Abstract
Our strategy to analyze the structures of natural amino acids with respect to the interaction of three different elements of chirality within the molecules was applied to the non-natural amino acid (S)-α-phenylglycine, its analogue (S)-α-phenylpropionic acid, and the drug (S)-ibuprofen. The three chirality elements are the configuration at Cα, the conformation at the Cα-C’ bond, and the distortion of the planar carboxylic group to a flat asymmetric tetrahedron. In all three compounds, a given (S) configuration at Cα predominantly induces (M) conformation at the Cα-C’ bond, which in turn preferentially distorts the carboxylic group to a tetrahedron with (R) configuration. Both steps of this chirality chain display high selectivities. Due to varying co-crystallization partners, in all the structures the molecules are in different environments with respect to packing and hydrogen bonding. Nevertheless, the structural pattern and the diaselectivities of the chirality chain persist. For phenylglycine, DFT (Density Functional Theory) calculations confirm the structural results.
1. Introduction
In amino acids there is chirality and selectivity beyond the configuration at the α-carbon atom. Previously, we established an unprecedented chirality chain in the amino acids alanine [1] and valine [2]. Then, we showed that 1179 natural amino acids H3NCαHRC’(Ocis)OtransX [3] and 420 glycine derivatives H3NCαH2C’(Ocis)OtransX [4] follow the chirality chain. X indicates that the carboxylic group may be protonated, esterified or coordinated. The chirality chain consists of three different elements of chirality: (1) the Cα configuration, which in natural amino acids is L, (2) the conformation concerning rotation around the central C’-Cα bond, and (3) the distortion of the planar carboxylic group CαC’OcisOtrans to a flat asymmetric tetrahedron with four different corners. The chirality chain involves two induction steps. The L configuration at Cα induces preferably (Mψ) conformation at the C’-Cα bond with small negative rotation angles ψ = OcisC’CαN and the (Mψ) conformation distorts the planar carboxylic group preferentially to a flat tetrahedron OcisOtransC’Cα with (Rθ) configuration. Both steps show high diastereoselectivities. The arrows in Figure 1 point to the three different elements of chirality.
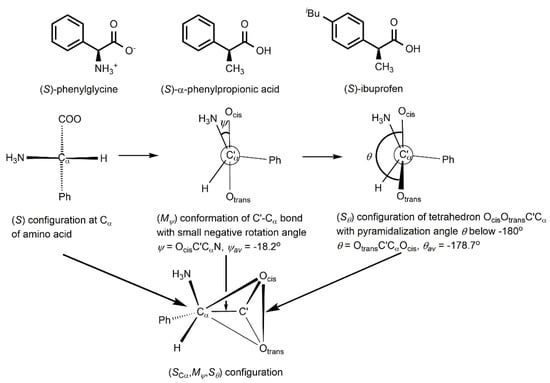
Figure 1.
Formulas of (S)-phenylglycine, (S)-α-phenylpropionic acid, and (S)-ibuprofen and chirality chain of the amino acid (S)-phenylglycine: for a given (S) configuration at Cα, chiral induction favors (Mψ) conformation of the C’-Cα bond and (Sθ) configuration of the flat tetrahedron OcisOtransC’Cα.
Worldwide, international companies collect data to establish profiles for commercial use. Data compiled in the Cambridge Structural Database (CSD) have been used to find chemical relationships for a long time. Seminal contributions identified energy minima and even reaction pathways using single crystal X-ray data [5,6,7]. Stimulated by the progress in X-ray crystallography, today, a wealth of CSD structures is available for exploitation. We used this information for an analysis of phenylglycine, α-phenylpropionic acid, and ibuprofen with respect to chiral selectivities within the molecules.
2. Results and Discussion
2.1. Analysis of (S)-Phenylglycine Structures
After having analyzed the natural amino acids [1,2,3,4], we turned to the non-natural amino acid phenylglycine H3N-CHPh-C’(O)O and its derivatives H3N-CHPh-COO(H/R/M) protonated, esterified or coordinated at the carboxylic group. A search in the Cambridge Structural Database and seven new structure determinations, added from our side, gave 25 hits. Eight structures contain independent molecules, differentiated by numbers in parentheses after the CSD symbol. Thus, 33 different structures are available for analysis, summarized in Table S1 (Supplementary Materials). (R)-Phenylglycine configurations were inverted to (S) by inverting the rotation and pyramidalization angles, and in the following discussion, we only use (S)-phenylglycine configurations.
