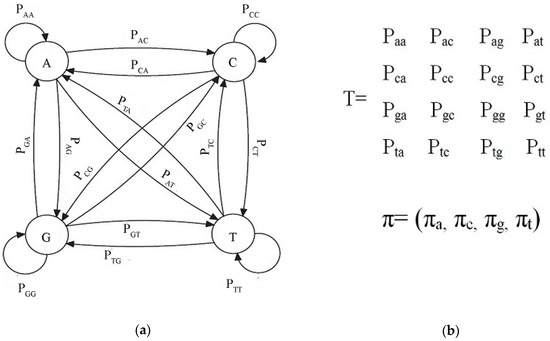
Figure 2.
Markov chain and transition probability: (a) Markov chain; (b) Transition probability matrix along with start probability vector.
The mathematical definition of entries in the transition matrix is as follows:
This means that the probability of moving from state x to state y is equal to the conditional probability of having state y given that the previous state was x. The probability of the entire sequence can be expressed as a joint probability:
Simplification of calculation of this probability can be made by factorizing it:
Or
where Π(s1 = x) means the probability of having x in location s1. As a result, we can conclude that the probability of a symbol occurring is dependent only on the previous symbol to simplify the calculation of the joint probability of the entire sequence.
3. Proposed Markov Chain Based Median String Algorithm
Let us consider a database with 10 sample DNA sequences (S1, S2,…, S10). Each sample is composed of 80 nucleotides as in Table 1. Samples are different from each other. The problem is to find whether there is any common motif that exists among the sequences.

Table 1.
Sample DNA sequence database.
From this database, we will extract the consensus string using the proposed system. A schematic layout of the proposed system is given in Figure 3.
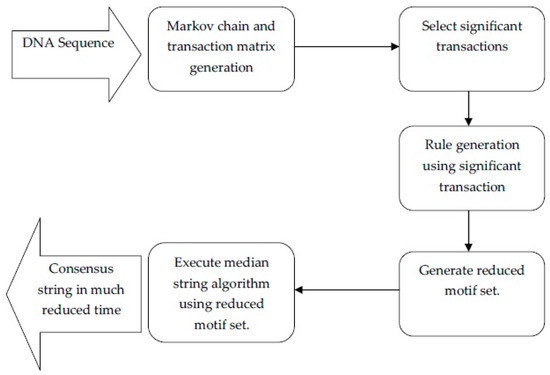
Figure 3.
Schematic layout of the proposed system.
3.1. Markov Chain Generation
In the database, there are 10 sequences. Each sequence contains 80 nucleotides. There are 42 = 16 possible two-nucleotide sequences i.e., aa,ac,ag,at,ca,cc,cg,ct,ga,gc,gg,gt,ta,tc,tg, and tt. We first count the frequencies of the occurrence of two-nucleotide sequences in the database. From the counts of the two nucleotide sequences, we constructed a Markov chain, which is depicted in Figure 4. According to the following Markov chain, the frequency of aa is 49, cc is 46, etc., in the database. Now, we have the counts of 16 possible two nucleotide sequences or di-grams in the database.
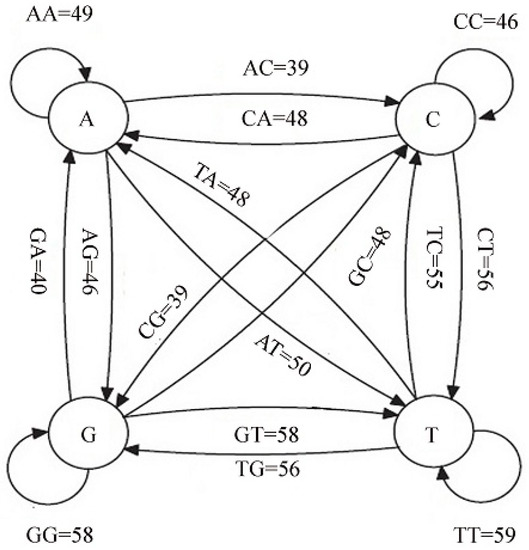
Figure 4.
Markov chain generated using data in Table 1.
