Tripodal Podand Ligand with a Superhalogen Nature as an Effective Molecular Trap


Abstract
:1. Introduction
2. Computational Methods
3. Results
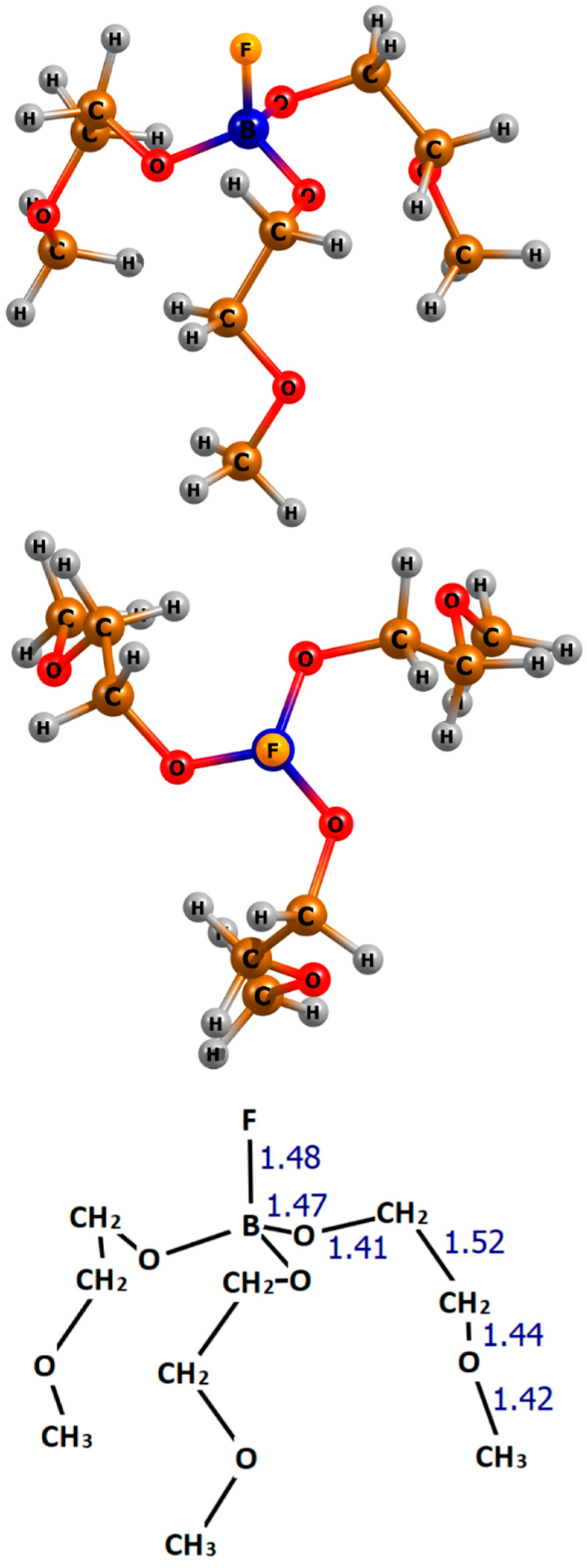
3.1. Structure and Stability of TMEFA
3.2. Complexes of TMEFA with Li+ and Mg2+ Ions
4. Conclusions
- The TMEFA system is a strongly electronically bound anionic compound with a superhalogen nature, thermodynamically stable against decomposition reactions, whose vertical electron detachment energy was found to be 5.72 eV.
- The equilibrium structure of TMEFA resembles those adopted by many well-known tripodal podand ligands, with a negatively charged cavity created by three open-ended 2-methoxyethoxy groups which allows for binding of selected metal ions (Li+ and Mg2+) inside the molecular “arms”.
- The examined ionic complexes formed by TMEFA, i.e., [F-B(O-CH2-CH2-O-CH3)3]−/Li+ and [F-B(O-CH2-CH2-O-CH3)3]−/Mg2+ systems, are characterized by large ligand–metal binding energies (180.1 and 457.3 kcal/mol, respectively) that far exceed those predicted for the coordination complexes formed by other representative podands (either tetradentate or tridentate).
