1. Introduction
Amidation is regarded as a change in organic molecules where, instead of the carboxyl group (–COOH), the amide group (–NH2) is incorporated in the molecule [
1,
2]. Amidated peptides have a longer half-life in the blood and are less susceptible to proteolysis. When a carboxyl group becomes an amide group, which may be a proton or a deproton, the peptide’s properties become less susceptible to physiological pH changes. Besides, the binding of peptide to G-protein-associated receptors is highly influenced by amidation [
3,
4]. In certain cases, the C-terminus of the amidated peptide is closely aligned with the GPCR transmembrane, resulting in enhanced coordination and signal transmission. Moreover, peptides’ biological activity such as vasopressin, oxytocin, and TRH is substantially decreased in the absence of a C-terminus amide moiety [
5,
6]. Alpha-amides in the C-terminus comprise about half of the physiologically active peptides and peptide hormones. This is important for complete bioactivity. Amidation occurs through a sequential reaction of two enzymes encoded with a single-function peptide glycine
-amidated monooxygenase (PAM or
-amide) [
7,
8,
9]. PAM catalyzes the formation of peptide amides from precursors of C-terminal glycine-containing peptides and requires copper, ascorbic acid, and molecular oxygen. PAM is the only enzyme in the body, which generates peptide amides.
Nonetheless, various strategies have been developed using PAM, carboxypeptidase Y enzyme, and chemical synthesis to produce peptide amides in vitro [
10,
11]. The growing demand and the importance of peptide amide medicines indicate the necessity for effective industrial in-vitro amidation systems. In recent years, there have been questions about the synthesis of peptide hormones such as calcitonin and oxytocin in recombinant enzymatic amidation systems [
12,
13]. All this requires the study of the mechanism of amidation. However, in vivo, ex vivo, in vitro studies are tedious, time-consuming, and expensive. Therefore, the research community has devised in silico approaches using advances in machine learning to solve prediction problems in the fields of computational biology and bioinformatics [
14,
15,
16,
17,
18,
19,
20,
21,
22]. Scarce research is available on the prediction of Valine amidation, which is an important phenomenon in the amidation mechanism study. Notable contributions for predicting sites of amidation are proposed in [
8,
23], which use machine learning to develop in-silico predictors of amidation sites. Current models of protein prediction are limited by their functionality, as they depend on the quality of features used to develop the model. Yau et al. [
24] proposed a 2-D graphical representation for protein sequences, constructed the moment vectors for protein sequences, and showed one-to-one correspondence between moment vectors and protein sequences. Yu et al. [
25] proposed an evolutionary protein map by incorporating physicochemical properties of amino acids to achieve greater evolutionary significance of protein classification at amino acid sequence level. Although these feature extraction approaches are promising, they are calculated independently of the learning system, and their quality cannot be determined in advance, as there is no feedback mechanism between feature selection and learning subsystems. Another limitation of these approaches is the requirement of expert human intervention and domain knowledge for extraction and selection of features which can produce prediction models with improved performance.
Advances in machine learning have led to the emergent discipline of deep learning, which is related to the study of different deep neural network architectures developed using neurologically inspired mathematical functions, dubbed as neurons, for learning tasks [
26]. Deep learning has enabled breakthroughs in different research areas, including computer vision [
27,
28], natural language processing [
29], and information security [
30,
31] to mention a few. In essence, all models of deep learning are composed of multilayer neural networks. These models are developed by stacking multiple layers of neurons in a manner that each layer receives the inputs from preceding layer, transform it using neurons to create the output of layer, and provide this output as input to following layers. All DNNs contain an input layer, which serves as the entry point of input and an output layer which transform the input of preceding layers to predictions. Transformations performed by layers of DNN are nonlinear and enable the creation of abstract, task-specific representations of input data in a hierarchical manner which ignore trivial deviations but retain the imperative features of input to enable effective predictions [
32]. After adequate training of the neural network on input/output pairs of the peptide sequences, resultant output label is given by last fully connected layer of the model using classifiers such as logistic regression and softmax to predict the outputs. Current models of deep learning offer a very powerful structure for solving learning problems. DNN based models can automatically learn the optimal low-dimensional hierarchical representation from the raw PseAAC sequences. The Gradient descent optimizer of the DNN model uses the loss score between actual and predicted labels as the feedback to adjust the subsequent weights of neurons in DNN layers, enabling better representations, resulting in accurate predictions [
31].
