
Vibrational Analysis of Paraelectric–Ferroelectric Transition of LiNbO3: An Ab-Initio Quantum Mechanical Treatment

 ,
, 
 ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. Computational Methods
3. Results
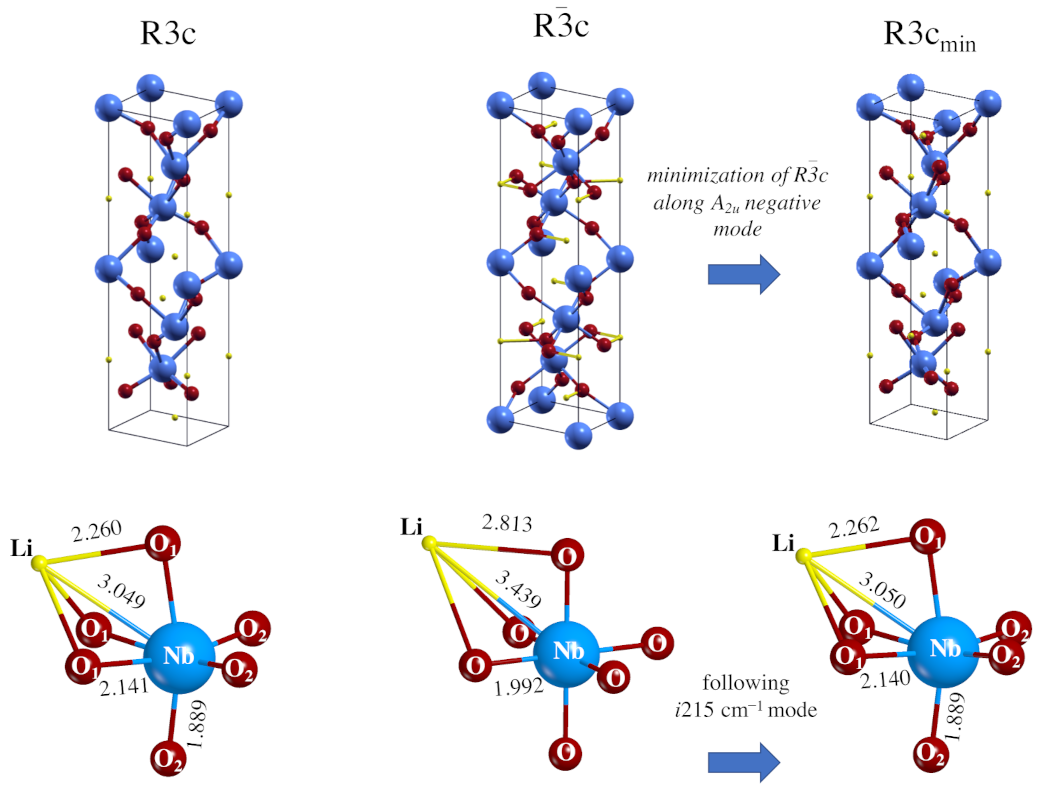