In the present paper we truncated Table S1 to Table 1 with eight entries, which show the relevant trends. Ordering is done according to decreasing negative rotation angles ψ. Table 1 includes the first and the last entry of Table S1 and all the multiples of 5. As structures, determined several times, should have the same weight as structures determined only once, we average rotation angles ψ and pyramidalization angles θ for multiple structure determinations, indicating the number of independent structure determinations in the footnotes of the Tables.

Table 1.
Phenylglycine (S)-NH3CH(Ph)COO and its derivatives (S)-NH3CH(Ph)COO(H/R/M) protonated, esterified, and coordinated at the carboxylic group.
The rotation angle is defined as the torsion angle ψ = Ocis-C’-Cα-N (Figure 1, top middle). We differentiate the oxygen atoms of the carboxylic group as Ocis and Otrans according to their orientation with regard to the nitrogen atom. Four-atom-systems, such as OcisC’CαN, are chiral, unless the torsion angle ψ is 0° or 180°. According to the helicity rule of the CIP system [8,9], positive rotation angles ψ result in (Pψ) conformation and negative in (Mψ) conformation (Figure 1, top middle).
The carboxylic group CαC’OcisOtrans with its sp2-hybridized C’ atom should be planar and achiral. Deviation from planarity results in a flat asymmetric tetrahedron with four different corners: Cα, C’, Ocis, and Otrans. The pyramidalization angle θ = Otrans-C’-Cα-Ocis measure the deviation of Ocis from the plane OtransC’Cα (Figure 1, top right). The configuration of the flat tetrahedron is specified by the (R)/(S) symbols of the CIP system [8,9], following the priority sequence Ocis > Otrans > C’ > Cα (Figure 1, top right). Pyramidalization angles θ below −180° define (Rθ) configuration and pyramidalization angles θ above −180° indicate (Sθ) configuration at the flat tetrahedron.
The rotation angles ψ in Table 1 and Table S1 extend between −34.2° and −4.0°. Cα-C’ bond conformations ψ in the range −40° to −90° are absent. Small negative rotation angles ψ had been typical for all the natural amino acids, favoring (SCα,Mψ) compared to a (SCα,Pψ) configuration [1,2,3,4]. For a given (SCα) configuration in phenylglycine, there are no positive rotation angles. Thus, the diastereoselectivity (SCα→Mψ):(SCα→Pψ) = 33:0 in the first step of the chirality chain from the (SCα) configuration to the (Mψ) conformation is 100%. The predominance of small negative rotation angles for a given (S) configuration is also expressed in the average ψav = −17.9° of the 33 structures in Table S1. Figure 2 shows a plot of the rotation angles ψ on the abscissa versus the frequency of occurrence of the rotation angles ψ in percent on the ordinate. In each of the 33 entries of Table S1, the (S)-phenylglycine molecule is in completely different environments with respect to lattice effects and hydrogen bond networks. This is demonstrated in the Comments of Table 1 and Table S1, in which its co-crystallization partners are listed. The appearance of a common structural motif, irrespective of the changing environment, indicates an energy minimum, represented by the envelope in Figure 2, which covers the columns representing the frequency of occurrence. Obviously, such a robust pattern is competitive with packing forces and intermolecular hydrogen bonding.
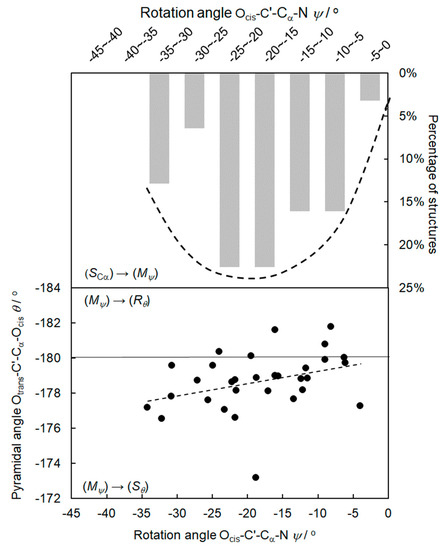
Figure 2.
