
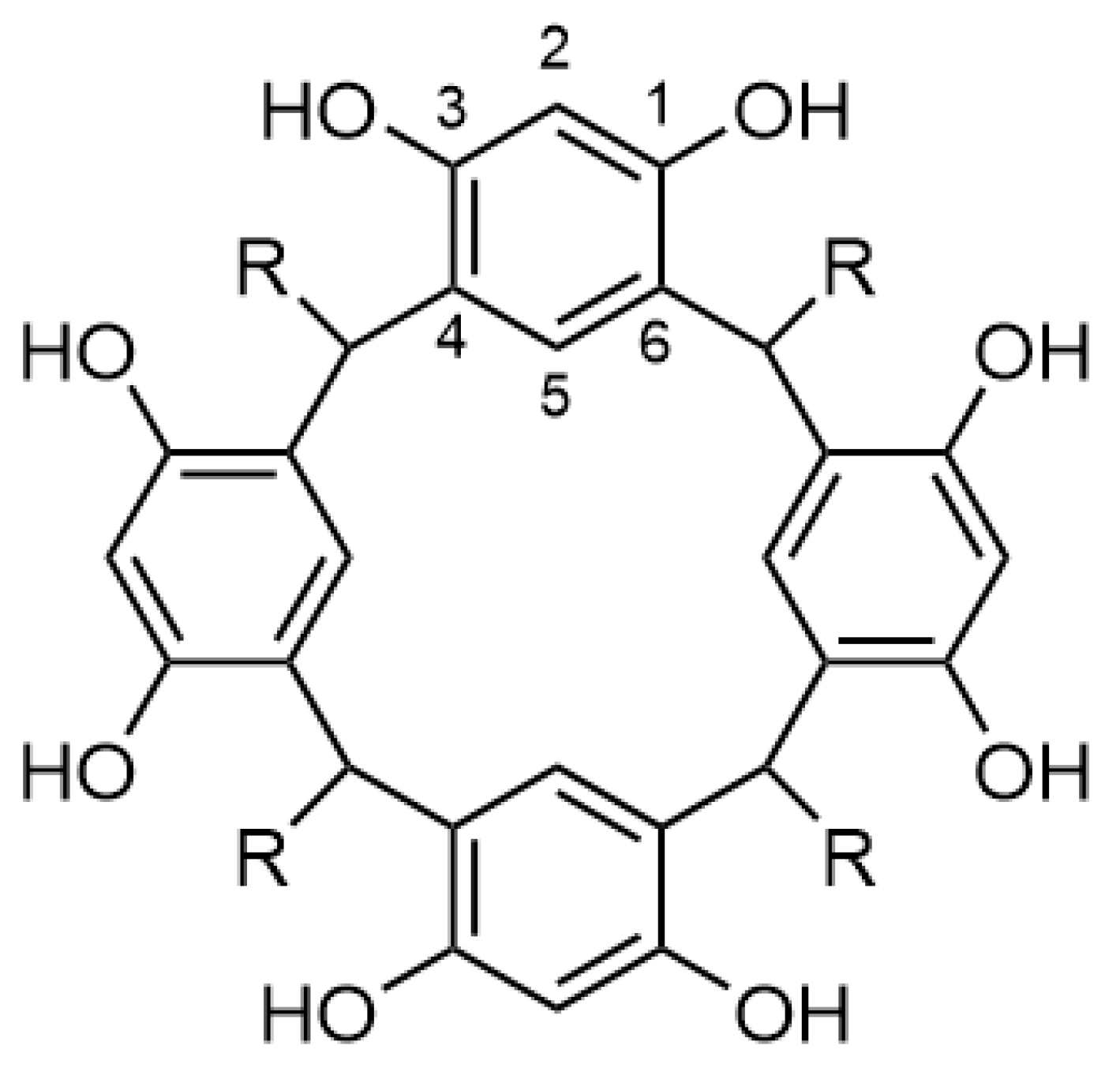
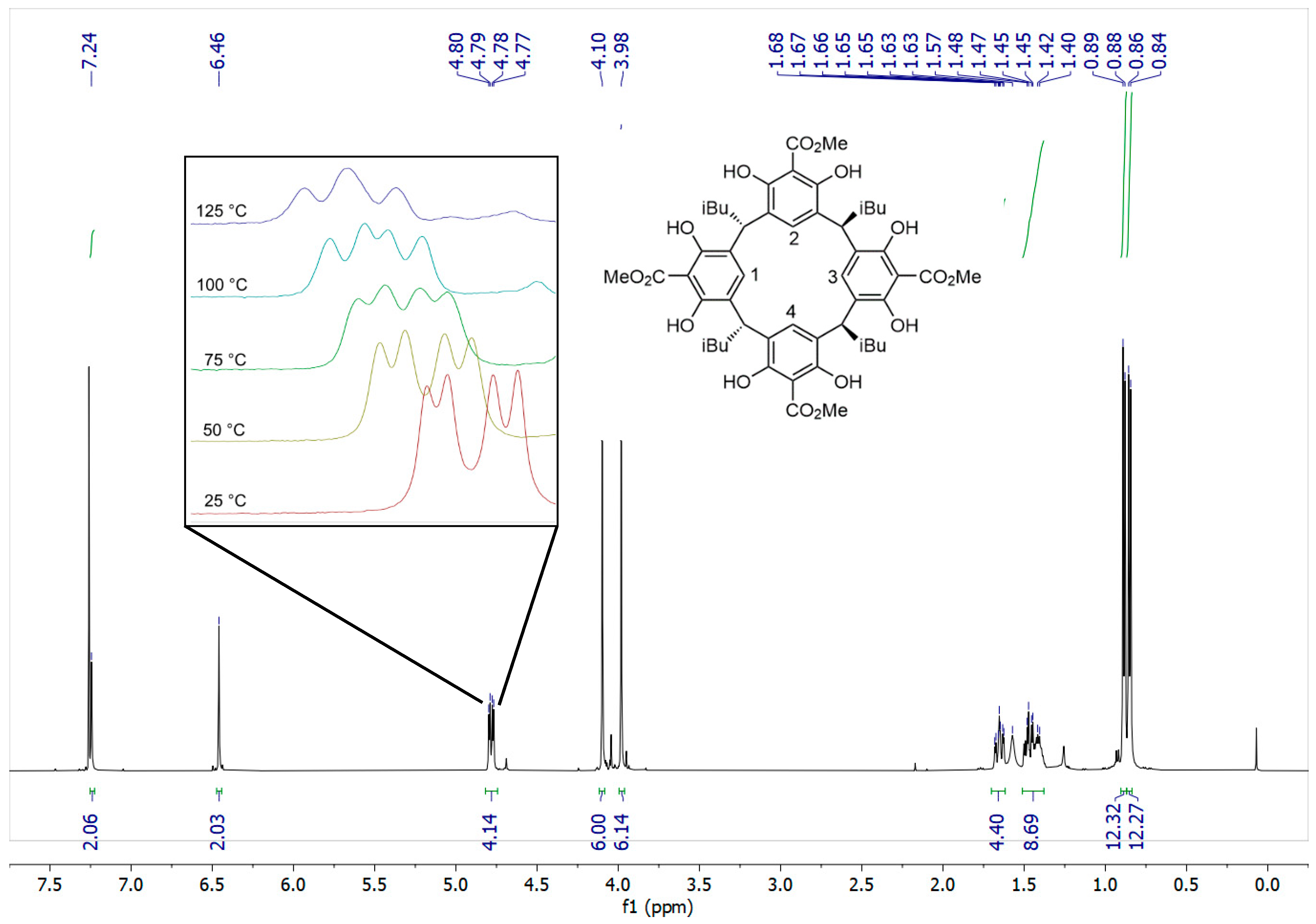
A Concise Synthesis of a Methyl Ester 2-Resorcinarene: A Chair-Conformation Macrocycle


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
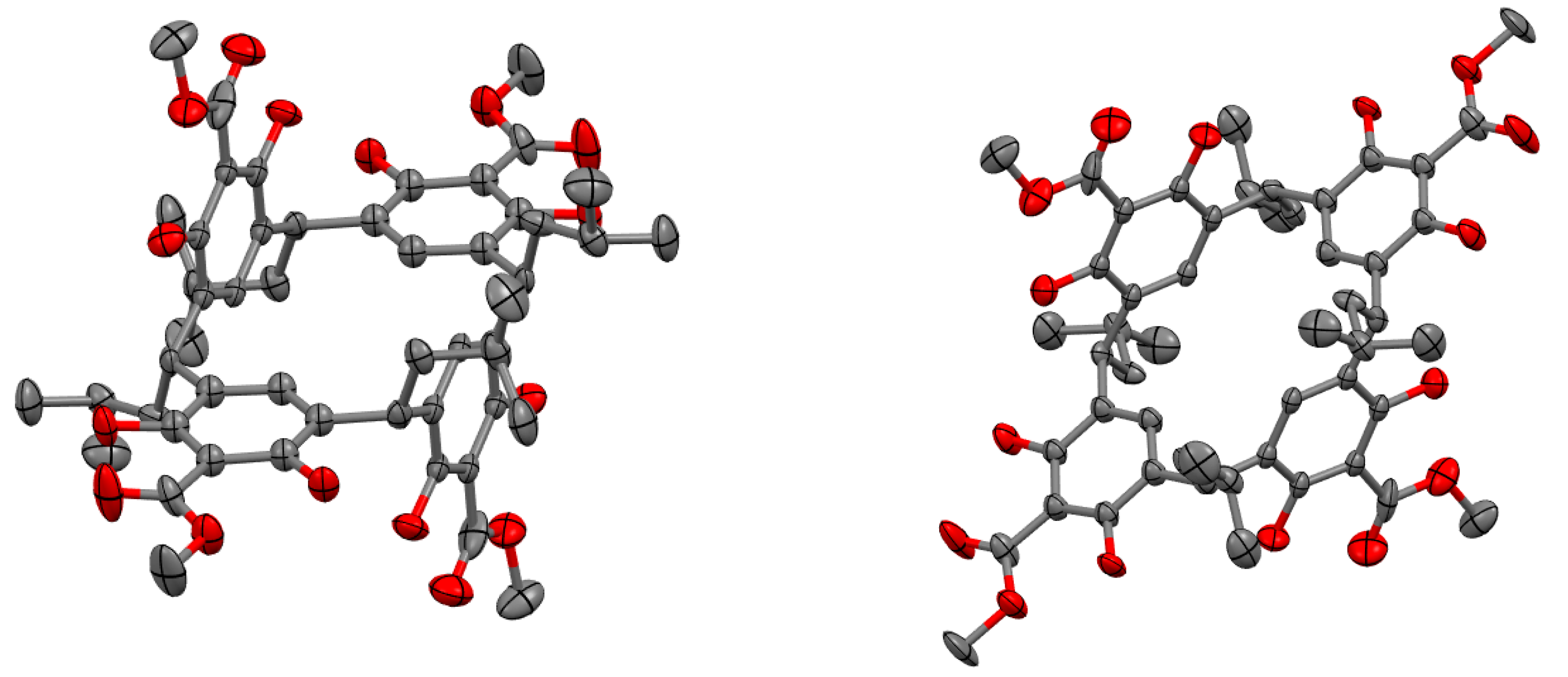
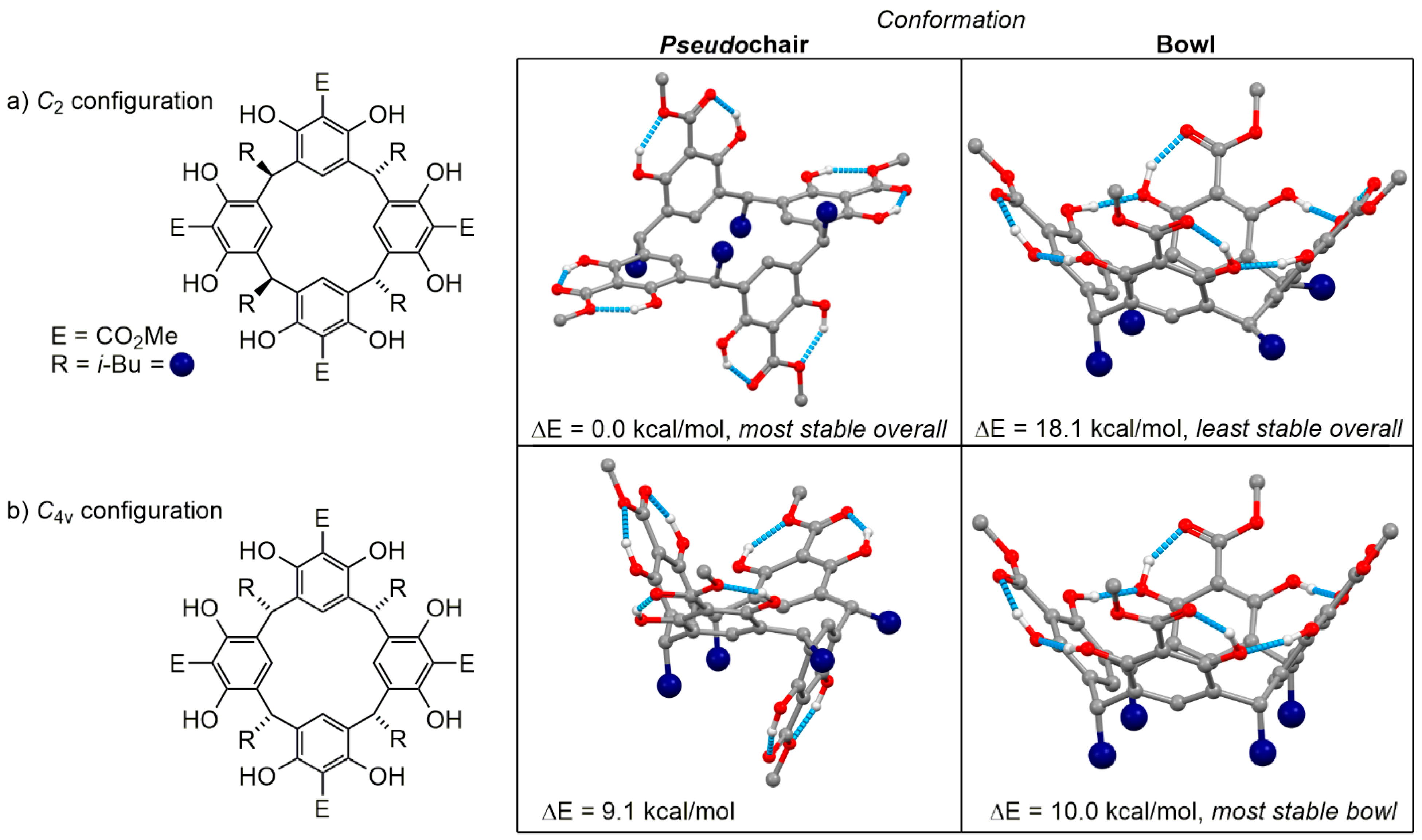
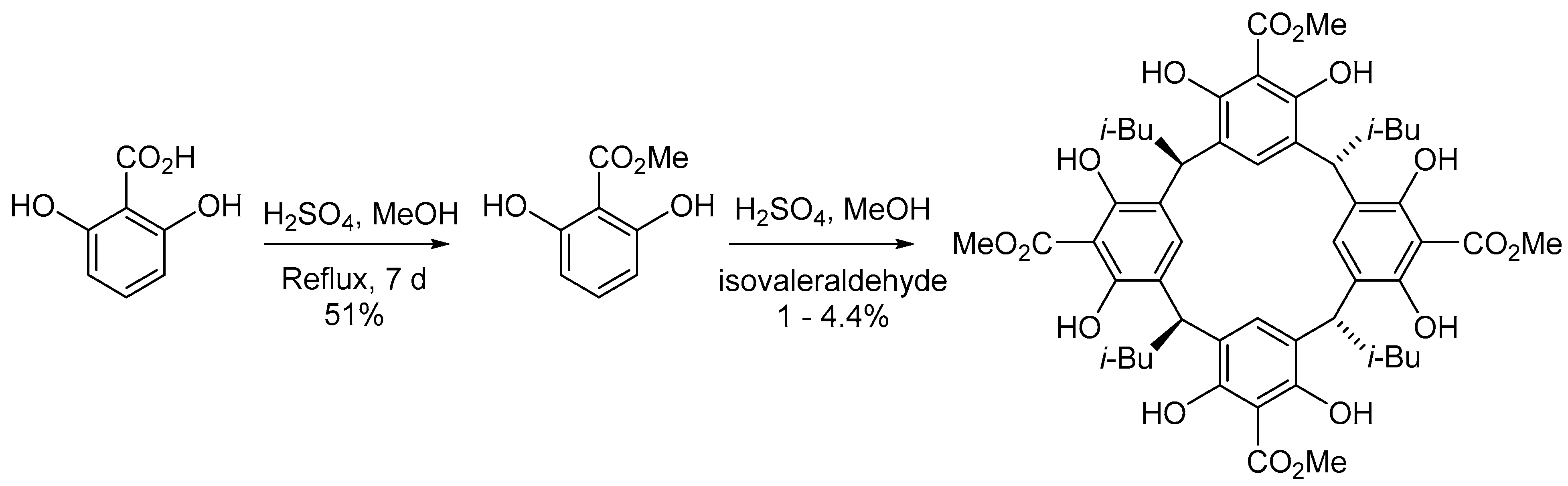
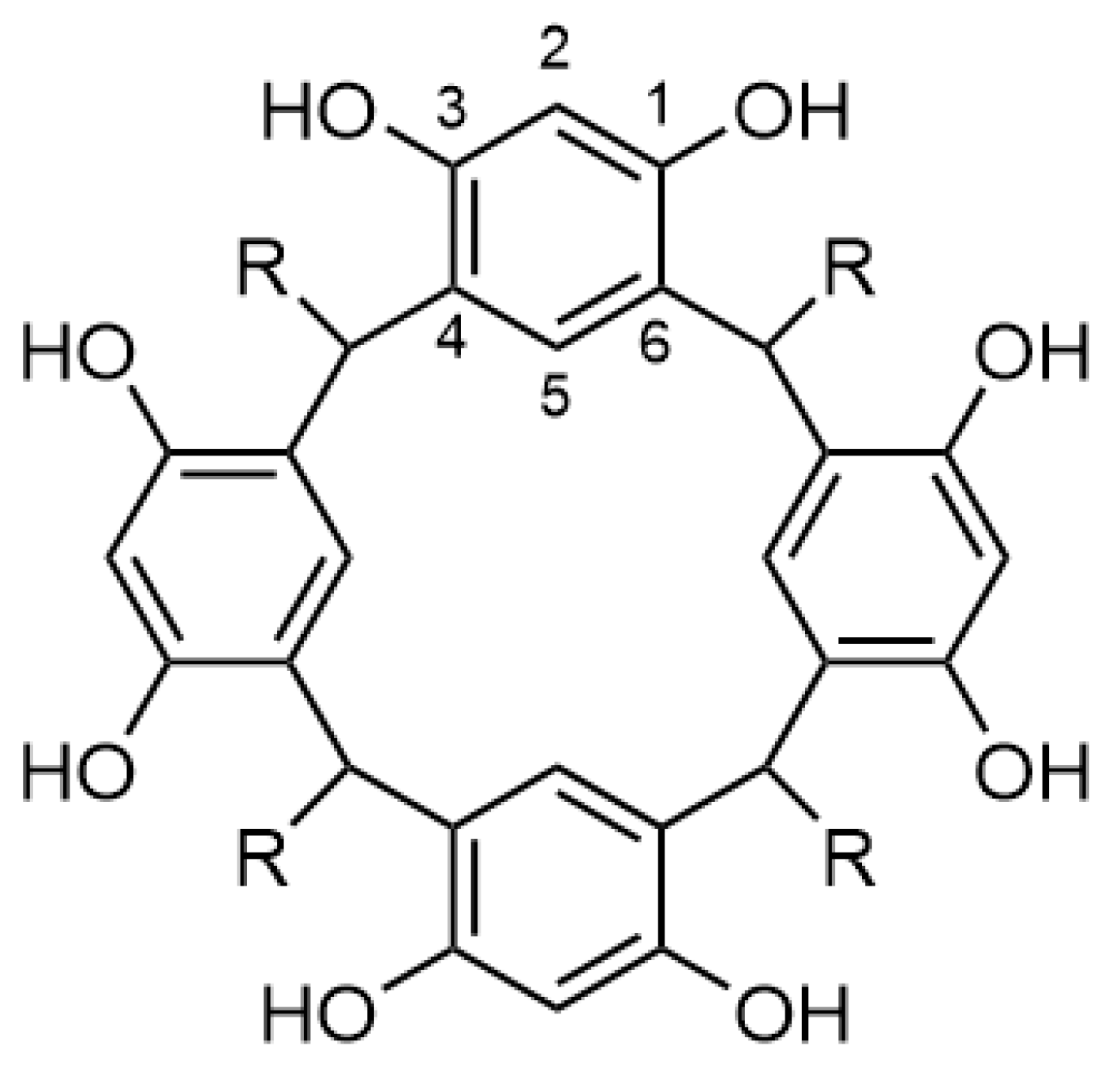
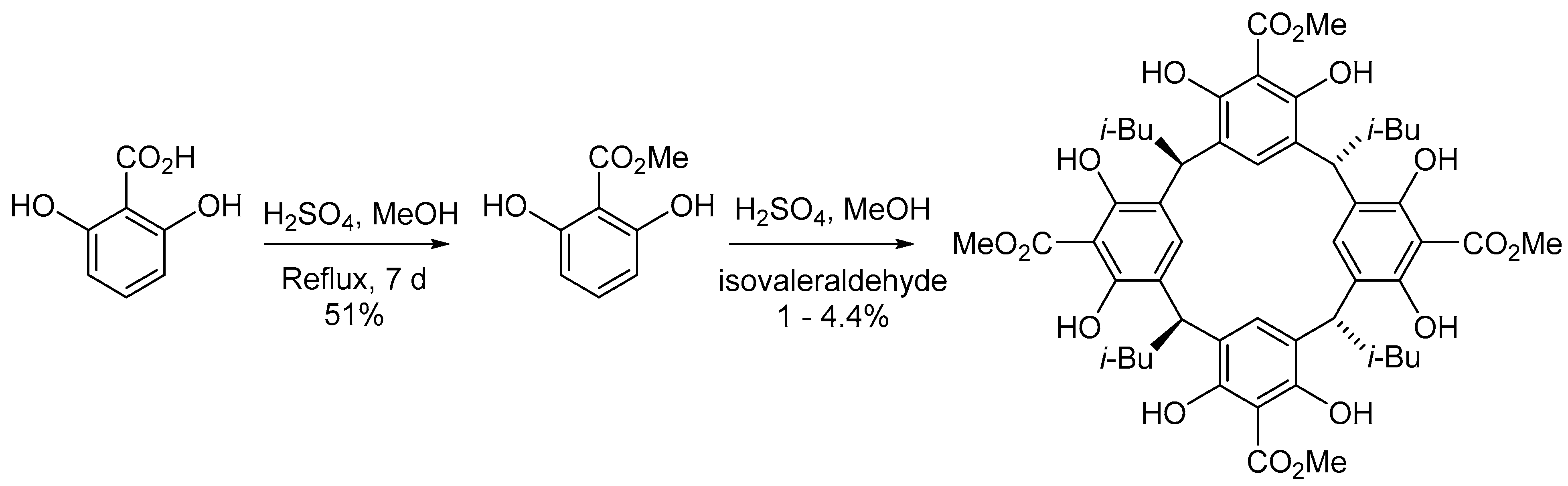
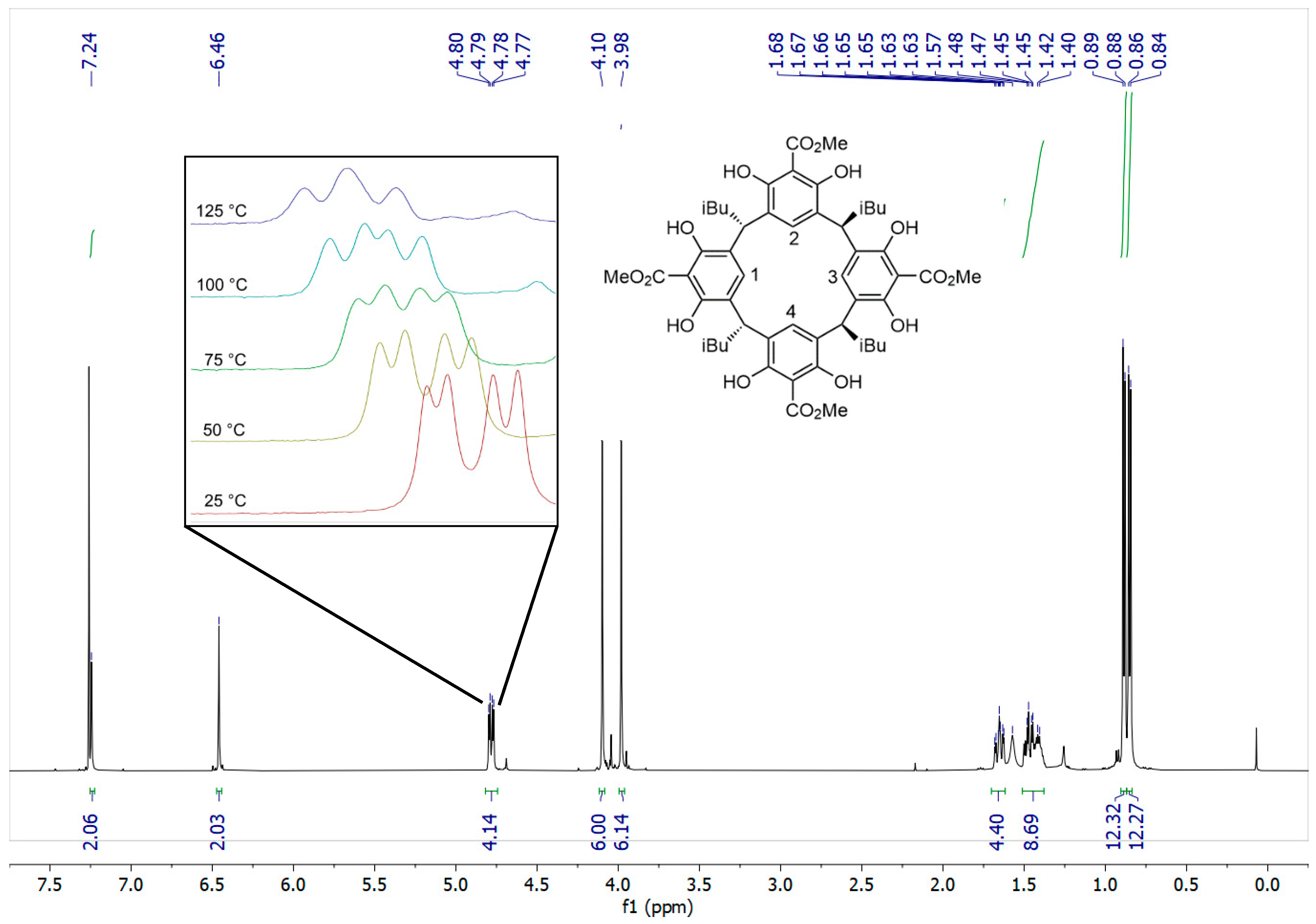
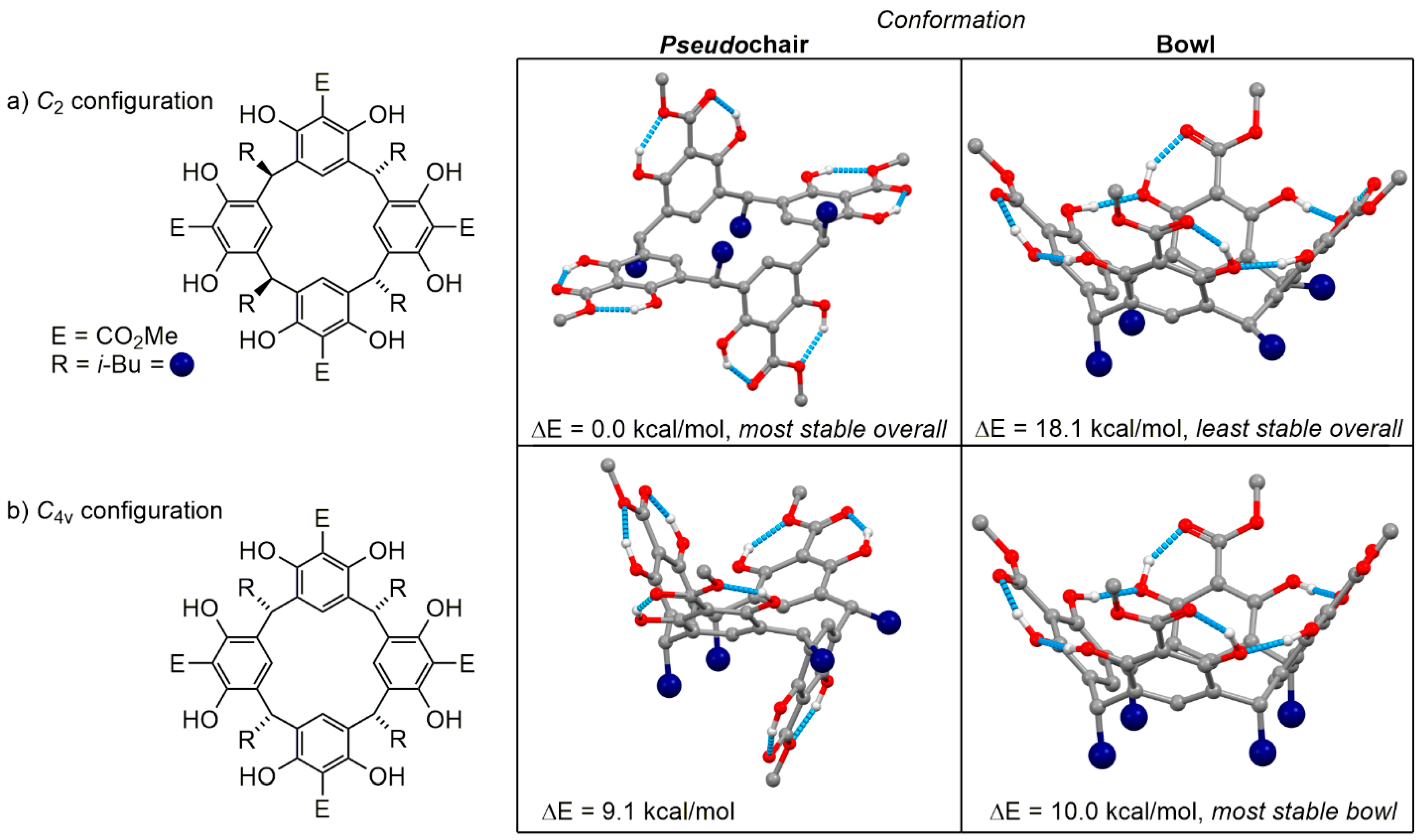
2. Results and Discussion
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sliwa, W.; Kozlowski, C. Calixarenes and Resorcinarenes; Wiley: Weinheim, Germany, 2009. [Google Scholar]
- Timmerman, P.; Verboom, W.; Reinhoudt, D.N. Resorcinarenes. Tetrahedron 1996, 52, 2663–2704. [Google Scholar] [CrossRef] [Green Version]
- Böhmer, V. Calixarenes, macrocycles with (almost) unlimited possibilities. Angew. Chem. Int. Ed. 1995, 34, 713–745. [Google Scholar] [CrossRef]
- Rissanen, K. Very large container molecules. Angew. Chem. Int. Ed. 2005, 44, 3652–3654. [Google Scholar] [CrossRef]