- Due to the anticipated ease of its synthesis and expected high affinity to various metal cations, TMEFA represents a promising novel podand ligand which might be utilized as an effective molecular trap and/or a steric shielding agent with respect to selected ions.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hotop, H.; Lineberger, W.C. Binding energies in atomic negative ions: II. J. Phys. Chem. Ref. Data 1985, 14, 731–750. [Google Scholar] [CrossRef] [Green Version]
- Gutsev, G.L.; Boldyrev, A.I. DVM-Xα calculations on the ionization potentials of MXk+1− complex anions and the electron affinities of MXk+1 “superhalogens”. Chem. Phys. 1981, 56, 277–283. [Google Scholar] [CrossRef]
- Gutsev, G.L.; Boldyrev, A.I. The electronic structure of superhalogens and superalkalies. Russ. Chem. Rev. 1987, 56, 519–531. [Google Scholar] [CrossRef]
- Sikorska, C.; Skurski, P. The saturation of the excess electron binding energy in AlnF3n+1− (n = 1−5) anions. Chem. Phys. Lett. 2012, 536, 34–38. [Google Scholar] [CrossRef]
- Sobczyk, M.; Sawicka, A.; Skurski, P. Theoretical search for anions possessing large electron binding energies. Eur. J. Inorg. Chem. 2003, 2003, 3790–3797. [Google Scholar] [CrossRef]
- Freza, S.; Skurski, P. Enormously large (approaching 14 eV!) electron binding energies of [HnFn+1]− (n = 1−5, 7, 9, 12) anions. Chem. Phys. Lett. 2010, 487, 19–23. [Google Scholar] [CrossRef]
- Czapla, M.; Ciepła, O.; Brzeski, J.; Skurski, P. Formation of enormously strongly bound anionic clusters predicted in binary superacids. J. Phys. Chem. A 2018, 122, 8539–8548. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-B.; Ding, C.-F.; Wang, L.-S.; Boldyrev, A.I.; Simons, J. First experimental photoelectron spectra of superhalogens and their theoretical interpretations. J. Chem. Phys. 1999, 110, 4763–4771. [Google Scholar] [CrossRef] [Green Version]
- Alexandrova, A.N.; Boldyrev, A.I.; Fu, Y.-J.; Yang, X.; Wang, X.-B.; Wang, L.-S. Structure of the NaxClx+1− (x = 1−4) clusters via ab initio genetic algorithm and photoelectron spectroscopy. J. Chem. Phys. 2004, 121, 5709–5719. [Google Scholar] [CrossRef] [Green Version]
- Anusiewicz, I. Mg2Cl5− and Mg3Cl7− superhalogen anions. Aust. J. Chem. 2008, 61, 712–717. [Google Scholar] [CrossRef]
- Smuczyńska, S.; Skurski, P. Is hydrogen capable of playing a central atom role in superhalogen anions? Chem. Phys. Lett. 2007, 443, 190–193. [Google Scholar] [CrossRef]
- Gutsev, G.L. A theoretical investigation on the structure of the hypervalent carbon and silicon pentahalogenides as well as their singly charged anions. Chem. Phys. 1992, 166, 57–68. [Google Scholar] [CrossRef]
- Smuczyńska, S.; Skurski, P. Halogenoids as ligands in superhalogen anions. Inorg. Chem. 2009, 48, 10231–10238. [Google Scholar] [CrossRef] [PubMed]
- Anusiewicz, I. Superhalogen anions utilizing acidic functional groups as ligands. J. Phys. Chem. A 2009, 113, 11429–11434. [Google Scholar] [CrossRef] [PubMed]
- Anusiewicz, I. Electrophilic substituents as ligands in superhalogen anions. J. Phys. Chem. A 2009, 113, 6511–6516. [Google Scholar] [CrossRef]
- Paduani, C.; Wu, M.M.; Willis, M.; Jena, P. Theoretical study of the stability and electronic structure of Al(BH4)n=1→4 and Al-(BF4)n=1→4 and their hyperhalogen behavior. J. Phys. Chem. A 2011, 115, 10237–10243. [Google Scholar] [CrossRef]
- Czapla, M. Dinuclear superhalogen anions containing two different central atoms. J. Fluor. Chem. 2017, 199, 97–102. [Google Scholar] [CrossRef]