Upper part: frequency of occurrence of (S)-phenylglycine structures in percent as a function of the rotation angle ψ. Each of the columns represents a 5° interval of ψ such as 0° to −5°, −5° to −10°, etc. Lower part: rotation angles ψ versus pyramidalization angles θ of the 33 structures of (S)-phenylglycine of Table S1 and the best-fit line.
Table S1 and Figure 3 show that the pyramidalization angles θ concentrate below −180°, in particular for small negative rotation angles ψ. The preference of pyramidalization angles θ below −180° also shows up in the average θav = −178.7° of the 33 structures in Table S1.
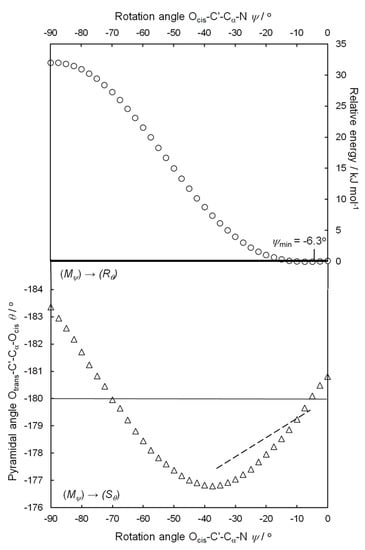
Figure 3.
Upper part: rotation angle ψcalc as a function of relative energy (○), calculated at the B3LYP/def2TZVP level in steps of 2.5°. Lower part: rotation angle ψcalc as a function of pyramidalization angle θcalc (△). The dashed line is the best-fit line of the structures in Figure 2.
The distortion of the carboxylic group to a flat asymmetric tetrahedron is induced by the (S) configuration at Cα and the (Mψ) conformation of the Cα-C’ bond. As for (S)-phenylglycine, there are no positive rotation angles ψ; the selectivities (SCα)→(Rθ,Sθ) and (Mψ)→(Rθ,Sθ) are identical. In numbers, the overall selectivity for (S)-phenylglycine is (Mψ)→(Rθ):(Mψ)→(Sθ) = 27:6 = 4.6. Thus, there is a considerable selectivity for the side of the (Rθ) configuration of the flat tetrahedron OcisOtransC’Cα.
The height of the columns, the ψ/θ values, and the selectivity ratios in Figure 2 are experimental results, based on the available CSD structures. The best-fit line of the X-ray structural data in Figure 2, lower part, shows that the pyramidalization of the carboxylic group increases, when the rotation angles increase from ψ = −4.0° to higher negative rotation angles.
2.2. DFT Calculations of (S)-Phenylglycine
DFT calculations of the zwitterion of (S)-phenylglycine H3N-CαHPh-C’(Ocis)Otrans were carried out, using the B3LYP/def2TZVP level, including dispersion (GD3BJ) and solvent effects (PCM; water) [10,11,12,13,14,15,16,17,18]. Details of the calculations and Table S2 with the numerical parameters are given in the Supplementary Materials. Figure 3, upper part, shows the rotation angle ψcalc as a function of the relative energy. In 2.5° steps, the calculation extends from ψcalc = −90° to 0°.
The relative energy of (S)-phenylglycine (open circles) is zero at ψcalc = −6.3°, which compares well with the average ψav = −17.9° of the 33 structures in Table S1. The small negative rotation angles are the result of the chiral induction from the (S) configuration at Cα to the (Mψ) conformation of the Cα-C’ bond in the first step of the chirality chain [1,2,3,4]. With increasing negative rotation angles, it rises slowly up to ψcalc angles of −30°. Then, the increase becomes steeper, before it levels off at highly negative rotation angles ψcalc. At ψcalc = −90°, the relative energy is 32.0 kJ/mol higher than in the minimum, a remarkable energy difference for a rotation around a single bond. On the side of positive rotation angles, the curve continues almost symmetrically.