- Atwood, J.L.; Szumna, A. Anion-sealed single-molecule capsules. Chem. Commun. 2003, 8, 940–941. [Google Scholar] [CrossRef]
- Atwood, J.L.; Barbour, L.J.; Jerga, A. Organization of the interior of molecular capsules by hydrogen bonding. Proc. Natl. Acad. Sci. USA 2002, 99, 4837–4841. [Google Scholar] [CrossRef] [Green Version]
- MacGillivray, L.; Atwood, J. Unique guest inclusion within multi-component, extended-cavity resorcin[4]arenes. Chem. Commun. 1999, 2, 181–182. [Google Scholar] [CrossRef]
- Beyeh, N.K.; Kogej, M.; Åhman, A.; Rissanen, K.; Schalley, C.A. Flying capsules: Mass spectrometric detection of pyrogallarene and resorcinarene hexamers. Angew. Chem. Int. Ed. 2006, 45, 5214–5218. [Google Scholar] [CrossRef]
- Shivanyuk, A.; Paulus, E.F.; Rissanen, K.; Kolehmainen, E.; Böhmer, V. Resorcarenes in the boat conformation as building blocks for hydrogen-bonded assemblies including two ammonium cations. Chem. Eur. J. 2001, 7, 1944–1951. [Google Scholar] [CrossRef]