3.2. Transaction Matrix Creation
We now construct a transition matrix from the Markov chain as in Figure 5. Rows in the matrix indicate the first nucleotide and columns in the matrix indicate the second nucleotide of the two-nucleotide sequences. The first row is for sequences starting with a. The first element in this row indicates the counts of aa in the database. The second element indicates the counts of ac, etc. The next row is for sequences starting with the nucleotide c, the third row is for sequences starting with the nucleotide g, etc. The transition matrix contains counts for all the 16 two nucleotide sequences in the database.
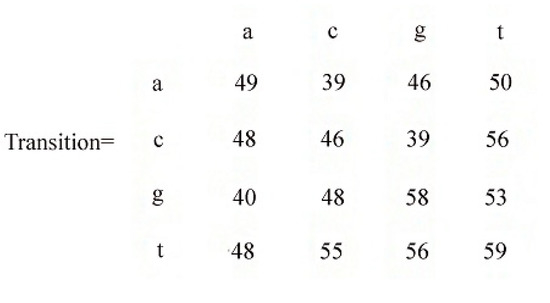
Figure 5.
Transaction matrix using Markov chain.
This matrix helps to infer about the next nucleotide given the staring nucleotide. For example, if we start from nucleotide a, then we can infer P(t|a) > P(a|a) > P(g|a) > P(c|a).
3.3. Rule Generation
In this stage, we tried to find the most frequent transitions as well as the most probable transitions. For this purpose, we sum up all the counts in the transaction matrix. The sum of all the elements of the transaction matrix is 790. To cover 50% of elements, we set the threshold as 790/2 = 395. We start with the highest value in the matrix. The highest value is 59, which represents P(t|t). This element value is less than the threshold value. We select this element and set the threshold as 395 − 59 = 336. The next highest value in the matrix is 58, which represents P(g|g). This value is less than the threshold value. We select this element and set the threshold as 336 − 58 = 278. The process continues until the element value is greater than the threshold value. This process produces the seven most frequent transitions representing the following probabilities P(t|t),P(g|g),P(c|t),P(t|g),P(t|c),P(g|t),P(a|t). At the end of the process, we will have a matrix with some encircled values as in Figure 6.
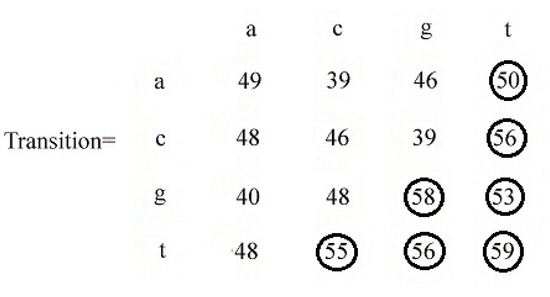
Figure 6.
Identification of significant elements in the transition matrix.
The encircled elements are significant; that is, two-nucleotide sequences (tt,ct,gg,gt,tc,tg,at) are significant. If we randomly pick any two-nucleotide sequence from the database, there is a more than 50% probability that the sequence will be in the set of (tt, ct, gg, gt, tc, tg, at). We now generate rules from this significant combination as in Table 2.
Table 2.
Rule generation.
3.4. Reduced l-mer Set Generation
According to the rules, we will now generate l-mers. For example, let us suppose l = 4, then we will generate 4-mers according to these rules. 4-mers can start with any nucleotide at the beginning. If it starts from a, we will have a tree generating all the possible 4-mers starting with a that follow the rules in Table 2, as in Figure 7. According to rules recorded in Table 2, the next nucleotide after a will be t; i.e., a two-nucleotide sequence starting with a is at. After at, the next nucleotide can be t or c or g, according to the rules. As a result, we will have 3 three-nucleotide sequences (att, atc, atg), which start with nucleotide a and follow the rules. After atc, the next nucleotide can be t or c or g, which produces the 4-mers attt, attc, and attg. Symmetrically, from atc we have the 4-mer atct and from atg we have the 4-mers atgg and atgt. 4-mers also can start with the other three nucleotides c,g, and t. Symmetrically, we will have three other trees for 4-mers starting with c,g, and t. From these trees, we will have all the 4-mers that follow the rules. These are the motifs that we will use instead of all possible 4l numbers of motifs.
