Molecular, Supramolecular Structures Combined with Hirshfeld and DFT Studies of Centrosymmetric M(II)-azido {M=Ni(II), Fe(II) or Zn(II)} Complexes of 4-Benzoylpyridine


 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Instrumentation
2.2. Syntheses
2.2.1. [Fe(4bzpy)4(N3)2]; 1
2.2.2. [Ni(4bzpy)4(N3)2]; 2
2.2.3. [Zn(4bzpy)2(N3)2]n; 3
2.3. X-ray Structure Determination
2.4. Computational Details
3. Results and Discussion
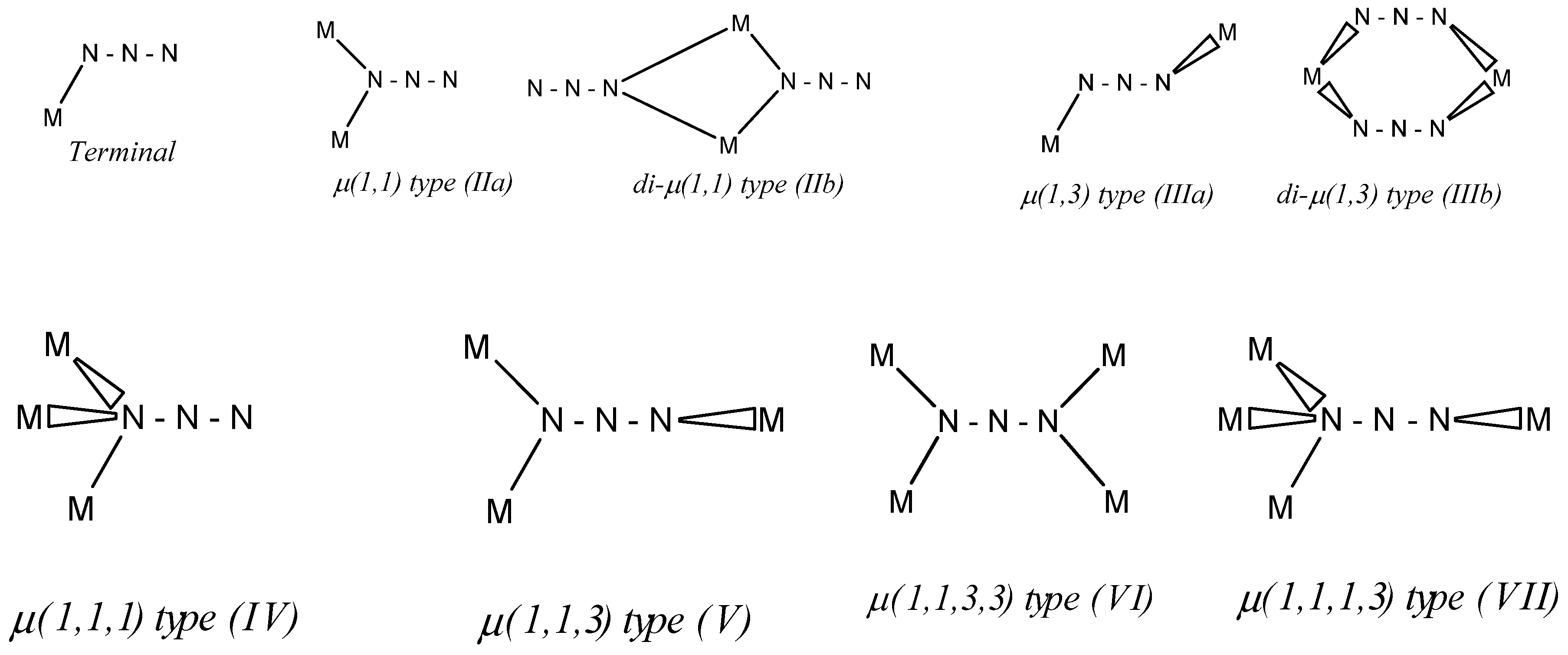
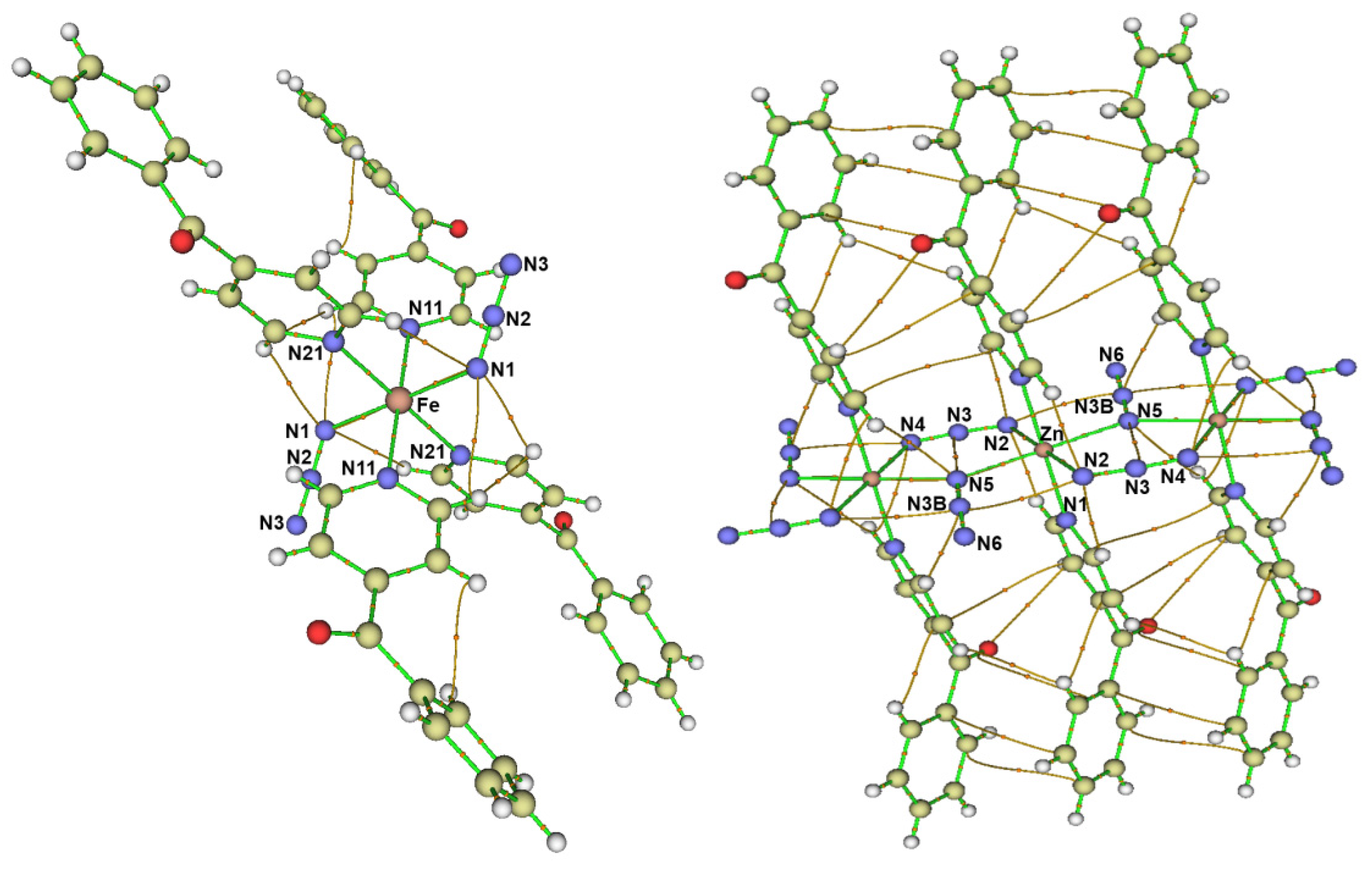
3.1. Structure Description
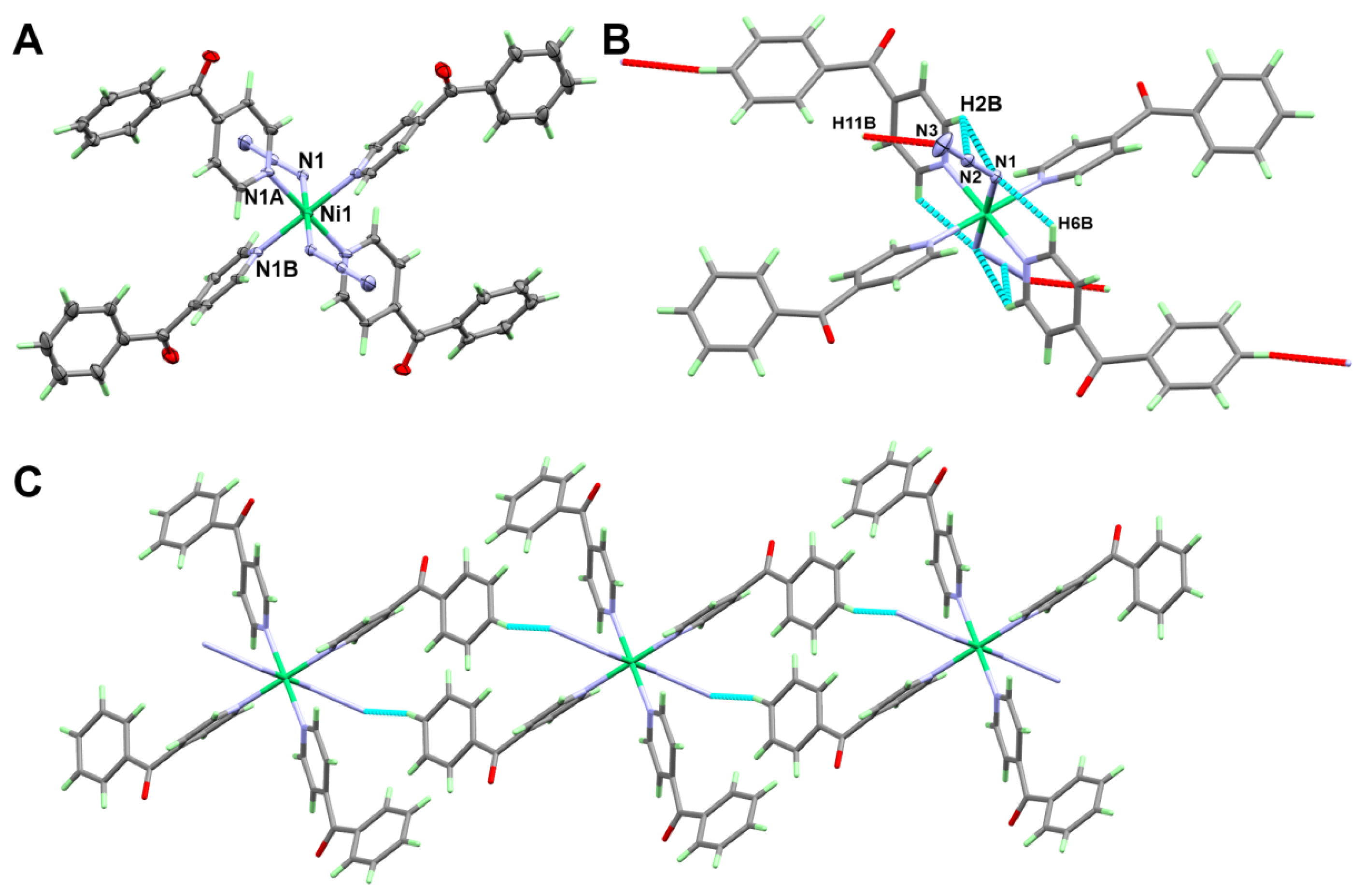
3.1.1. Structure of [Fe(4bzpy)4(N3)2]; 1
3.1.2. Structure of [Ni(4bzpy)4(N3)2]; 2
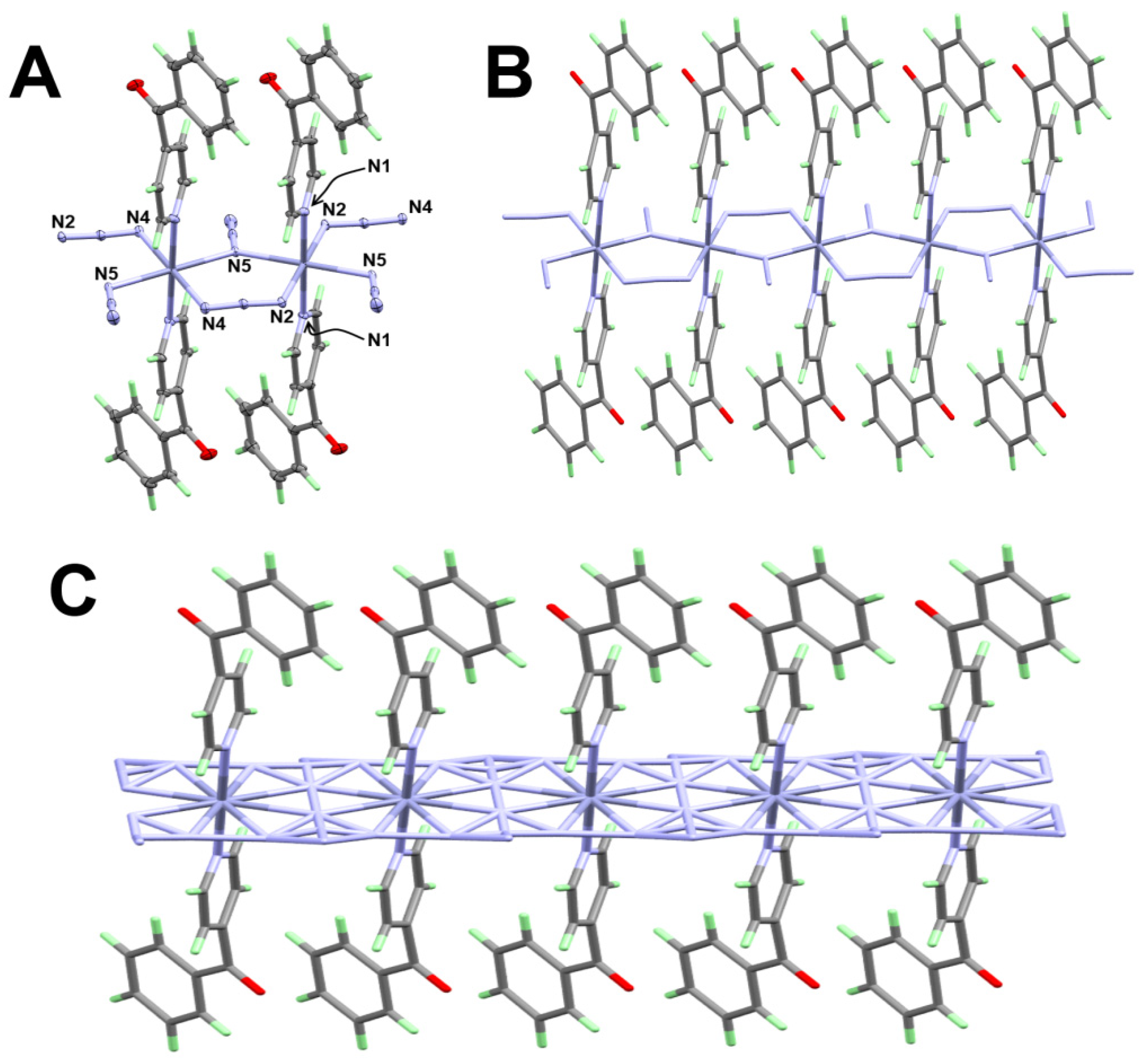
3.1.3. Structure of [Zn(4bzpy)2(N3)2]n; 3
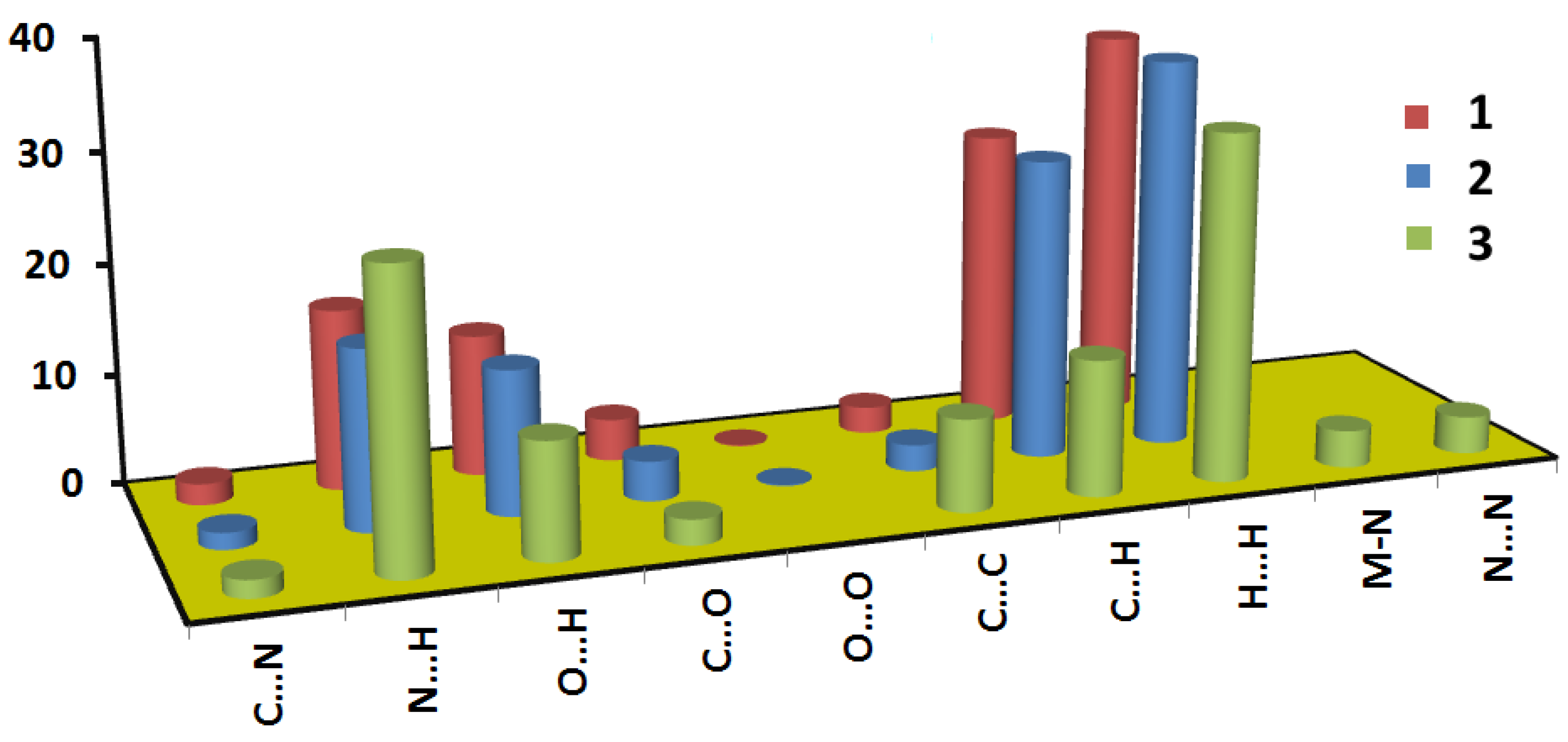
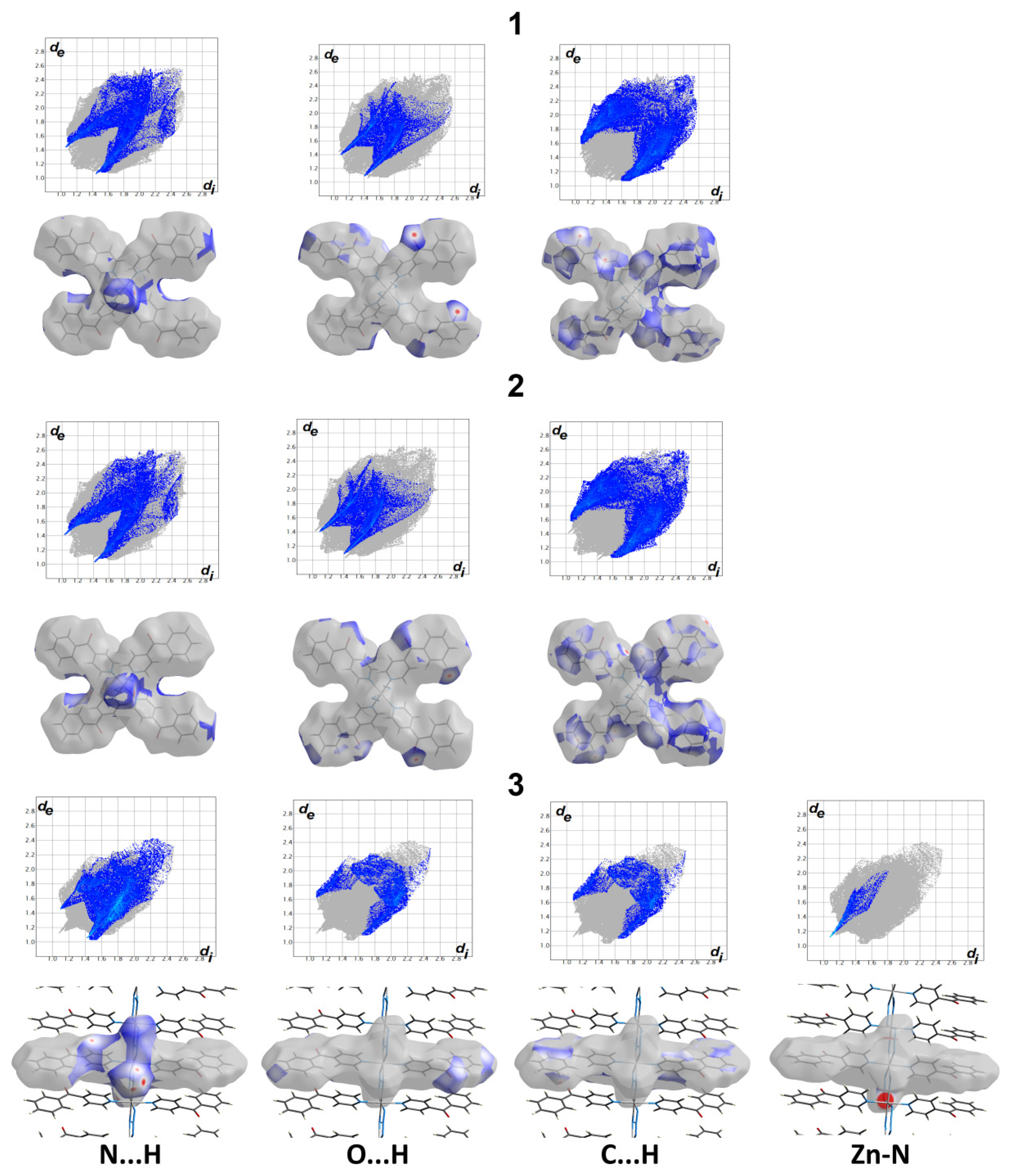
3.2. Hirshfeld Analysis
3.3. Natural Charges
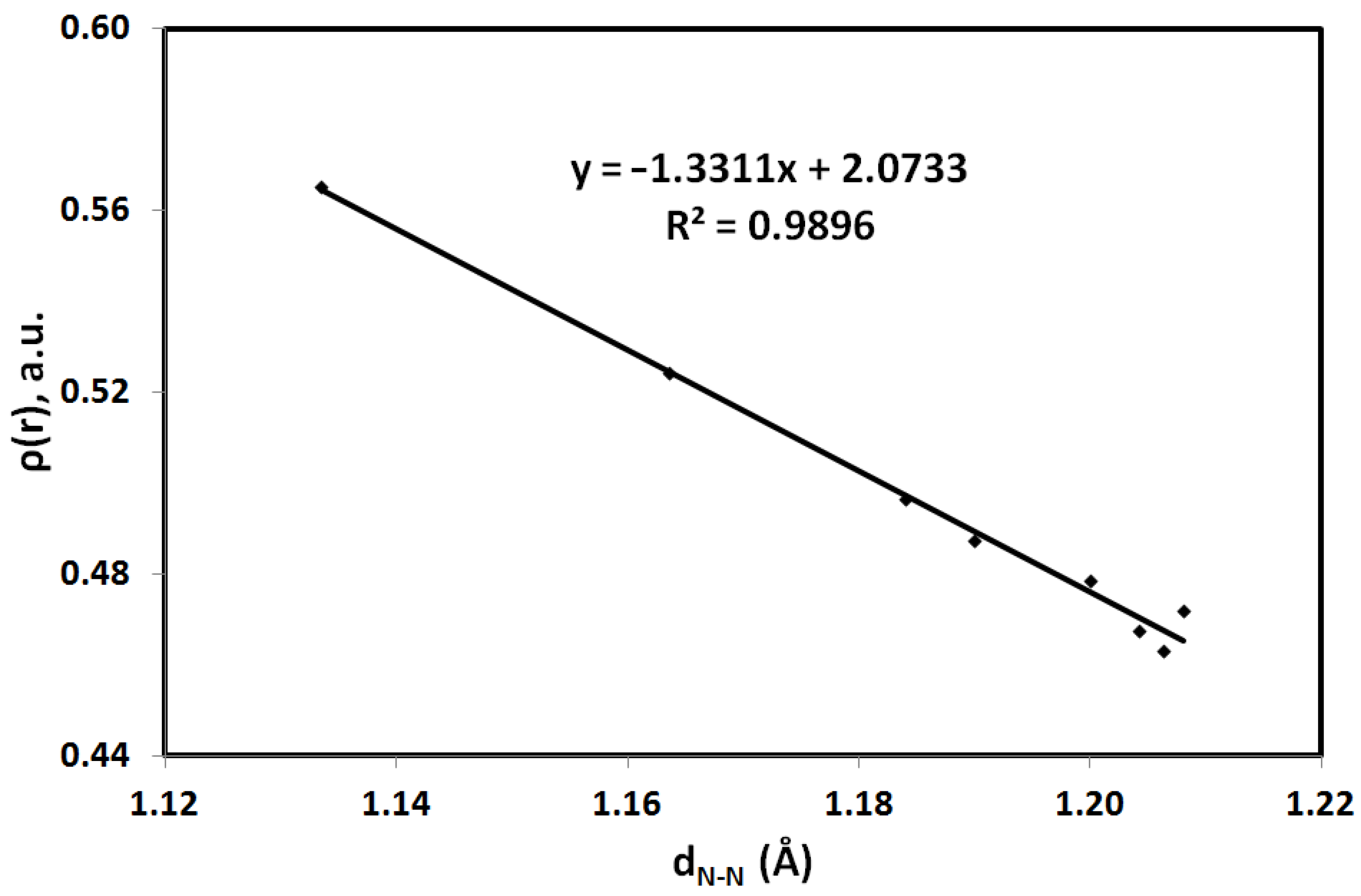
3.4. AIM Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Constable, E.E.C. Novel oligopyridines for metallosupramolecular chemistry. Pure Appl. Chem. 1996, 68, 253–260. [Google Scholar] [CrossRef]
- Constable, E.C.; Lehn, J.-M.; Atwood, L.; Davis, J.E.D.; MacNicol, D.D.; Vögtle, F. Comprehensive Supramolecular Chemistry; Pergamon: New York, NY, USA; Elsevier Science Ltd.: Amsterdam, The Netherlands; Oxford: Oxford, UK, 1996. [Google Scholar]
- Champness, N.R. Coordination frameworks—Where next? Dalton Trans. 2006, 877–880. [Google Scholar] [CrossRef]
- Moulton, B.; Zaworotko, M.J. From Molecules to Crystal Engineering: Supramolecular Isomerism and Polymorphism in Network Solids. Chem. Rev. 2001, 101, 1629–1658. [Google Scholar] [CrossRef]
- Wuest, J.D. Engineering crystals by the strategy of molecular tectonics. Chem. Commun. 2005, 5830–5873. [Google Scholar] [CrossRef]