- Beyeh, N.K.; Weimann, D.P.; Kaufmann, L.; Schalley, C.A.; Rissanen, K. Ion-pair recognition of tetramethylammonium salts by halogenated resorcinarenes. Chem. Eur. J. 2012, 18, 5552–5557. [Google Scholar] [CrossRef] [PubMed]
- Avram, L.; Cohen, Y.; Rebek, J., Jr. Recent advances in hydrogen-bonded hexameric encapsulation complexes. Chem. Commun. 2011, 47, 5368–5375. [Google Scholar] [CrossRef] [PubMed]
- Koshets, I.A.; Kazantseva, Z.I.; Belyaev, A.E.; Kalchenko, V.I. Sensitivity of resorcinarene films towards aliphatic alcohols. Sens. Actuators B 2009, 140, 104–108. [Google Scholar] [CrossRef]
- Ugono, O.; Holman, K.T. An achiral form of the hexameric resorcin[4]arene capsule sustained by hydrogen bonding with alcohols. Chem. Commun. 2006, 20, 2144–2146. [Google Scholar] [CrossRef] [PubMed]
- Fox, O.D.; Leung, J.F.Y.; Hunter, J.M.; Dalley, N.K.; Harrison, R.G. Metal-assembled cobalt(II) resorc[4]arene-based cage molecules that reversibly capture organic molecules from water and act as NMR shift reagents. Inorg. Chem 2000, 39, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Kalenius, E.; Kekäläinen, T.; Neitola, R.; Beyeh, K.; Rissanen, K.; Vainiotalo, P. Size- and structure-selective noncovalent recognition of saccharides by tetraethyl and tetraphenyl resorcinarenes in the gas phase. Chem. Eur. J. 2008, 14, 5220–5228. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Johnson, R.J.; Escobedo, J.O.; Beck, P.A.; Kim, K.K.; St. Luce, N.N.; Davis, C.J.; Lewis, P.T.; Fronczek, F.R.; Melancon, B.J.; et al. Chromophore formation in resorcinarene solutions and the visual detection of mono- and oligosaccharides. J. Am. Chem. Soc. 2002, 124, 5000–5009. [Google Scholar] [CrossRef] [Green Version]
- Evan-Salem, T.; Baruch, I.; Avram, L.; Cohen, Y.; Palmer, L.C.; Rebek, J. Resorcinarenes are hexameric capsules in solution. Proc. Natl. Acad. Sci. USA 2006, 103, 12296. [Google Scholar] [CrossRef] [Green Version]
- Rhlalou, T.; Ferhat, M.; Frouji, M.A.; Langevin, D.; Métayer, M.; Verchère, J.F. Facilitated transport of sugars by a resorcinarene through a supported liquid membrane. J. Membr. Sci. 2000, 168, 63–73. [Google Scholar] [CrossRef]