- Yang, H.; Li, Y.; He, H.-M.; Tong, J.; Wu, D.; Li, Z.-R. Superhalogen properties of hetero-binuclear anions MM′F4− and MM″F5− (M = Li, Na, M′ = Be, Mg, Ca; M″ = B, Al, Ga). Chem. Phys. Lett. 2017, 684, 273–278. [Google Scholar] [CrossRef]
- Sikorska, C.; Skurski, P. Moderately reactive molecules forming stable ionic compounds with superhalogens. Inorg. Chem. 2011, 50, 6384–6391. [Google Scholar] [CrossRef]
- Marchaj, M.; Freza, S.; Rybacka, O.; Skurski, P. Superhalogen oxidizers capable of ionizing water molecules. Chem. Phys. Lett. 2013, 574, 13–17. [Google Scholar] [CrossRef]
- Czapla, M.; Freza, S.; Skurski, P. Ionizing benzene with superhalogens. Chem. Phys. Lett. 2015, 619, 32–35. [Google Scholar] [CrossRef]
- Czapla, M.; Skurski, P. Oxidizing CO2 with superhalogens. Phys. Chem. Chem. Phys. 2017, 19, 5435–5440. [Google Scholar] [CrossRef] [PubMed]
- Sikorska, C. Oxidizing metal oxides with polynuclear superhalogen: An ab initio study. J. Phys. Chem. A 2018, 122, 7328–7338. [Google Scholar] [CrossRef] [PubMed]
- Czapla, M.; Skurski, P. The existence and gas phase acidity of the HAlnF3n+1 superacids (n = 1−4). Chem. Phys. Lett. 2015, 630, 1–5. [Google Scholar] [CrossRef]
- Czapla, M.; Skurski, P. Strength of the Lewis−Brønsted superacids containing In, Sn, and Sb and the electron binding energies of their corresponding superhalogen anions. J. Phys. Chem. A 2015, 119, 12868–12875. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.K.; Misra, N. Hydrogenated superhalogens behave as superacids. Polyhedron 2015, 102, 711–714. [Google Scholar] [CrossRef]
- Czapla, M.; Anusiewicz, I.; Skurski, P. Does the protonation of superhalogen anions always lead to superacids? Chem. Phys. 2016, 465−466, 46–51. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Kumar, A.; Misra, N. Superhalogens as building blocks of a new series of superacids. New J. Chem. 2017, 41, 5445–5449. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Kumar, A.; Misra, N. A path to design stronger superacids by using superhalogens. J. Fluor. Chem. 2017, 197, 59–62. [Google Scholar] [CrossRef]
- Zhou, F.-Q.; Xu, W.-H.; Li, J.-F.; Zhao, R.-F.; Yin, B. The combination of superhalogens and Brønsted acids HX (X = F, Cl, Br): An effective strategy for designing strong superacids. Inorg. Chem. 2017, 56, 11787–11797. [Google Scholar] [CrossRef] [PubMed]
- Anusiewicz, I.; Freza, S.; Skurski, P. Stability of the TinF4n+1− and GenF4n+1− superhalogen anions and the acidity of the HTinF4n+1 and HGenF4n+1 (n = 1−3) superacids. Polyhedron 2018, 144, 125–130. [Google Scholar] [CrossRef]
- Zhao, R.F.; Zhou, F.-Q.; Xu, W.; Li, J.F.; Li, C.-C.; Li, J.; Yin, B. Superhalogen-based composite with strong acidity: A crossing point between two topics. Inorg. Chem. Front. 2018, 5, 2934–2947. [Google Scholar] [CrossRef]
- Brzeski, J.; Czapla, M.; Skurski, P. Icosahedral carborane superacids and their conjugate bases comprising H, F, Cl, and CN substituents: A theoretical investigation of monomeric and dimeric cages. ChemPlusChem 2020, 85, 312–318. [Google Scholar] [CrossRef]
- Sikorska, C. Magnesium-based clusters as building blocks of electrolytes in lithium-ion batteries. ChemPhysChem 2019, 20, 2236–2246. [Google Scholar] [CrossRef] [PubMed]
- Giri, S.; Behera, S.; Jena, P. Superhalogens as building blocks of halogen-free electrolytes in lithium-ion batteries. Angew. Chem. Int. Ed. 2014, 53, 13916–13919. [Google Scholar] [CrossRef]
- Jena, P. Superhalogens: A bridge between complex metal hydrides and Li ion batteries. J. Phys. Chem. Lett. 2015, 6, 1119–1125. [Google Scholar] [CrossRef]