- Biradha, K.; Sarkar, M.; Rajput, L. Crystal engineering of coordination polymers using 4,4′-bipyridine as a bond between transition metal atoms. Chem. Commun. 2006, 4169–4179. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Hong, M.; Wu, X. Lanthanide–transition metal coordination polymers based on multiple N- and O-donor ligand. Chem. Commun. 2006, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Madalan, M.; Avarvari, N.; Andruh, M. Metal complexes as second-sphere ligands. New J. Chem. 2006, 521–523. [Google Scholar] [CrossRef]
- Abu-Youssef, M.A.M.; Langer, V.; Öhrstrom, L. A unique example of a high symmetry three- and four-connected hydrogen bonded 3D-network. Chem. Comm. 2006, 10, 1082–1084. [Google Scholar] [CrossRef] [PubMed]
- Abu-Youssef, M.A.M.; Langer, V. 1D, 2D and 3D cadmium(II) polymeric complexes with quinoline-4-carboxylato anion, quinazoline and 2,5-dimethylpyrazine. Polyhedron 2005, 25, 1187–1194. [Google Scholar] [CrossRef]
- Kahn, O. Chemistry and Physics of Supramolecular Magnetic Materials. Acc. Chem. Res. 2000, 33, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Eddaoudi, M.; Moler, D.B.; Li, H.; Chen, B.; Reineke, T.M.; O’Keeffe, M.; Yaghi, O.M. Modular chemistry: Secondary building units as a basis for the design of highly porous and robust metal-organic carboxylate frameworks. Acc. Chem. Res. 2001, 34, 319–330. [Google Scholar] [CrossRef]
- Kitagawa, S.; Kitaura, R.; Noro, S. Functional porous coordination polymers. Angew. Chem. Int. Ed. 2004, 43, 2334–2375. [Google Scholar] [CrossRef]
- Kepert, J. Advanced functional properties in nanoporous coordination framework materials. Chem. Commun. 2006, 695–700. [Google Scholar] [CrossRef]