- Shivanyuk, A.; Rebek, J.J. Hydrogen-bonded capsules in polar, protic solvents. Chem. Commun. 2001, 22, 2374–2375. [Google Scholar] [CrossRef]
- Faull, J.D.; Gupta, V.K. Chemical selectivity of self-assembled monolayers of calix[4]resorcinarene. Thin Solid Films 2003, 440, 129–137. [Google Scholar] [CrossRef]
- Faull, J.D.; Gupta, V.K. Impact of host structure on guest−host recognition at self-assembled surfaces of tetrathiol and tetrasulfide derivatives of calix[4]resorcinarene. Langmuir 2002, 18, 6584–6592. [Google Scholar] [CrossRef]
- Puttreddy, R.; Beyeh, N.K.; Jurček, P.; Turunen, L.; Trant, J.F.; Ras, R.H.A.; Rissanen, K. Host-guest complexes of C-propyl-2-bromoresorcinarene with aromatic N-oxides. Supramol. Chem. 2018, 30, 445–454. [Google Scholar] [CrossRef]
- Puttreddy, R.; Beyeh, N.K.; Taimoory, S.M.; Meister, D.; Trant, J.F.; Rissanen, K. Host–guest complexes of conformationally flexible C-hexyl-2-bromoresorcinarene and aromatic