Figure 3, lower part, shows the pyramidalization angles θcalc as a function of the rotation angles ψcalc (triangles). For the rotation angle ψcalc = 0°, the pyramidal angle θcalc = −180.8° is in the regime of (Rθ) configuration of the flat tetrahedron OcisOtransC’Cα. Going to negative rotation angles, the curve steeply drops down to the minimum around ψcalc = −35° in the area of (Sθ) configuration. At higher negative rotation angles, the curve passes about ψcalc = −70°, the state of a planar carboxylic group with a pyramidalization angle θ = −180°. Then, the curve continues into the area of the inverted (Rθ) configuration of the carboxylic group. However, it must be kept in mind that structures with rotation angles between ψ = −40° and −90° have high relative energies, and for (S)-phenylglycine structures, such high rotation angles are absent. The remarkable and unexpected changes, which the pyramidalization of the carboxylic group does undergo during rotation around the Cα-C’ bond, comes as a surprise. The region between 0° and +90° is almost symmetrical to the region between 0° and −90°.
In Figure 3, we added the best-fit line of the structural data of (S)-phenylglycine. It follows the decrease of the calculated curve of negative rotation angles in remarkable agreement.
2.3. Analysis of (S)-α-Phenylpropionic Acid Structures
Replacement of the NH3 group in (S)-phenylglycine by a CH3 group results in (S)-α-phenylpropionate, which is the parent compound of important drugs. Is the two-step chirality chain, operative in amino acids, also working for (S)-α-phenylpropionic acid? We analyzed α-phenylpropionic acid (S)-H3C-CHPh-C’(O)OH and its derivatives (S)-H3NC-CHPh-COO(R/M) esterified or coordinated at the carboxylic group in a CSD search. Similar to Table S1 of (S)-α-phenylglycine, Table S3 contains 87 structures of (S)-α-phenylpropionic acids, ordered according to decreasing negative rotation angles ψ. Structures with (R) configurations were inverted to (S) configurations.
The rotation angles ψ of (S)-α-phenylpropionic acid in Table S3 range between −84.2° and 81.6°. Similar to (S)-phenylglycine, the rotation angles ψ accumulate in the area of small negative values. However, in contrast to (S)-phenylglycine, there are some highly negative and also some positive rotation angles. Therefore, the induction field (SCα)→(Mψ) of (S)-phenylglycine in Figure 2 doubles to the two halves (SCα)→(Mψ) and (SCα)→(Pψ) in Figure 4, upper part. The preference of small negative rotation angles for a given (S) configuration shows up in the average ψav = −22.1° of the 87 structures in Table S3. In the upper part of Figure 4 the rotation angles ψ are given as a function of their frequency of occurrence in percent in a column representation with the envelope corresponding to the energy minimum.
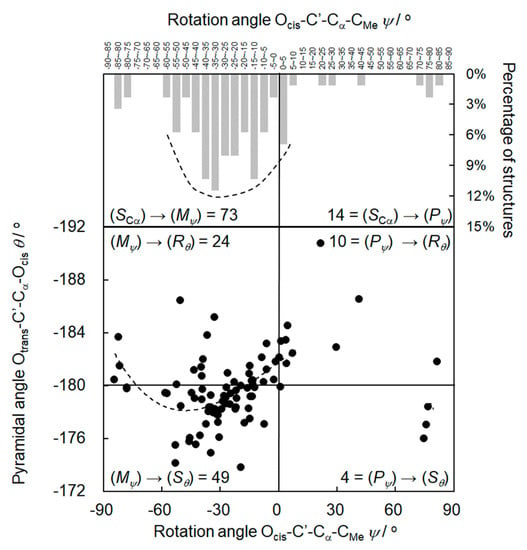
Figure 4.
Upper part: the rotation angles ψ of the (S)-α-phenylpropionic acids in Table S3 as a function of their frequency of occurrence in percent in a column representation with the envelope corresponding to the energy minimum. Lower part: the rotation angles ψ as a function of the pyramidalization angles θ, including the best-fit curve.
Table S3 shows that the (S) configuration of α-phenylpropionic acid confers a high chiral induction to the torsional system ψ = OC’CαN. The columns in Figure 4, upper part, visualize the high (SCα)→(Mψ) selectivity. Thus, 73 of a total of 87 structures adopt (Mψ) conformation with respect to the Cα-C’ bond. Only 14 structures have (Pψ) conformation, resulting in a selectivity (SCα)→(Mψ):(SCα)→(Pψ) = 73:14 = 5.2 in the first step of the chirality chain.