- Kitagawa, S.; Noro, S.; Nakamura, T. Pore surface engineering of microporous coordination polymers. Chem. Commun. 2006, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Evans, O.R.; Lin, W. Crystal Engineering of NLO Materials Based on Metal−Organic Coordination Networks. Acc. Chem. Res. 2002, 35, 511–522. [Google Scholar] [CrossRef]
- Coronado, E.; Galán-Mascarós, J.-R. Hybrid molecular conductors. J. Mater. Chem. 2005, 15, 66–74. [Google Scholar] [CrossRef]
- Bradshaw, D.; Claridge, J.B.; Cussen, E.J.; Prior, T.J.; Rosseinsky, M.J. Design, chirality, and flexibility in nanoporous molecule-based materials. Acc. Chem. Res. 2005, 38, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Coronado, E.; Day, P. Magnetic molecular conductors. Chem. Rev. 2004, 104, 5419–5448. [Google Scholar] [CrossRef] [PubMed]
- Farhoud, M.; Abu-Youssef, M.A.M.; El-Ayan, U. Kerr Effect and Thermal Analysis of Some 3D and 1D polymeric Cadmium (II) and Zinc (II) Azido Complexes. Nonlinear Opt. Quantum Opt. 2006, 36, 1–15. [Google Scholar]
- Lehn, J.-M. Supramolecular Chemistry—Concepts and Perspectives; VCH: Weinheim, Germany, 1995. [Google Scholar]
- Lehn, J.-M. Toward self-organization and complex matter. Science 2002, 295, 2400–2403. [Google Scholar] [CrossRef] [Green Version]
- Evans, B.L.; Yofee, A.D.; Gray, P. Physics and Chemistry of the Inorganic Azides. Chem. Rev. 1959, 59, 515–568. [Google Scholar] [CrossRef]
- Dori, Z.; Ziolo, R.F. Chemistry of coordinated azides. Chem. Rev. 1973, 73, 247–254. [Google Scholar] [CrossRef]
- Ribas, J.; Escure, A.; Monfort, M.; Vicente, R.; Cortes, R.; Lezama, L.; Rojo, T. Polynuclear NiII and MnII azido bridging complexes. Structural trends and magnetic behaviour. Coord. Chem. Rev. 1999, 1027, 193–195. [Google Scholar]
- Palenik, G.J. The Structure of Coordination Compounds I. The Crystal and Molecular Structure of Azidopentamminecobalt(III) Azide. Acta Crystallogr. 1964, 17, 360–367. [Google Scholar] [CrossRef] [Green Version]