N-oxides: Solid-state, solution and computational studies. Beilstein J. Org. Chem. 2018, 14, 1723–1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puttreddy, R.; Beyeh, N.K.; Ras, R.H.A.; Trant, J.; Rissanen, K. Endo-/exo- and halogen bonded complexes of conformationally rigid C-ethyl-2-bromoresorcinarene and aromatic N-oxides. CrystEngComm 2017, 19, 4312–4320. [Google Scholar] [CrossRef] [Green Version]
- Nissinen, M.; Rissanen, K. Crystal engineering studies of the complexes of ethyl resorcinarene with aromatic nitrogen heterocycles. Supramol. Chem. 2003, 15, 581–590. [Google Scholar] [CrossRef]
- Nissinen, M.; Wegelius, E.; Falábu, D.; Rissanen, K. Melamine induced conformational change of ethyl resorcinarene in solid state. CrystEngComm 2000, 2, 151–153. [Google Scholar] [CrossRef]
- Taimoory, S.M.; Twum, K.; Dashti, M.; Pan, F.; Lahtinen, M.; Rissanen, K.; Puttreddy, R.; Trant, J.F.; Beyeh, N.K. Bringing a molecular plus one: Synergistic binding creates guest-mediated three-component complexes. J. Org. Chem. 2020, 85, 5884–5894. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.-L.; Gembicky, M.; Messerschmidt, M.; Dominiak, P.M.; Coppens, P. Effect of the environment on molecular properties: Synthesis, structure, and photoluminescence of Cu(I) bis(2,9-dimethyl-1,10-phenanthroline) nanoclusters in eight different supramolecular frameworks. Inorg. Chem. 2006, 45, 9281–9289. [Google Scholar] [CrossRef]
- Murayama, K. Resorcin[4]arene dimer linked by eight water molecules and incorporating a tetraethylammonium ion: Guest-driven capsule formation via cation–π interactions. Chem. Commun. 1998, 5, 607–608. [Google Scholar] [CrossRef]
- Atwood, J.L.; Barbour, L.J.; Hardie, M.J.; Lygris, E.; Raston, C.L.; Webb, H.R. Inclusion complexes of 18-crown-6 and (Na+⊂[2.2.2]cryptand) in [C-methylcalix[4]resorcinarene-Hn], n⊕=⊕0, 1. CrystEngComm 2001, 3, 41–43. [Google Scholar] [CrossRef]
- Högberg, A.G.S. Two stereoisomeric macrocyclic resorcinol-acetaldehyde condensation products. J. Org. Chem. 1980, 45, 4498–4500. [Google Scholar] [CrossRef]