According to Table S3 and Figure 4, lower part, the pyramidalization angles θ concentrate below −180°, indicating (Sθ) configuration in the flat tetrahedron OcisOtransC’Cα, in particular for small negative rotation angles ψ. The concentration of the pyramidalization angles θ below −180° appears in the average θav = −178.9° of the 87 structures of Table S3. In the lower part of Figure 4, the (ψ)/(θ) representation expands to a four-field system with four different chiral inductions. The two fields on the left side of Figure 4 show the two diastereomers (Mψ,Sθ) and (Mψ,Rθ). The induction according to the second step of the chirality chain results in a diastereoselectivity (Mψ)→(Sθ):( Mψ)→(Rθ) = 49:24 = 2.0. (Mψ,Sθ) in the bottom left field and (Pψ,Rθ) in the top right field are enantiomers. Therefore, the inductions (Mψ)→(Sθ) and (Pψ)→(Rθ) should be the same. However, the ratio (Mψ)→(Sθ):(Pψ)→(Rθ) is 49:10 = 4.9, the deviation from 1:1 reflecting the contribution of the (S) configuration at Cα.
The best-fit curve in Figure 4, lower part, starts in the field (Mψ,Rθ) with (Rθ) configuration of the flat tetrahedron OcisOtransC’Cα. Then, the majority of structures with small negative rotation angles adopts (Sθ) configuration. At rotation angles above ψ = −70°, the curve again enters the area of (Rθ) configuration of the flat tetrahedron. Thus, the curve progression parallels the curve of the DFT calculation of (S)-phenylglycine in Figure 3.
The 87 structures of (S)-α-phenylpropionic acids in Table S3 accumulate at small negative rotation angles around the average ψav = −22.1°, indicating an energy minimum. For (S)-phenylglycine, there had been no CSD structures with higher negative rotation angles than ψ = −40°, which increasingly rise in energy content according to Figure 3. However, Figure 4 shows that a few structures of (S)-α-phenylpropionic acids accept high-energy conformations with rotation angles of ψav = −80° to −90°, probably due to strong packing forces and/or hydrogen bond networks. Interestingly, at these high conformational energies, the configuration of the flat tetrahedron tends to move from (Mψ)→(Sθ) to the regime of (Mψ)→(Rθ).
2.4. Analysis of the (S)-Ibuprofen Structures
Ibuprofen is the 4-isobutyl derivative of α-phenylpropionic acid. We searched for H3C-CH(4-iBuC6H4)-C’(O)OH and its derivatives H3C-CH(4-iBuC6H4)-COO(R/M) esterified or coordinated at the carboxylic group. Table S4 comprises 80 structures, in which (S)-ibuprofen crystallizes with different co-crystallization partners. Ordering is done according to decreasing negative rotation angles ψ. Structures with (R) configurations were inverted to (S) configurations.
Similar to the (S)-α-phenylpropionic acids, the rotation angles ψ of the (S)-ibuprofens in Table S4 span a wide range from −92.7° to 71.6°. The rotation angles ψ accumulate in the area of small negative values. As for the (S)-α-phenylpropionic acids, there are some positive and also some highly negative rotation angles. The average ψav = −28.0° of the 80 points in Table S4 emphasizes the preference of small negative rotation angles for a given (S) configuration. The upper part of Figure 5 shows the rotation angles ψ versus their frequency of occurrence in percent. The envelope enwrapping the columns corresponds to the energy minimum.
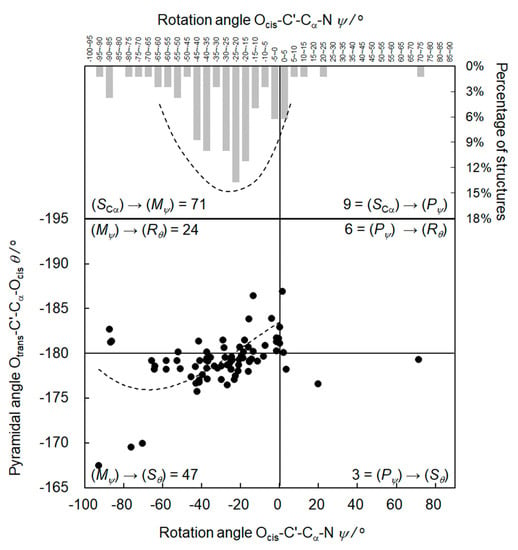
Figure 5.