- SAINT. Siemens Analytical X-ray Instruments Inc.; SAINT: Madison, WI, USA, 1995. [Google Scholar]
- Sheldrick, G.M. SADABS; University of Göttingen: Göttingen, Germany, 1996. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. Crystal Explorer17 (2017) University of Western Australia. Available online: http://hirshfeldsurface.net (accessed on 12 June 2017).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. GAUSSIAN 09; Revision A02; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Glendening, E.D.; Reed, A.E.; Carpenter, J.E.; Weinhold, F. NBO; Version 3.1 CI; University of Wisconsin: Madison, WI, USA, 1998. [Google Scholar]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamo, C.; Barone, V. Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The mPW and mPW1PW models. J. Chem. Phys. 1998, 108, 664–675. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comp. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta. 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. Cryst. Eng. Comm. 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Spackman, M.A.; McKinnon, J.J. Fingerprinting intermolecular interactions in molecular crystals. Cryst. Eng. Comm. 2002, 4, 378–392. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces. Chem. Commun. 2007, 3814–3816. [Google Scholar] [CrossRef]
- Pauling, L. The Natural of the Chemical Bond; Cornell University Press: Ithaca, NY, USA, 1967. [Google Scholar]
- Matta, C.F.; Hernandez-Trujillo, J.; Tang, T.-H.; Bader, R.F.W. Hydrogen–Hydrogen Bonding: A Stabilizing Interaction in Molecules and Crystals. Chem. Eur. J. 2003, 9, 1940–1951. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J.; Pfitzner, A.; Zabel, M.; Dubis, A.T.; Palusiak, M. Intramolecular H...H Interactions for the Crystal Structures of [4-((E)-But-1-enyl)-2,6-dimethoxyphenyl]pyridine-3-carboxylate and [4-((E)-Pent-1-enyl)-2,6-dimethoxyphenyl]pyridine-3-carboxylate; DFT Calculations on Modeled Styrene Derivatives. J. Phys. Chem. B 2004, 108, 1831–1837. [Google Scholar] [CrossRef]