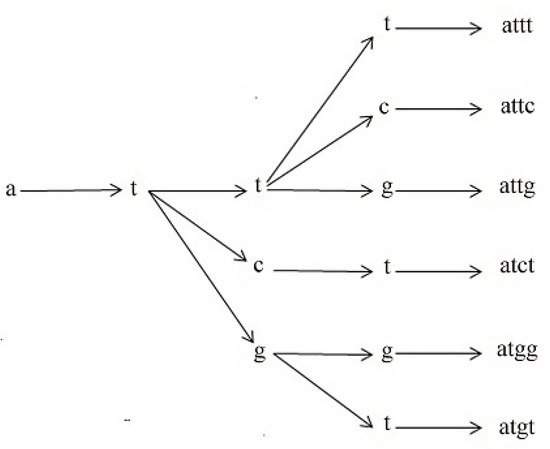
Figure 7.
Motif generation tree.
3.5. Proposed Algorithm
| Algorithm 2: Markow chain based median string algorithm(DNA,l) |
| { input:DNA,l Output: consensus sequence Modified_Median_String (DNA,l) { bestFit_Motif ←AAA…A bestFit_Score ←∞ reduced_motif_set = Reduced_Motif_Set_Generator(DNA,l) for each l-mer in reduced_motif_set if TotalFit_Score(s, DNA) < bestFit_Score bestFit_Score←TotalFit_Score(s, DNA) bestFit_Motif ← s return bestFit_Motif } Transaction_Matrix_Generator(DNA) {input:DNA output:Transition_Matrix ngram_dict←new dictionary() i←1 for each sequence in DNA for each character in sequence up to sequence length-1 if character not in ngram_dict.keys() ngram_dict[character]←Null NextCharacter←Sequence[i + 1] ngram_dict[character].append(NextCharacter) ←i + 1 TM←empty matrix for each key in ngram_dict TM ← Counter(ngram_dict(key)) Return TM } Rule_Generator(Transition_Matrix) {input:Transition_Matrix output:rule_list total= ΣTransition_matrixij s ← 0 rule_list ← empty list() for each element in Transition_Matrix in descending order s ← s+ Transition_matrix element if s < total/2 append a rule in rule_list else break return rule_list } Reduced_Motif_Set_Generator(DNA, l) {input:DNA, length of l-mer output:Reduced_Motif_Set TM ← Transition_Matrix_Generator(DNA) rules ← Rule_Generator(TM) S1,tmp,tmp2 ← empty list For each character in {‘a’, ‘c’, ‘g’, ‘t’} if rules.key ==character S1 ← character + rules.value tmp ← S1 for i = 1 to length of l-mer-2 for element in tmp if rules.key ==last character of element tmp2 ← element + rules.value tmp ← tmp2 tmp2 ← null return tmp } |
4. Result and Discussion
In this section, we have analyzed the performance of the proposed method and compared it with state-of-the-art methods in terms of Jupyter Notebook as a Python programming language. We developed the existing method using Algorithm 1. and the proposed method using Algorithm 2. We compared the performance of the proposed system with that of the median string algorithm and voting algorithm. The system configuration is
- Processor: Intel Core i5 CPU
- Clock rate:2.6 GHz
- HardDisk:1000GB
- RAM:4GB
4.1. Comparison with Median String Algorithm
In comparison with the median string algorithm, the proposed algorithm expedites the whole process easier and faster. In this illustration, we have analyzed the number of motifs generated by the Markov chain-based median string algorithm and the median string algorithm for different l-mer sizes. A comparison between the proposed algorithm and median string algorithm in terms of the number of generated motifs is listed in Table 3.
Table 3.
Comparative interpretation between the proposed algorithm with the median string algorithm in terms of motif generation.