- Fang, H.; Jena, P. Super-ion inspired colorful hybrid perovskite solar cells. J. Mater. Chem. A 2016, 4, 4728–4737. [Google Scholar] [CrossRef]
- Fang, H.; Jena, P. Molecular origin of properties of organic–inorganic hybrid perovskites: The big picture from small clusters. J. Phys. Chem. Lett. 2016, 7, 1596–1603. [Google Scholar] [CrossRef]
- Welton, T. Room-temperature ionic liquids. Solvents for synthesis and catalysis. Chem. Rev. 1999, 99, 2071–2084. [Google Scholar] [CrossRef]
- Czapla, M. Polynuclear Li12F13− anion as a steric shielding agent with respect to selected metal ions. Theor. Chem. Acc. 2016, 135, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Blair, S.M.; Brodbelt, J.S.; Marchand, A.P.; Chong, H.S.; Alihodžić, S. Evaluation of alkali metal binding selectivities of caged aza-crown ether ligands by microelectrospray ionization/quadrupole ion trap mass spectrometry. J. Am. Soc. Mass Spectrom. 2000, 11, 884–891. [Google Scholar] [CrossRef]
- Kumondai, K.; Toyoda, M.; Ishihara, M.; Katakuse, I.; Takeuchi, T.; Ikeda, M.; Iwamoto, K. Experimental and theoretical study on gas-phase ion/molecule reactions of silver trimer cation, Ag+3, with 12-crown-4. J. Chem. Phys. 2005, 123, 024314. [Google Scholar] [CrossRef] [PubMed]
- Inokuchi, Y.; Ebata, T.; Ikeda, T.; Haino, T.; Kimura, T.; Guo, H.; Furutani, Y. New insights into metal ion–crown ether complexes revealed by SEIRA spectroscopy. New J. Chem. 2015, 39, 8673–8680. [Google Scholar] [CrossRef] [Green Version]
- Allen, O.R.; Field, L.D.; Magill, A.M.; Vuong, K.Q.; Bhadbhade, M.M.; Dalgarno, S.J. Ruthenium complexes of CP3: A new carbon-centered polydentate podand ligand. Organometallics 2011, 30, 6433–6440. [Google Scholar] [CrossRef]
- Inoue, Y. Cation Binding by Macrocycles: Complexation of Cationic Species by Crown Ethers; CRC Press: New York, NY, USA, 1990. [Google Scholar]
- Weber, E. Encyclopedia of Supramolecular Chemistry; Atwood, J.L., Steed, J.W., Eds.; Taylor & Francis: Boca Raton, MA, USA, 2004; p. 1106. [Google Scholar]
- Weber, E.; Vögtle, F. Classification and nomenclature of coronands, cryptands, podands, and of their complexes. Inorg. Chim. Acta 1980, 45, L65–L67. [Google Scholar] [CrossRef]
- Gierczyk, B.; Pankiewicz, R. Supramolecular complexes of podand ligands with xenon. Cent. Eur. J. Chem. 2014, 12, 624–634. [Google Scholar] [CrossRef]
- Filby, M.H.; Steed, J.W. A modular approach to organic, coordination complex and polymer based podand hosts for anions. Coord. Chem. Rev. 2006, 250, 3200−3218. [Google Scholar] [CrossRef]
- Head-Gordon, M.; Pople, J.A.; Frisch, M.J. MP2 energy evaluation by direct methods. Chem. Phys. Lett. 1988, 153, 503–506. [Google Scholar] [CrossRef]
- Frisch, M.J.; Head-Gordon, M.; Pople, J.A. Direct MP2 gradient method. Chem. Phys. Lett. 1990, 166, 275–280. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H., Jr. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef] [Green Version]
- Woon, D.E.; Dunning, T.H., Jr. Gaussian-basis sets for use in correlated molecular calculations. 3. The atoms aluminum through argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef] [Green Version]
- Petersson, G.A.; Bennett, A.; Tensfeldt, T.G.; Al-Laham, M.A.; Shirley, W.A.; Mantzaris, J. A complete basis set model chemistry. I. The total energies of closed-shell atoms and hydrides of the first-row atoms. J. Chem. Phys. 1998, 89, 2193–2218. [Google Scholar] [CrossRef]