3.1. Geometrical and Electronic Properties of the Two Phases
3.2. Infrared Spectra
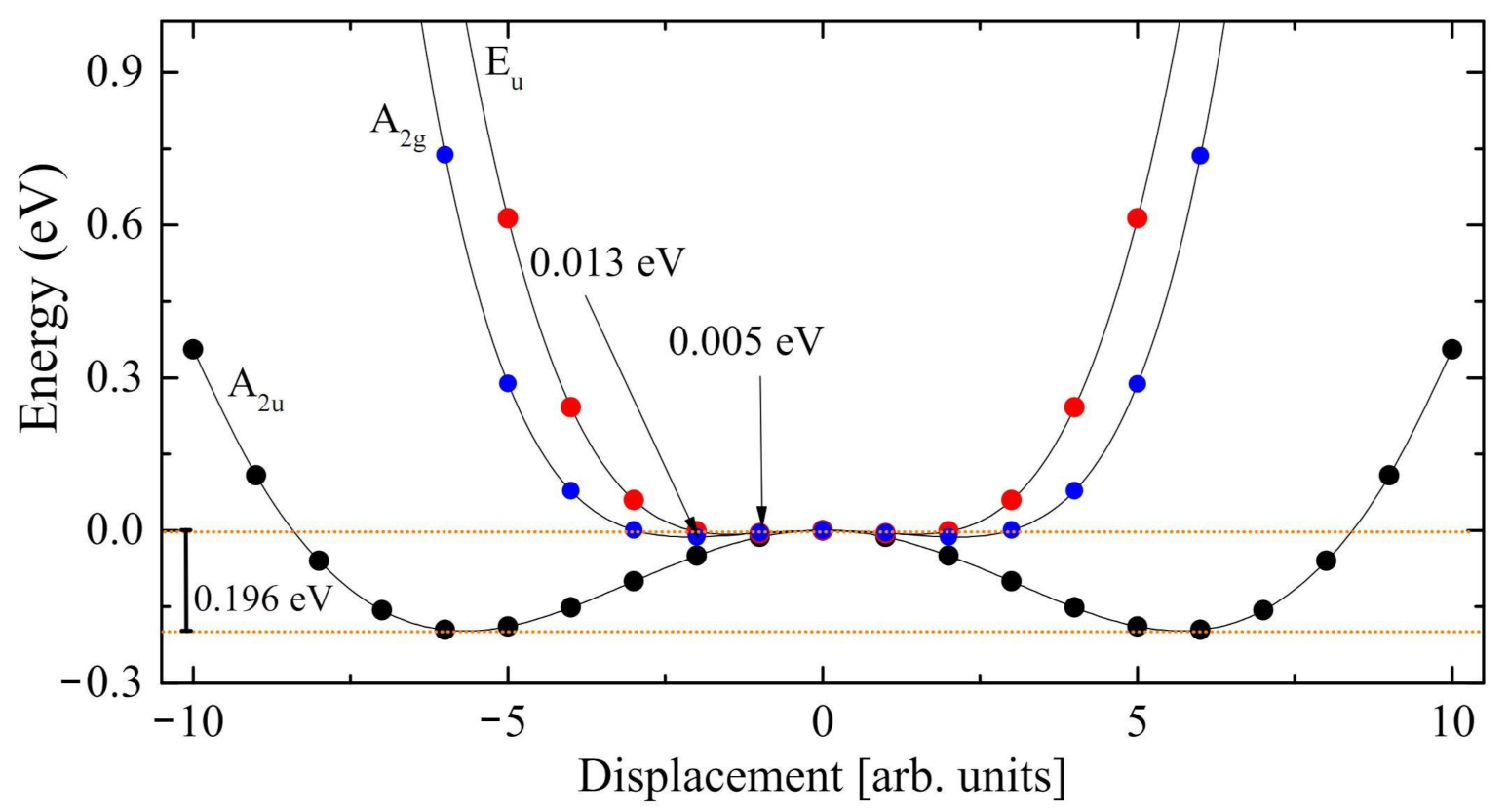
3.3. The Modes with Imaginary Wavenumber
- (i)
- the SCANMODE option, which allows us to