- Beyeh, N.K.; Pan, F.; Rissanen, K. A halogen-bonded dimeric resorcinarene capsule. Angew. Chem. Int. Ed. 2015, 54, 7303–7307. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reynolds, M.R.; Pick, F.S.; Hayward, J.J.; Trant, J.F. A Concise Synthesis of a Methyl Ester 2-Resorcinarene: A Chair-Conformation Macrocycle. Symmetry 2021, 13, 627. https://doi.org/10.3390/sym13040627
Reynolds MR, Pick FS, Hayward JJ, Trant JF. A Concise Synthesis of a Methyl Ester 2-Resorcinarene: A Chair-Conformation Macrocycle. Symmetry. 2021; 13(4):627. https://doi.org/10.3390/sym13040627
Chicago/Turabian StyleReynolds, Michael R., Fraser S. Pick, John J. Hayward, and John F. Trant. 2021. "A Concise Synthesis of a Methyl Ester 2-Resorcinarene: A Chair-Conformation Macrocycle" Symmetry 13, no. 4: 627. https://doi.org/10.3390/sym13040627
APA StyleReynolds, M. R., Pick, F. S., Hayward, J. J., & Trant, J. F. (2021). A Concise Synthesis of a Methyl Ester 2-Resorcinarene: A Chair-Conformation Macrocycle. Symmetry, 13(4), 627. https://doi.org/10.3390/sym13040627