In this study, we propose a new predictor for determining sites of valine amide (V-amide) in proteins by integrating Chou’s Pseudo Amine Acid Composition (PseAAC) [
33,
34] with deep neural networks to learn deep representations resulting in better site identification. DNN based predictors were developed and compared using standard model evaluation parameters to identify the best performing model of V-amide site predictions. We adopted Chou’s 5-step Rule [
34] that is widely used in research contributions [
3,
35,
36,
37,
38,
39] and consists of five stages. i.e., (i) collection of benchmark dataset (ii) mathematical formulation of biological samples and feature selection (iii) implementation and training of prediction algorithm to create predictor (iv) cross-validation of results, and (v) development of webserver.
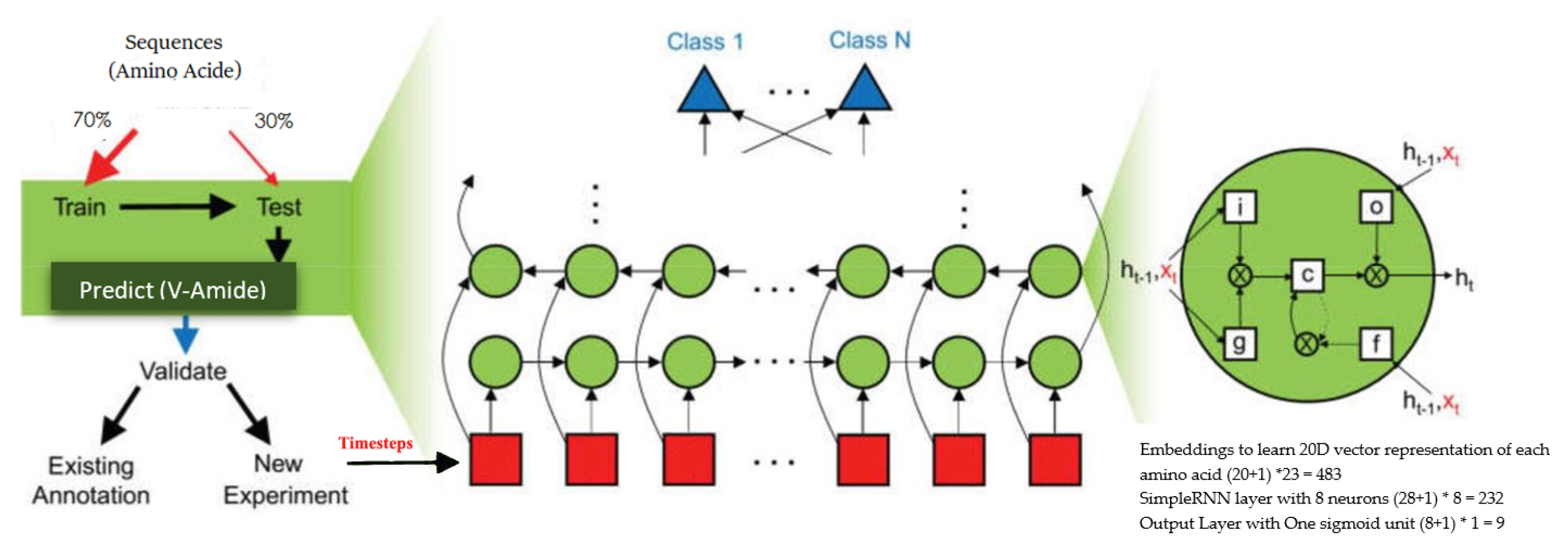
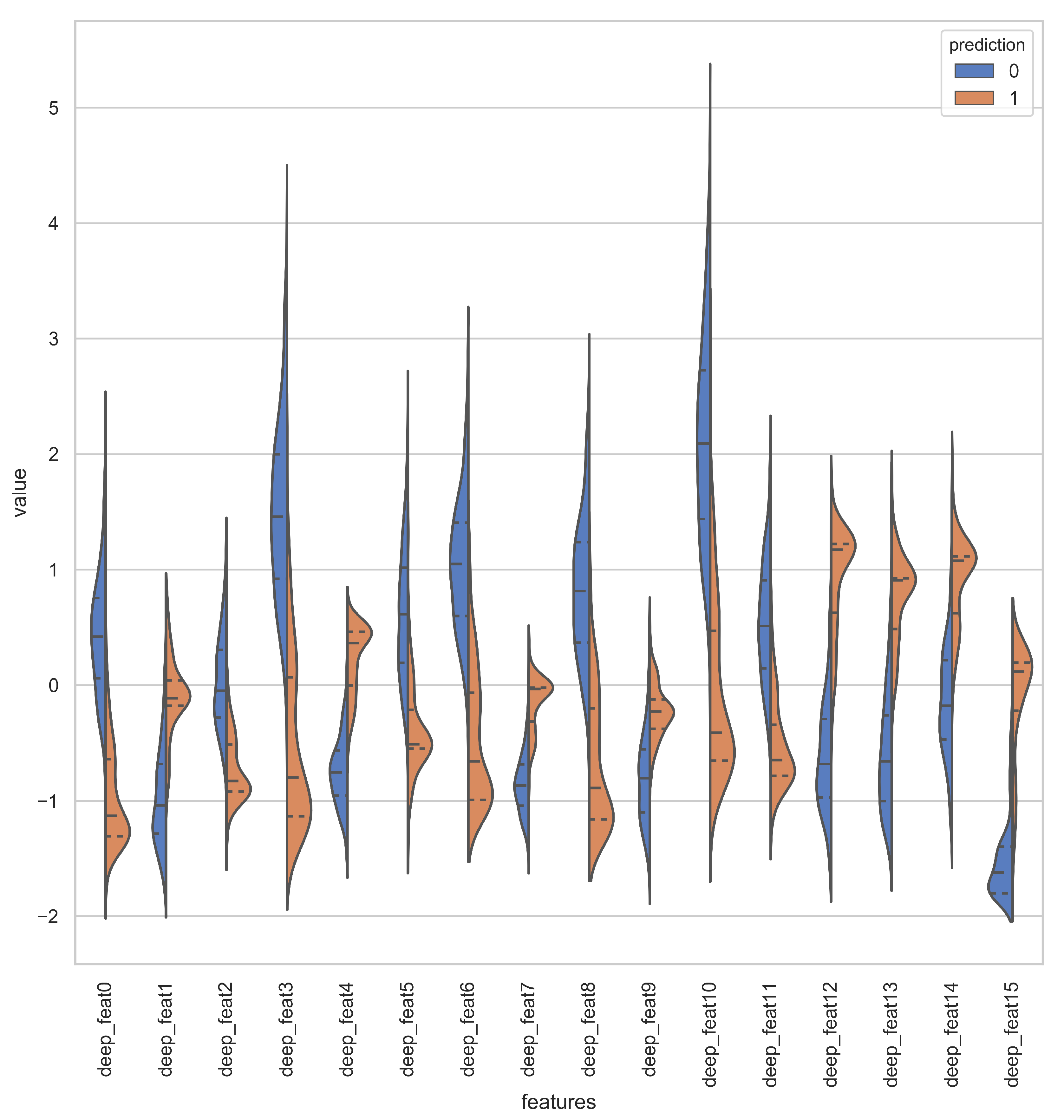
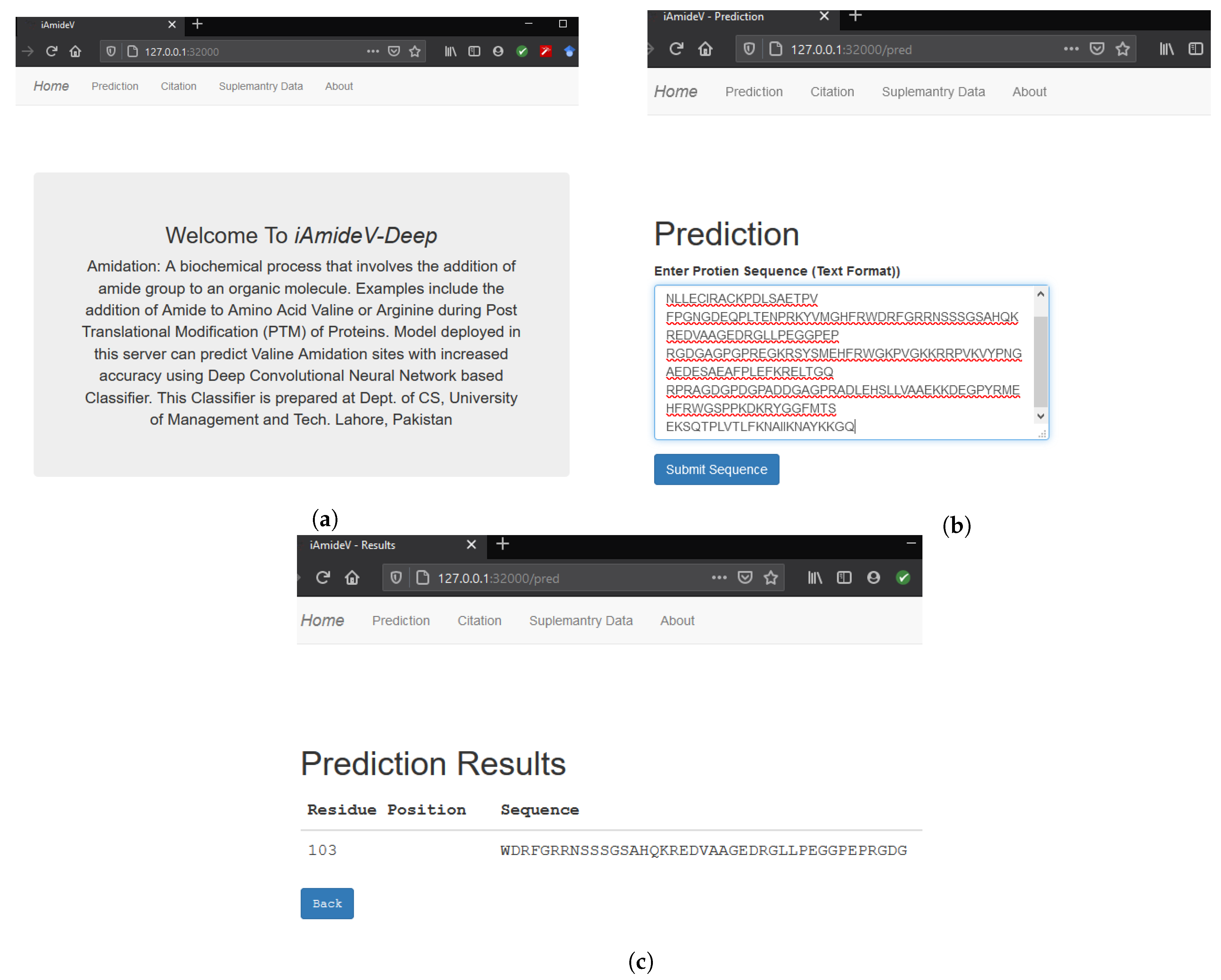
Figure 1 shows different phases of Chou’s 5-step rule. Our methodology is derived from Chou’s 5-step rule, but we combine the feature selection and model training step by employing deep neural networks (DNNs). The advantage of DNN is the automatic learning of meaningful and effective representations from raw PseAAC sequences. That is, no additional steps are required to extract or select the representations for developing a predictor model [
40]. To obtain the best V-Amide prediction model, several DNN-based prediction models are implemented using different DNN algorithms and evaluated against each other using the standard model evaluation parameters.