- Matta, C.F.; Castillo, N.; Boyd, R.J. Characterization of a Closed-Shell Fluorine−Fluorine Bonding Interaction in Aromatic Compounds on the Basis of the Electron Density. J. Phys. Chem. A 2005, 109, 3669–3681. [Google Scholar] [CrossRef] [PubMed]
- Pendás, A.M.; Francisco, E.; Blanco, M.A.; Gatti, C. Bond Paths as Privileged Exchange Channels. Chem. Eur. J. 2007, 13, 9362–9371. [Google Scholar] [CrossRef]
- Gibbs, G.V.; Cox, D.F.; Crawford, T.D.; Rosso, K.M.; Ross, N.L.; Downs, R.T. Classification of metal-oxide bonded interactions based on local potential- and kinetic-energy densities. J. Chem. Phys. 2006, 124, 084704. [Google Scholar] [CrossRef]
- Dinda, S.; Samuelson, A.G. The Nature of Bond Critical Points in Dinuclear Copper(I) Complexes. Chem. A Eur. J. 2012, 18, 3032–3042. [Google Scholar] [CrossRef]
- Cremer, D.; Kraka, E. Chemical Bonds without Bonding Electron Density—Does the Difference Electron-Density Analysis Suffice for a Description of the Chemical Bond? Angew. Chem. Int. Ed. Engl. 1984, 23, 627–628. [Google Scholar] [CrossRef]
- Jenkins, V.; Morrison, I. The chemical character of the intermolecular bonds of seven phases of ice as revealed by ab initio calculation of electron densities. Chem. Phys. Lett. 2000, 317, 97–102. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. From weak to strong interactions: A comprehensive analysis of the topological and energetic properties of the electron density distribution involving X–H…F–Y systems. J. Chem. Phys. 2002, 117, 5529–5542. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Marques, H.M. The physical chemistry of coordinated aqua-, ammine-, and mixed-ligand Co2+ complexes: DFT studies on the structure, energetics, and topological properties of the electron density. Phys. Chem. Chem. Phys. 2010, 12, 2126–2138. [Google Scholar] [CrossRef] [PubMed]