The table reports the results of both of the techniques adopted in this paper. In the case of l-mer size 2, the proposed method forms 7 motifs whereas the existing algorithm produces 16 motifs. In this case, the number of motifs generated by the proposed algorithm is 43.75% of that of the existing algorithm. The last row depicts that the introduced algorithm produces 427 and the existing algorithm returns 16,784 motifs of length 7. In this case, the proposed algorithm yields only 2.5% motifs while contrasted with that of the existing algorithm. Hence, our proposed algorithm originates fewer motifs in relation to the existing algorithm, which is depicted in Figure 8a. As a result, we claim that our proposed algorithm can reduce the search space significantly.
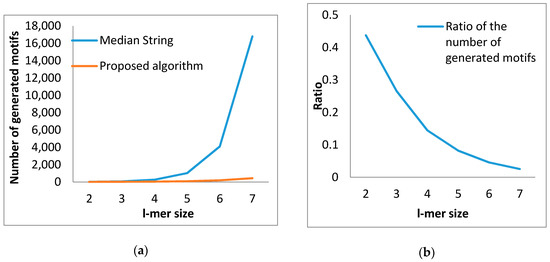
Figure 8.
Comparison in terms of motif generation: (a) Number of motifs generated by both algorithms; (b) The ratio of the number of generated motifs between the proposed algorithm and median string algorithms.
Furthermore, with the increase in l-mer size, the proposed algorithm appears to be superior to the median string algorithm in terms of the number of motifs produced. The ratio of the number of produced motifs between the proposed and the existing algorithm for various l-mer lengths is recorded in the rightmost column of Table 3. In the case of the l-mer of length 2, the ratio is 0.4375 and for l-mer of length 7, the ratio is 0.0254. This indicates, with the increase in l-mer size, that the ratio is gradually decreasing, which is depicted in Figure 8b. This finding means that the proposed algorithm produces fewer motifs than the existing algorithm. This lower number dramatically reduces with the increase in l-mer size (See Appendix A).
We have investigated every level of time consumed by both methods. The introduced approach necessitates a huge time for DNA consensus. The time consumed by these two approaches betokens the robustness of our proposed algorithm. The execution time of both algorithms for different l-mer size is listed in Table 4.
Table 4.
Comparative interpretation between the proposed algorithm and median string algorithm in terms of execution time.
Every row of the table demonstrates that the proposed algorithm spent less time than the existing orientation. In connection with the l-mer of length 2, the difference in execution time is (16.10 − 10.20) ms = 5.90 ms. It implies that for l-mer size 2, the execution time of the proposed method is only 63% of that of the existing algorithm. The time differences have been increased as the l-mer length increased. From the last tuple, we notice that for l-mer of length 7, the proposed algorithm consumed 587.44 ms, whereas the existing algorithm spent 24,400 ms. In this case, the proposed algorithm’s execution time is only 2.4% of the execution time of the median string algorithm. Now, we can claim our proposed algorithm to be faster than the median string algorithm, which is depicted in Figure 9a.
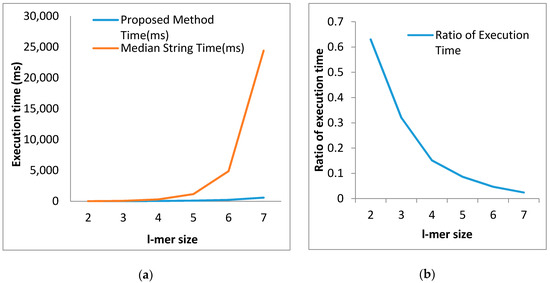
Figure 9.
Illustration in terms of execution time (a) Comparative illustration of execution time between the proposed algorithm and median string algorithm; (b) The ratio of execution time between the proposed algorithm and median string algorithm.
With the increase in the length of l-mer, the introduced algorithm appears to be superior to the existing algorithm. The ratios of execution time between the proposed and the existing algorithm for l-mer of different lengths are listed in the rightmost column of Table 4. In the case of the l-mer of length 2, the ratio is 0.63 and for l-mer of length 7, the ratio is 0.024. The ratio is gradually decreasing with the increase in l-mer size, which is depicted in Figure 9b. This finding means that the proposed algorithm is faster than the median string algorithm. Besides that, it also proves that the fastness of the proposed algorithm rises dramatically with the increase in l-mer size.