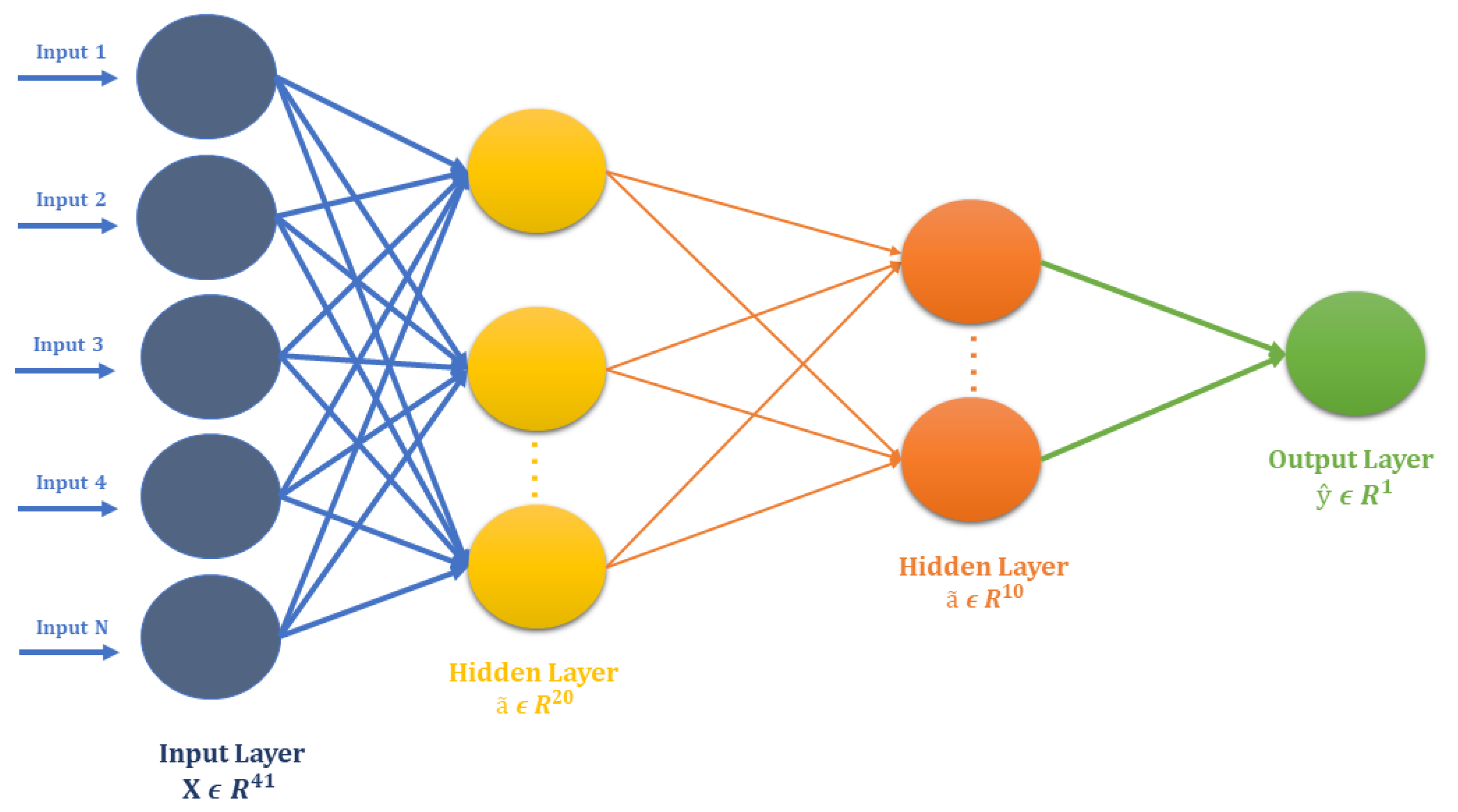
Instead of relying on human-engineered features, our methodology, as shown in
Figure 2, combines the feature extraction and model training step using DNNs. Once the DNN model is sufficiently trained, the intermediate layers of DNN transform raw peptide sequences of PseAAC to meaningful deep representations and an output layer of DNN perform prediction using the deep representation learned by earlier layers. Since both, the representation learning subsystem and prediction subsystem work in unison, the optimizer uses the loss score as the feedback signal to improve both the subsystems of DNN.