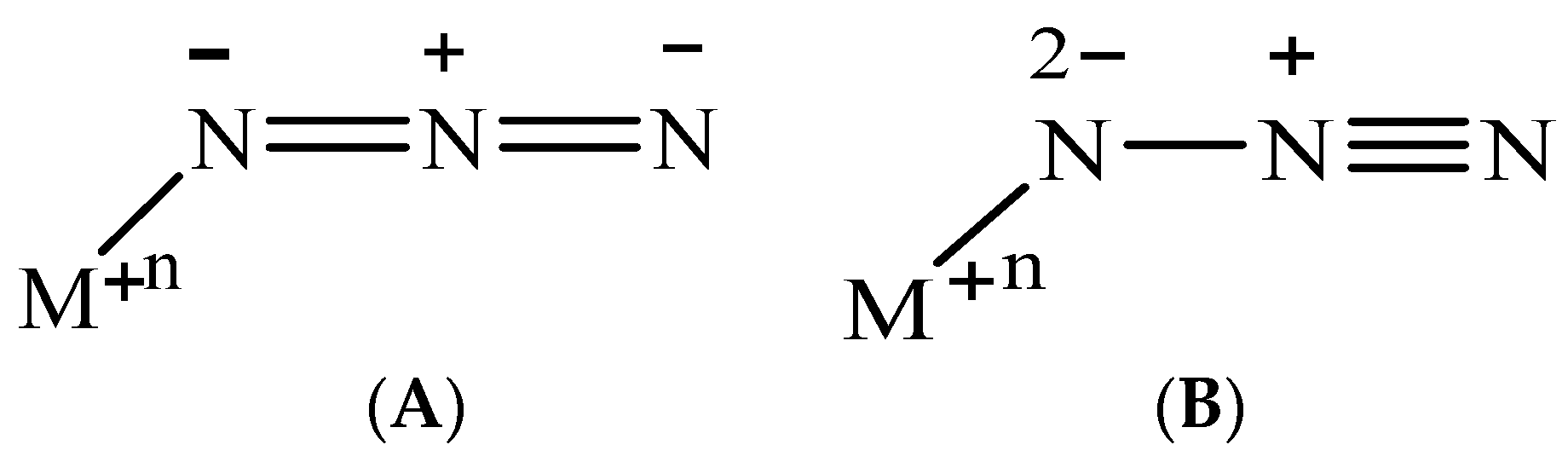

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | 3 | |
|---|---|---|---|
| Empirical formula | C48H36FeN10O4 | C48H36N10NiO4 | C24H18N8O2Zn |
| fw | 872.72 | 875.58 | 515.83 |
| temp (K) | 173(2) | 173(2) | 173(2) |
| λ(Å) | 0.71073 | 0.71073 | 0.71073 |
| cryst syst | Triclinic | Triclinic | Triclinic |
| space group | P | P | P?1 |
| a (Å) | 6.98090(10) | 6.9531(4) | 4.0405(1) |
| b (Å) | 12.16860(10) | 11.9596(7) | 8.30040(10) |
| c (Å) | 12.49150(10) | 12.4219(8) | 16.4323(2) |
| α (deg) | 92.7020(10) | 87.2790(10) | 89.0500(10) |
| β (deg) | 90.2030(10) | 89.6840(10) | 83.0480(10) |
| γ (deg) | 91.6290(10) | 88.3160(10) | 83.3630(10) |
| V (Å3) | 1059.504(19) | 1031.34(11) | 543.383(9) |
| Z | 1 | 1 | 1 |
| ρcalc (Mg/m3) | 1.368 | 1.410 | 1.576 |
| μ(Mo Kα) (mm−1) | 0.414 | 0.530 | 1.172 |
| No. reflns. | 18342 | 11297 | 9272 |
| Unique reflns. | 7211 | 3626 | 3768 |
| GOOF (F2) | 1.023 | 1.007 | 1.066 |
| Rint | 0.0378 | 0.0951 | 0.0307 |
| R1 a (I ≥ 2σ) | 0.0431 | 0.0725 | 0.0312 |
| wR2 b (I ≥ 2σ) | 0.0658 | 0.1776 | 0.0717 |
| Largest diff. peak and hole e.Å−3 | 0.404/−0.529 | 0.771/−0.619 | 0.524/−0.437 |
| CCDC | 2,110,424 | 2,110,425 | 2,110,426 |
| Bond | Distance Å | Bond | Distance Å |
|---|---|---|---|
| 1 | 2 | ||
| Fe1-N1 | 2.1080(12) | Ni1-N1 | 2.091(4) |
| Fe1-N11 | 2.2604(11) | Ni1-N1A | 2.133(4) |
| Fe1-N21 | 2.2929(11) | Ni1-N1B | 2.180(4) |
| Bonds | Angle ° | Bonds | Angle ° |
| N11 1-Fe1-N11 | 180.00(5) | N1 1-Ni1-N1B 1 | 91.81(14) |
| N11 1-Fe1-N21 1 | 92.03(4) | N1-Ni1-N1B 1 | 88.19(14) |
| N11-Fe1-N21 1 | 87.96(4) | N1 1-Ni1-N1 | 180.0 |