- Montgomery, J.A., Jr.; Ochterski, J.W.; Petersson, G.A. A complete basis set model chemistry. IV. An improved atomic pair natural orbital method. J. Chem. Phys. 1994, 101, 5900–5909. [Google Scholar] [CrossRef]
- Petersson, G.A.; Tensfeldt, T.G.; Montgomery, J.A., Jr. A complete basis set model chemistry. III. The complete basis set-quadratic configuration interaction family of methods. J. Chem. Phys. 1991, 94, 6091–6101. [Google Scholar] [CrossRef]
- Zakrzewski, V.G.; Ortiz, J.V.; Nichols, J.A.; Heryadi, D.; Yeager, D.L.; Golab, J.T. Comparison of perturbative and multiconfigurational electron propagator methods. Int. J. Quantum Chem. 1996, 60, 29–36. [Google Scholar] [CrossRef]
- Simons, J. Direct calculation of first- and second-order density matrices. The higher RPA method. J. Chem. Phys. 1971, 55, 1218–1230. [Google Scholar] [CrossRef]
- Ortiz, J.V. Electron binding energies of anionic alkali metal atoms from partial fourth order electron propagator theory calculations. J. Chem. Phys. 1988, 89, 6348–6352. [Google Scholar] [CrossRef]
- Cederbaum, L.S. One-body Green’s function for atoms and molecules: Theory and application. J. Phys. B At. Mol. Phys. 1975, 8, 290–303. [Google Scholar] [CrossRef]
- Zakrzewski, V.G.; Ortiz, J.V. Semidirect algorithms for thirdorder electron propagator calculations. Int. J. Quantum Chem. 1995, 53, 583–590. [Google Scholar] [CrossRef]
- Simons, J.; Smith, W.D. Theory of electron affinities of small molecules. J. Chem. Phys. 1973, 58, 4899–4907. [Google Scholar] [CrossRef]
- Sikorska, C.; Ignatowska, D.; Freza, S.; Skurski, P. The performance of selected ab initio methods in estimating electron binding energies of superhalogen anions. J. Theor. Comput. Chem. 2011, 10, 93–109. [Google Scholar] [CrossRef]
- Zakrzewski, V.G.; Dolgounitcheva, O.; Ortiz, J.V. Ionization energies of anthracene, phenanthrene, and naphthacene. J. Chem. Phys. 1996, 105, 8748–8753. [Google Scholar] [CrossRef]
- Besler, B.H.; Merz, K.M., Jr.; Kollman, P.A. Atomic charges derived from semiempirical methods. J. Comput. Chem. 1990, 11, 431–439. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Foresman, J.B.; Fox, D.J. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- Sikorska, C.; Smuczyńska, S.; Skurski, P.; Anusiewicz, I. BX4− and AlX4− superhalogen anions (X = F, Cl, Br): An ab initio study. Inorg. Chem. 2008, 47, 7348–7354. [Google Scholar] [CrossRef] [PubMed]
- Smuczyńska, S.; Skurski, P. Introducing various ligands into superhalogen anions reduces their electronic stabilities. Chem. Phys. Lett. 2008, 452, 44–48. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Species | Symmetry | BE (kcal/mol) |
|---|---|---|
| [F-B(O-CH2-CH2-O-CH3)3]–/Li+ | C3 | 180.1 |
| [F-B(O-CH2-CH2-O-CH3)3]–/Mg2+ | C3 | 457.3 |
| [N(O-CH2-CH2-O-CH3)3Li]+ | C3 | 81.5 |
| [N(O-CH2-CH2-O-CH3)3Mg]2+ | C3 | 246.5 |
| [N(CH2-CH2-N=CH-CH3)3Li]+ | C1 | 104.4 |
| [N(CH2-CH2-N=CH-CH3)3Mg]2+ | C1 | 297.6 |
| [B(CH2-CH2-N=CH-CH3)3Li]+ | C1 | 97.0 |
| [B(CH2-CH2-N=CH-CH3)3Mg]2+ | C1 | 273.0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cyraniak, A.; Czapla, M. Tripodal Podand Ligand with a Superhalogen Nature as an Effective Molecular Trap. Symmetry 2020, 12, 1441. https://doi.org/10.3390/sym12091441
Cyraniak A, Czapla M. Tripodal Podand Ligand with a Superhalogen Nature as an Effective Molecular Trap. Symmetry. 2020; 12(9):1441. https://doi.org/10.3390/sym12091441
Chicago/Turabian StyleCyraniak, Adrianna, and Marcin Czapla. 2020. "Tripodal Podand Ligand with a Superhalogen Nature as an Effective Molecular Trap" Symmetry 12, no. 9: 1441. https://doi.org/10.3390/sym12091441