Upper part: the rotation angles ψ of the (S)-ibuprofen structures in Table S4 versus their frequency of occurrence in percent in a column representation with the envelope corresponding to the energy minimum. Lower part: the rotation angles ψ versus the pyramidalization angles θ, including the best-fit curve.
Table S4 and Figure 5 confirm the high chiral induction from the (SCα) configuration of the (S)-ibuprofens to the (ψ) conformation of the Cα-C’ bond. The columns in Figure 5 visualize the high (SCα)→(Mψ) selectivity. Indeed, 71 of the 80 structures adopt (Mψ) conformation with respect to the Cα-C’ bond. Only nine structures have (Pψ) conformation, resulting in a selectivity of (SCα)→(Mψ):(SCα)→(Pψ) = 71:9 = 7.9 in the first step of the chirality chain.
As for the (S)-α-phenylpropionic acids, the pyramidalization angles θ in Table S5 and Figure 4, lower part, accumulate below −180°, indicating (Sθ) configuration in the flat tetrahedron OcisOtransC’Cα, in particular for small negative rotation angles ψ. Similar to the (S)-α-phenylpropionic acids, the diastereoselectivity in the second step of the chirality chain is (Mψ)→(Sθ):(Mψ)→(Rθ) = 49:24 = 2.0. Due to the chiral induction of the (S) configuration at Cα, the ratio of the two enantiomers (Mψ)→(Sθ):(Pψ)→(Rθ) is 47:6 = 7.8. The best-fit curve in Figure 4, lower part, resembles the curves of the (S)-α-phenylpropionic acid derivatives.
The results obtained for the (S)-ibuprofen structures closely resemble those of the (S)-α-phenylpropionic acids. The features established for the low-energy (Mψ) conformations of the Cα-C’ bond and the (Sθ) configurations of the flat tetrahedron OcisOtransC’Cα should persist in the solution, where ibuprofen performs its effectiveness.
3. Conclusions
(S)-Phenylglycine, (S)-α-phenylpropionic acid, and (S)-ibuprofen derivatives follow the two-step chirality chain, in which three elements of chirality selectively interact within the molecules. In the first step, (S) configuration at Cα preferentially induces (Mψ)-conformation at the Cα-C’ bond, tantamount to small negative rotation angles ψ. In the second step, there is a preferred formation of (Sθ) configuration in the four-atom system of the carboxylic group, which is distorted to a flat tetrahedron. The chirality chain in a drug such as ibuprofen reveals structural details, important for approach and binding to its receptor.
4. Materials and Methods
The Cambridge Structural Database ver. 5.41 [19] (update August 2020) was used for a search of phenylglycine, α-phenylpropionic acid, and ibuprofen derivatives. The programs OLEX2 [20], Mercury CSD ver. 2020.2.0 [21], and ConQuest ver. 2020.2.0 [22] were applied for the structure analyses. Single crystal preparation and X-ray diffraction analysis of (R)/(S)-, (R)-, and (S)-phenylglycine, (R)/(S)-, (R)-, and (S)-methyl phenylglycinate hydrochloride, as well as (R)-ethyl phenylglycinate hydrochloride are described in the Supplementary Materials.