| N21-Fe1-N21 1 | 180.0 | N1 1-Ni1-N1A | 88.42(15) |
| N1-Fe1-N11 1 | 89.46(4) | N1 1-Ni1-N1A 1 | 91.58(15) |
| N1-Fe1-N11 | 90.54(4) | N1A-Ni1-N1B | 92.62(14) |
| N1 1-Fe1-N21 | 88.42(4) | N1A 1-Ni1-N1B | 87.38(14) |
| N1-Fe1-N21 | 91.58(4) | N1A-Ni1-N1A 1 | 180.0 |
| Symm. code: 1 -X,1-Y,1-Z | Symm. code: 1 1-X,1-Y,1-Z | ||
| D-H...A | d(D-H) | d(H...A) | d(D...A) | <(DHA) |
|---|---|---|---|---|
| 1 | ||||
| C22-H22…N2 | 0.95 | 2.58 | 3.254(2) | 128 |
| C26-H26…N1 1 | 0.95 | 2.54 | 3.123(2) | 120 |
| C112-H112…O27 2 | 0.95 | 2.60 | 3.419(2) | 145 |
| Symm. Codes: 1 -x,1-y,1-z and 2 -1-x,1-y,2-z | ||||
| 2 | ||||
| C2B-H2B…N1 | 0.95 | 2.54 | 3.139(6) | 121 |
| C2B-H2B…N2 | 0.95 | 2.48 | 3.147(7) | 127 |
| C6B-H6B...N1 3 | 0.95 | 2.41 | 3.004(6) | 120 |
| C11B-H11B...N3 4 | 0.95 | 2.58 | 3.508(9) | 165 |
| Symm. Codes: | 3 1-x,1-y,1-z | 4 -x,1-y,2-z | ||
| Bond | Distance Å | Bond | Distance Å |
|---|---|---|---|
| Zn1-N5 1 | 2.177(2) | Zn1-N5 | 2.205(2) |
| Zn1-N1 | 2.198(1) | Zn1-N4 1 | 2.212(2) |
| Zn1-N2 | 2.199(2) | ||
| Bonds | Angle ° | Bonds | Angle ° |
| N5 1-Zn1-N1 | 91.00(6) | N1-Zn1-N2 | 89.60(7) |
| N5 2-Zn1-N1 | 89.00(6) | N2 3-Zn1-N2 | 180.00(7) |
| N1 3-Zn1-N2 3 | 89.60(7) | N1-Zn1-N5 | 88.69(6) |
| N1-Zn1-N2 3 | 90.40(7) | N1-Zn1-N5 3 | 91.31(6) |
| 1 | 2 | 3 | |||
|---|---|---|---|---|---|
| Fe | 1.004 (1.008) | Ni | 0.847 (0.864) | Zn | 1.147 (1.153) |
| 2(4bzpy) | 0.217 (0.228) | 2(4bzpy) | 0.298 (0.300) | 4bzpy | 0.093 (0.091) |
| N3¯ | −0.719 (−0.731) | N3¯ | −0.722 (−0.732) | N3¯μ(1,1) | −0.696 (−0.701) |
| N3¯μ(1,3) | −0.681 (0.687) | ||||
| CPX | dN-N | Δd | ρ(r), a.u | ∇2ρ(r) a |
|---|---|---|---|---|
| N1-N2 | 1.204 | 0.0405 | 0.4675 | −1.2160 |
| N2-N3 | 1.164 | 0.5242 | −1.3276 | |
| N1-N2 | 1.206 | 0.0728 | 0.4631 | −1.4209 |
| N2-N3 | 1.133 | 0.5653 | −1.8049 | |