This paper is organized as follows.
Section 2 describes the research methodology of the proposed study.
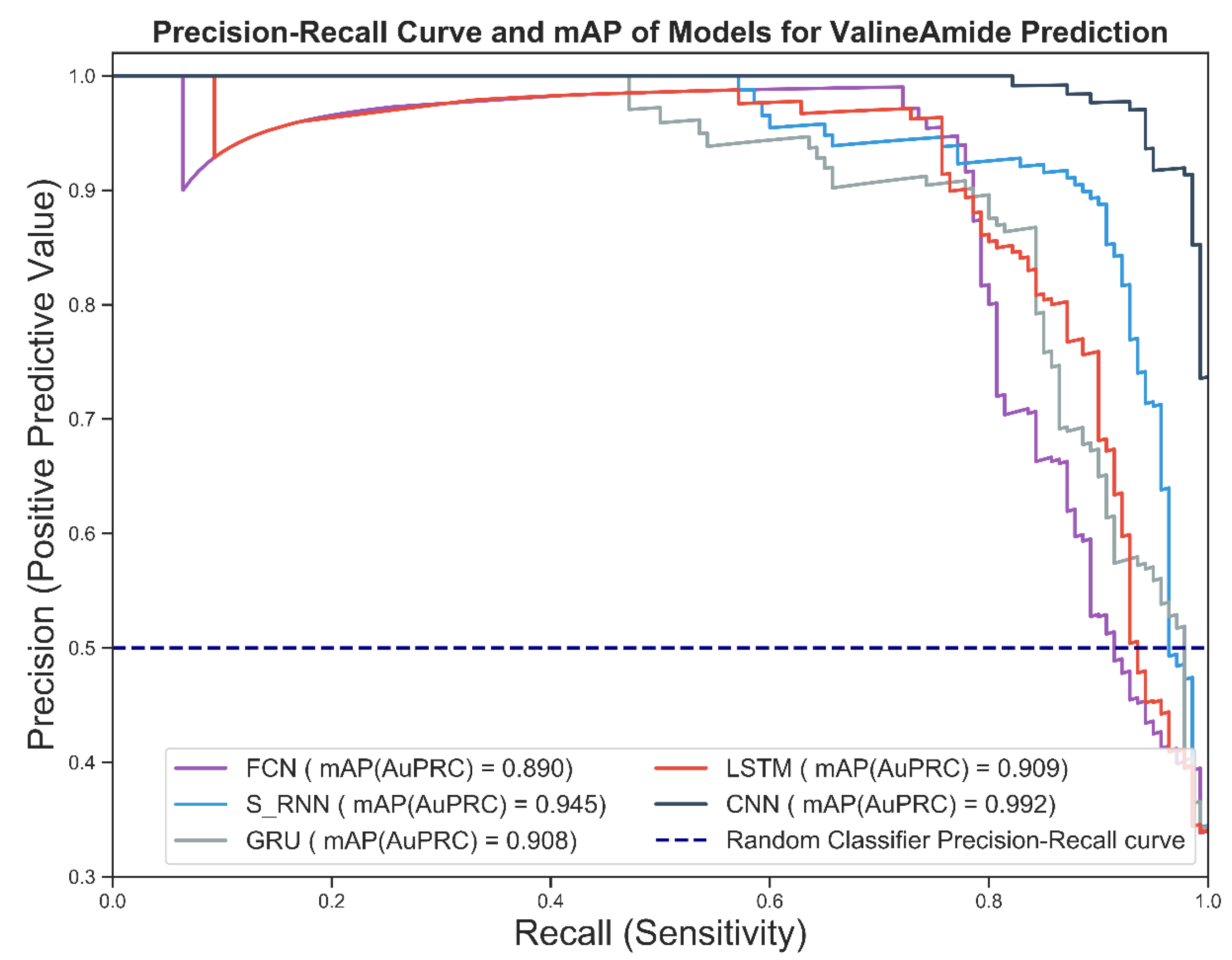
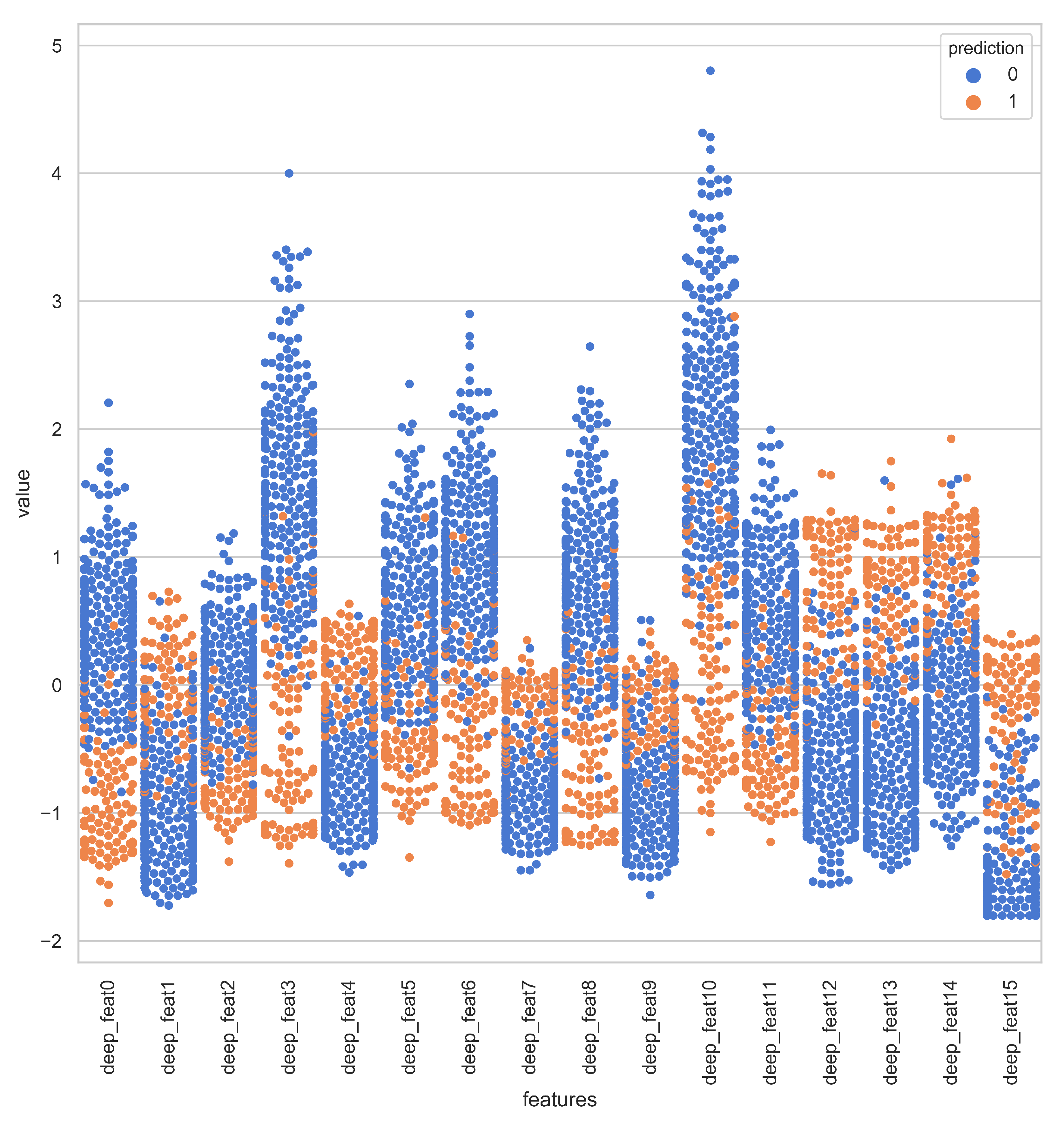
Section 3 provides the results and findings.
Section 4 provides the discussion while the conclusion and the future work are given in
Section 5.



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}