4.2. Comparison with the Voting Algorithm
Finally, we compared the execution time of our proposed algorithm with the execution time of the voting algorithm for l-mer of different lengths. The execution times for both algorithms, and their ratios, are given in Table 5.
Table 5.
Comparative illustration between the proposed algorithm and voting algorithm in terms of execution time.
From the table above, we can see for l-mer size 2, the execution time of our proposed algorithm is 5.23 times that of the voting algorithm. Though the ratio of execution time is gradually decreasing, it is still above 1 for l-mer size 5. This means that, up to l-mer size 5, the proposed algorithm remains slower than the voting algorithm;however, from l-mer size 6 upward, the execution time ratio becomes less than 1. From this finding, we can claim that our proposed algorithm is faster than the voting algorithm for longer l-mer sizes, which is depicted in Figure 10a.
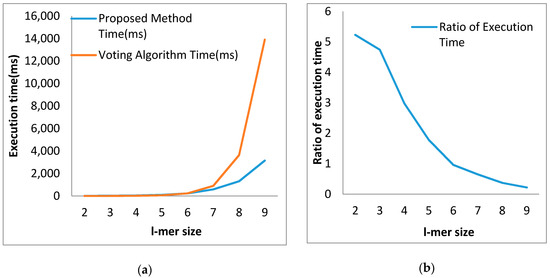
Figure 10.
Illustration in terms of execution time (a) Comparative illustration of execution time between the proposed algorithm and voting algorithm; (b) The ratio of execution time between the proposed algorithm and voting algorithm.
Figure 10b depicts the scenario well. The execution time ratio starts at 5.23 for l-mer size 2. For l-mer size 6, it decreases to less than 1, actually to 0.96. From this level, our proposed algorithm becomes faster. Decreasing gradually, the execution time ratio becomes 0.22 for l-mer size 9. From this finding, we can claim that the fastness of the proposed algorithm rises with the increase in l-mer size.
5. Conclusions
The median string is an enumeration-based algorithm. The algorithm exhaustively searches the entire search space to find the consensus sequence. As a result, it has become an exponential-time algorithm. The number of candidate l-mers increases with the rise of the l-mer size. As a result, the execution time also increases with l-mer size. As a result, the existing algorithm takes a long time to detect longer l and is inefficient for longer sequences. The only advantage is that it can guarantee that it must find the consensus. Our proposed Markov chain-based median string algorithm produces a subset of the 4l number of l-mers, and does the same operation as the existing algorithm with a reduced number of l-mers. Since the search has been reduced, this algorithm takes less time to execute. We compared the proposed algorithm with the recently developed voting algorithm. This comparison illustrates that the proposed phenomena can eradicate the inefficiency of the median string algorithm in the case of longer l-mer. The proposed algorithm can guarantee that it must find the consensus; however, it does not use an exhaustive search approach rather a stochastic approach. The execution time of the proposed algorithm does not grow exponentially with the rise of l-mer length. In the field of gene regulatory motif search, position weight metric-based algorithms take a shorter time to execute but can not guarantee that they can certainly find consensus. Enumeration-based algorithms can guarantee this, but take exponential time. The proposed algorithm combined both of these approaches and eradicated both method’s disadvantages. In this research, we calculated all the samples as a whole. In the future, however, we have the plan to execute an experiment by taking each sample’s Markov chain and transition matrix individually, then superimpose the transition matrices one after another. From that superimposed matrix some rules could be generated.
Author Contributions
All authors contributed equally to the conception of the idea, the design of experiments, the analysis and interpretation of results, and the writing and improvement of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A. Mathematical Proof That the System Will Work
From the transition matrix in the Figure 8, we have seven encircled elements. There are sixteen elements for sixteen possible transitions. The first element of the first row means there are 49 transitions from nucleotide a to a, the second element of the first row indicates that there is 39 transitions from nucleotide a to c. We have seven encircled transition frequencies. These are the top frequencies of transition. Based on these top frequencies, we can generate a probability matrix as follows. Using the encircled frequencies, we generated some transition rules. Using those rules, we generated some motifs. Let us consider our l-mer size as 5. Then, our system generates only 84 motifs of length 5. Let us put these motifs in set X = {set of motifs generated by the proposed system}. On the other hand, the median string algorithm generates all the 45 = 1024 motifs of length 5. Let us put these motifs in a set Y = {motifs generated by median string}. Now x is a subset of y. Basically,
where,
- X = {set of motifs generated by the proposed system},
- W = {set of motifs generated by the rules based on frequencies which are not encircled},
- Z = {set of motifs generated by the rules based on frequencies both encircled and not encircled}.