Supplementary Materials
Available online at https://www.mdpi.com/2073-8994/13/1/55/s1: Table S1: Phenylglycine (S)-NH3CH(Ph)COO and its derivatives (S)-NH3CH(Ph)COO(H/R/M) protonated, esterified, and coordinated at the carboxylic group (structures with R factors >10% excluded). Table S2: (S)-Phenylglycine: relative energy as a function of the rotation angle ψ = Ocis-C’-Cα-N and the pyramidalization angle θ = Otrans-C’-Cα-Ocis, calculated at the B3LYP/def2-TZVP level, including dispersion (GD3BJ) and solvent effects (PCM; water). Table S3: α-Phenylpropionic acid (S)-CH3CH(Ph)COOH and its derivatives (S)-CH3CH(Ph)COO(R/M) esterified and coordinated at the carboxylic group (structures with R factors >10% excluded). Table S4: Ibuprofen (S)-CH3CH(C6H4Bui-4)COOH and its derivatives (S)-CH3CH(C6H4Bui-4)COO(R/M) esterified and coordinated at the carboxylic group (structures with R factors >10% excluded). Table S5: Crystallographic data for (R)/(S)-, (R)-, and (S)-phenylglycine, (R)/(S)-, (R)-, and (S)-methyl phenylglycinate hydrochloride, and (R)-ethyl phenylglycinate hydrochloride. Figure S1: ORTEP drawing of conglomerate (R)/(S)-phenylglycine: CCDC1981269. Figure S2: ORTEP drawing of (R)-phenylglycine: CCDC1981270. Figure S3: ORTEP drawing of (S)-phenylglycine: CCDC1981271. Figure S4: ORTEP drawing of racemate (R)/(S)-methyl α-phenylglycinate hydrochloride: CCDC1981272. Figure S5: ORTEP drawing of (R)-methyl α-phenylglycinate hydrochloride: CCDC1981273. Figure S6: ORTEP drawing of (S)-methyl α-phenylglycinate hydrochloride: CCDC1981274. Figure S7: ORTEP drawing of (R)-ethyl α-phenylglycinate hydrochloride: CCDC2049622. The crystallographic data for this paper (CCDC1981269, 1981270, 1981271, 1981272, 1981273, 1981274, and 2049622) can also be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, by emailingdata_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033.
Author Contributions
Conceptualization, methodology, investigation, writing—original draft preparation, writing—review and editing: H.B. and T.T. DFT Calculations: G.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Research Institute of Industrial Technology, Nihon University.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in supplementary material here.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Brunner, H.; Tsuno, T. Selective distortion of the planar group CαC’(O)O to a chiral flat tetrahedron in the amino acid alanine. Chirality 2019, 31, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Brunner, H.; Tsuno, T. The chirality chain in valine: How the configuration at the Cα position through the OcisC’CαN torsional system leads to distortion of the planar group CαC’(Ocis)Otrans to a flat tetrahedron. ChemistryOpen 2018, 7, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Brunner, H.; Tsuno, T. Chirality in amino acids beyond the Cα-configuration. Chirality 2019, 31, 635–640. [Google Scholar] [CrossRef] [PubMed]
- Brunner, H.; Tsuno, T.; Balázs, G. Chiral selectivity in the achiral amino acid glycine. J. Org. Chem. 2019, 85, 16199–16203. [Google Scholar] [CrossRef] [PubMed]
- Bürgi, H.-B. Chemical reaction coordinates from crystal structure data. J. Inorg. Chem. 1973, 12, 2321–2325. [Google Scholar] [CrossRef]
- Murray-Rust, P.; Bürgi, H.B.; Dunitz, J.D. Chemical reaction paths. V. SN1 reaction of tetrahedral molecules. J. Am. Chem. Soc. 1975, 97, 921–922. [Google Scholar] [CrossRef]
- Bye, E.; Schweizer, W.B.; Dunitz, J.D. Chemical reaction paths. 8. Stereoisomerization path for triphenylphosphine oxide and related molecules: Indirect observation of the structure of the transition state. J. Am. Chem. Soc. 1982, 104, 5893–5898. [Google Scholar] [CrossRef]
- Cahn, R.S.; Ingold, C.; Prelog, V. Spezifikation der molekularen Chiralität. Angew. Chem. 1966, 78, 413–447. [Google Scholar] [CrossRef]
- Cahn, R.S.; Ingold, C.; Prelog, V. Specification of molecular chirality. Angew. Chem. Int. Ed. Engl. 1966, 5, 385–415. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision, E.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B. 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Scalmani, G.; Frisch, M.J. Continuous surface charge polarizable continuum models of solvation. I. General formalism. J. Chem. Phys. 2010, 132, 114110. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B 2016, B72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H.J. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez Monge, L.; Taylor, R.; van de Streek, R.J.; Wood, P.A. Mercury CSD 2.0—new features for the visualization and investigation of crystal structures. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Bruno, I.J.; Cole, J.C.; Edgington, P.R.; Kessler, M.; Marcrae, C.F.; McCabe, P.; Pearson, J.; Taylor, R. New software for searching the Cambridge Structural Database and visualizing crystal structures. Acta Crystallogr. Sect. B 2002, B58, 389–397. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).