| N6-N3B | 1.207 | 0.017 | 0.4721 | −1.0357 |
| N5-N3B | 1.190 | 0.4873 | −1.3938 | |
| N2-N3 | 1.199 | 0.015 | 0.4787 | −1.0351 |
| N3-N4 | 1.184 | 0.4967 | −1.3545 |
| Bond | ρ(r); a.u. | H(r) b; a.u. | V(r)/G(r) c | ∇ρ(r) d |
|---|---|---|---|---|
| Complex 1 | ||||
| Fe1-N11 | 0.0346 | 0.0010 | 0.985 | 0.2646 |
| Fe1-N21 | 0.0319 | 0.0011 | 0.981 | 0.2422 |
| Fe1-N1 | 0.0501 | −0.0006 | 1.007 | 0.3528 |
| Complex 2 | ||||
| Ni1-N1B | 0.0357 | 0.0021 | 0.973 | 0.3252 |
| Ni1-N1A | 0.0408 | 0.0017 | 0.981 | 0.3720 |
| Ni1-N5 | 0.0612 | −0.0095 | 1.102 | 0.3347 |
| Complex 3 | ||||
| Zn1-N1 | 0.0389 | −0.0647 | 0.941 | 0.2770 |
| Zn1-N41 | 0.0369 | −0.0626 | 0.938 | 0.2770 |
| Zn1-N5 | 0.0437 | −0.0658 | 1.010 | 0.2575 |
| Zn1-N51 | 0.0525 | −0.0785 | 1.062 | 0.2770 |
| Zn1-N2 | 0.0297 | −0.0497 | 0.924 | 0.2316 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abu-Youssef, M.A.M.; Langer, V.; Barakat, A.; Haukka, M.; Soliman, S.M. Molecular, Supramolecular Structures Combined with Hirshfeld and DFT Studies of Centrosymmetric M(II)-azido {M=Ni(II), Fe(II) or Zn(II)} Complexes of 4-Benzoylpyridine. Symmetry 2021, 13, 2026. https://doi.org/10.3390/sym13112026
Abu-Youssef MAM, Langer V, Barakat A, Haukka M, Soliman SM. Molecular, Supramolecular Structures Combined with Hirshfeld and DFT Studies of Centrosymmetric M(II)-azido {M=Ni(II), Fe(II) or Zn(II)} Complexes of 4-Benzoylpyridine. Symmetry. 2021; 13(11):2026. https://doi.org/10.3390/sym13112026
Chicago/Turabian StyleAbu-Youssef, Morsy A. M., Vratislav Langer, Assem Barakat, Matti Haukka, and Saied M. Soliman. 2021. "Molecular, Supramolecular Structures Combined with Hirshfeld and DFT Studies of Centrosymmetric M(II)-azido {M=Ni(II), Fe(II) or Zn(II)} Complexes of 4-Benzoylpyridine" Symmetry 13, no. 11: 2026. https://doi.org/10.3390/sym13112026