We can calculate the probability of a particular motif of length 5 using the theory of conditional probability
For l-mer of length 5 we can write,
Since this equation is a multiplication, and the frequencies that are not encircled are smaller than those that are encircled, the conditional probability of occurrence of any motif from the set W must be less than the probability of occurrence of the motif with the highest conditional probability from the set X. In addition, the conditional probability of occurrences of any motif from the set Z will be smaller than the probability of occurrences of the motif with highest conditional probability from the set X. The motif with the highest conditional probability will be the consensus. Since the motifs in set X are generated using the most frequent di-grams, the motif with the highest conditional probability will always be found in set X.
In our test data set, we found ctggt as the consensus for l-mer size 5 using both methods. Now, according to our claim, ctggt should have the highest conditional probability. Using Equation (2), we found the conditional probability of ctggt as follows:
This is the highest conditional probability, among all the 1024 motifs generated by the median string algorithm. The proposed system generates only 84 motifs including the consensus motif ctggt.
References
- Kellis, M.; Patterson, N.; Birren, B.; Berger, B.; Lander, E.S. Methods in Comparative Genomics: Genome Correspondence, Gene Identification and Regulatory Motif Discovery. J. Comput. Boil. 2004, 11, 319–355. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Schneider, T.D. Consensus sequence Zen. Appl. Bioinform. 2002, 1, 111–119. [Google Scholar]
- Lawrence, C.E.; Altschul, S.F.; Boguski, M.S.; Liu, J.S.; Neuwald, A.F.; Wootton, J.C. Detecting subtle sequence signals: A Gibbs sampling strategy for multiple alignment. Science 1993, 262, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. In Proceedings of the International Conference on Intelligent Systems for Molecular Biology, Stanford, CA, USA, 14–17 August 1994; pp. 28–36. [Google Scholar]
- Zhang, Y.; Huo, H.; Yu, Q. A heuristic cluster-based em algorithm for the planted (l, d) problem. J. Bioinform. Comput. Boil. 2013, 11, 1350009. [Google Scholar] [CrossRef] [PubMed]
- Kuksa, P.; Pavlovic, V. Efficient motif finding algorithms for large-alphabet inputs. BMC Bioinform. 2010, 11 (Suppl. S8), S1. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.; Lipman, D. Trees, stars, and multiple sequence alignment. SIAM J. Appl. Math. 1989, 49, 197–209. [Google Scholar] [CrossRef]
- Gramm, J.; Hüffner, F.; Niedermeier, R. Closest strings, primer design, and motif search. In Proceedings of the 6th Annual International Conference on Computational Biology(RECOMB 2002), Washington, DC, USA, 18–21 April 2002; pp. 74–75. [Google Scholar]
- Gramm, J.; Niedermeier, R.; Rossmanith, P. Exact solutions for closest string and related problems. In Proceedings of the 12th International Symposium on Algorithms and Computation, Christchurch, New Zealand, 19–21 December 2001; pp. 441–453. [Google Scholar]
- Karp, R.M. Mapping the genome: Some combinatorial problems arising in molecular biology. In Proceedings of the 25th Annual ACM Symposium on Theory of Computing, San Diego, CA, USA, 16–18 May 1993; pp. 278–285. [Google Scholar]
- Li, M.; Ma, B.; Wang, L. On the closest string and substring problems. J. ACM 2002, 49, 157–171. [Google Scholar] [CrossRef]
- Mauch, H.; Melzer, M.J.; Hu, J.S. Genetic algorithm approach for the closest string problem. In Proceedings of the 2nd IEEE Computer Society Bioinformatics Conference, Stanford, CA, USA, 11–14 August 2003; pp. 560–561. [Google Scholar]
- Meneses, C.N.; Lu, Z.; Oliveira, C.A.S.; Pardalos, P.M. Optimal Solutions for the Closest-String Problem via Integer Programming. INFORMS J. Comput. 2004, 16, 419–429. [Google Scholar] [CrossRef]
- Nicolas, F.; Rivals, E. Complexities of the centre and median string problems. In Proceedings of the 14th Symposium on Combinatorial Pattern Matching, Michoacan, Mexico, 25–27 June 2003; pp. 315–327. [Google Scholar]
- Gramm, J.; Niedermeier, R.; Rossmanith, P. Fixed-Parameter Algorithms for Closest String and Related Problems. Algorithmica 2003, 37, 25–42. [Google Scholar] [CrossRef]
- Ma, B.; Sun, X. More efficient algorithms for closest string and substring problems. In Proceedings of the 12th Annual International Conference on Research in Computational Molecular Biology, Singapore, 30 March–2 April 2008; pp. 396–409. [Google Scholar]
- Stojanovic, N.; Berman, P.; Gumucio, D.; Hardison, R.; Miller, W. A linear-time algorithm for the 1-mismatch problem. In Proceedings of the 5th International Workshop on Algorithms and Data Structures, NS, Canada, 6–8 August 1997; pp. 126–135. [Google Scholar]
- Ben-Dor, A.; Lancia, G.; Perone, J.; Ravi, R. Banishing bias from consensus sequences. In Proceedings of the 8th Symposium on Combinatorial Pattern Matching, Aarhus, Denmark, 30 June–2 July 1997; pp. 247–261. [Google Scholar]
- Gasieniec, L.; Jansson, J.; Lingas, A. Efficient approximation algorithms for the Hamming center problem. In Proceedings of the 10th ACM-SIAM Symposium on Discrete Algorithms, Baltimore, MD, USA, 17–19 January 1999; pp. 905–906. [Google Scholar]
- Gąsieniec, L.; Jansson, J.; Lingas, A. Approximation algorithms for Hamming clustering problems. J. Discret. Algorithms 2004, 2, 289–301. [Google Scholar] [CrossRef]
- Lanctot, J.K.; Li, M.; Ma, B.; Wang, S.; Zhang, L. Distinguishing string selection problems. Inf. Comput. 2003, 185, 41–55. [Google Scholar] [CrossRef]
- Boucher, C.; Brown, D.; Durocher, S. On the structure of small motif recognition instances. In Proceedings of the 15th Symposium on String Processing and Information Retrieval, Melbourne, Australia, 11–12 November 2008; pp. 269–281. [Google Scholar]
- Sze, S.; Lu, S.; Chen, J. Integrating sample-driven and pattern-driven approaches in motif finding. In Proceedings of the 4th Workshop on Algorithms in Bioinformatics, Bargen, Norway, 17–21 September 2004; pp. 438–449. [Google Scholar]
- Fatma, A.H.; Mai, S.M.; Walid, A.A. Review of different sequence motif finding algorithms. Avicenna J. Med. Biotechnol. 2019, 11, 130–148. [Google Scholar]
- Sun, H.Q.; Low, M.Y.H.; Hsu, W.J.; Tan, C.W.; Rajapakse, J.C. Tree-structured algorithm for long weak motif discovery. Bioinformatics 2011, 27, 2641–2647. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.; Carson, M.B.; Wang, Y.; Lin, Y.; Lu, H. A New Exhaustive Method and Strategy for Finding Motifs in ChIP-Enriched Regions. PLoS ONE 2014, 9, e86044. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, S.; Sahni, S.; Rajasekaran, S. PMS6: A fast algorithm for motif discovery. Int. J. Bioinform. Res. Appl. 2014, 10, 369. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S. Improved Exact Enumerative Algorithms for the Planted (l, d)-Motif Search Problem. IEEE/ACM Trans. Comput. Boil. Bioinform. 2014, 11, 361–374. [Google Scholar] [CrossRef] [PubMed][Green Version]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).