follow the PES along a particular vibrational eigenvalue;
- (ii)
- the isotopic substitution;
- (iii)
- the graphical animation of the modes of interest.
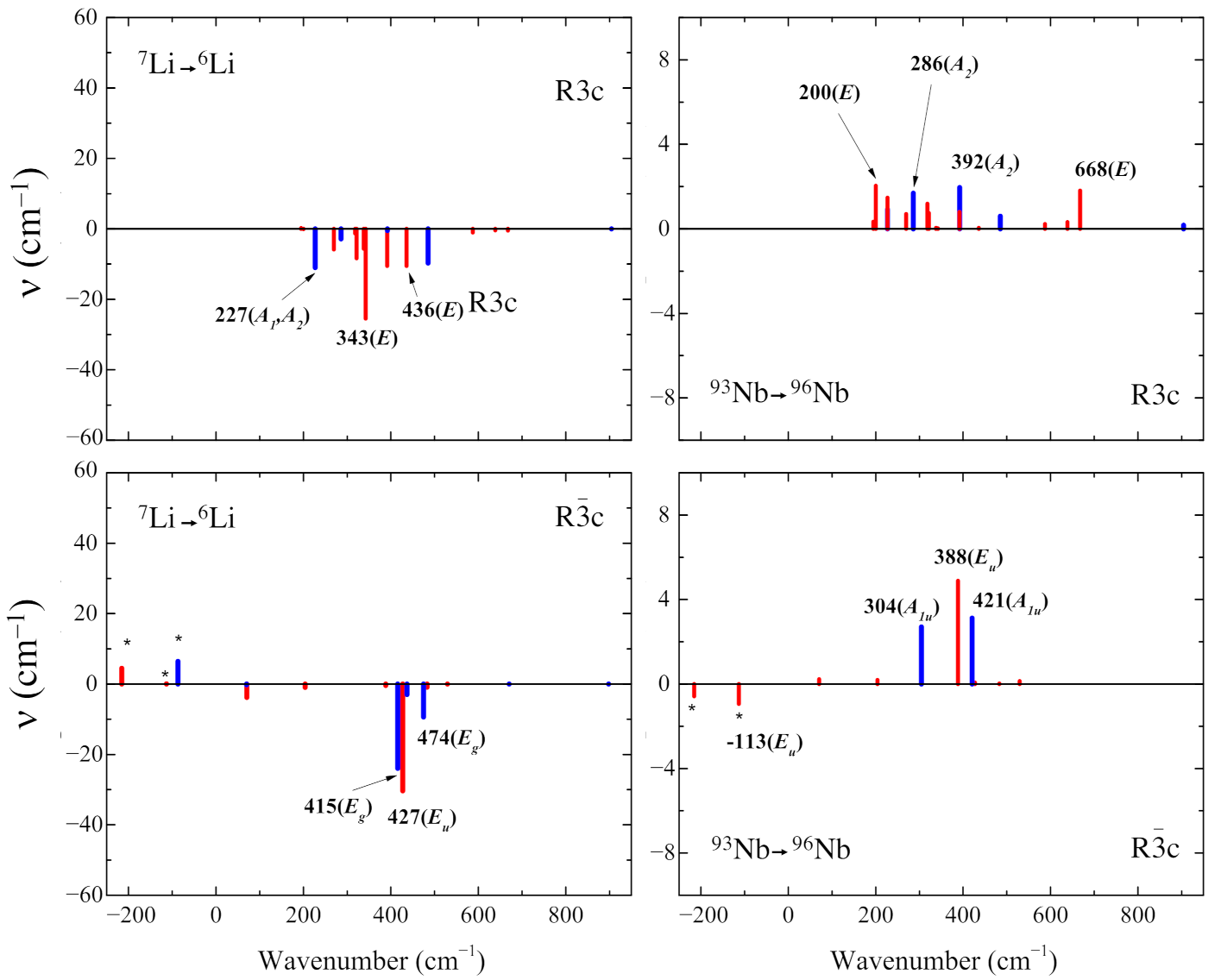
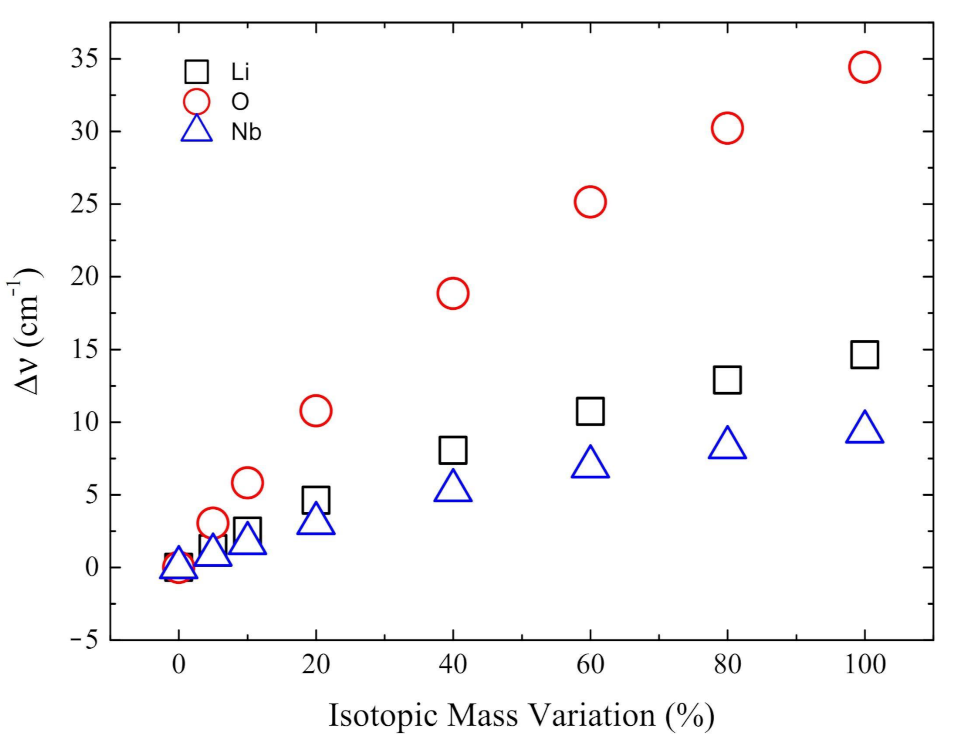
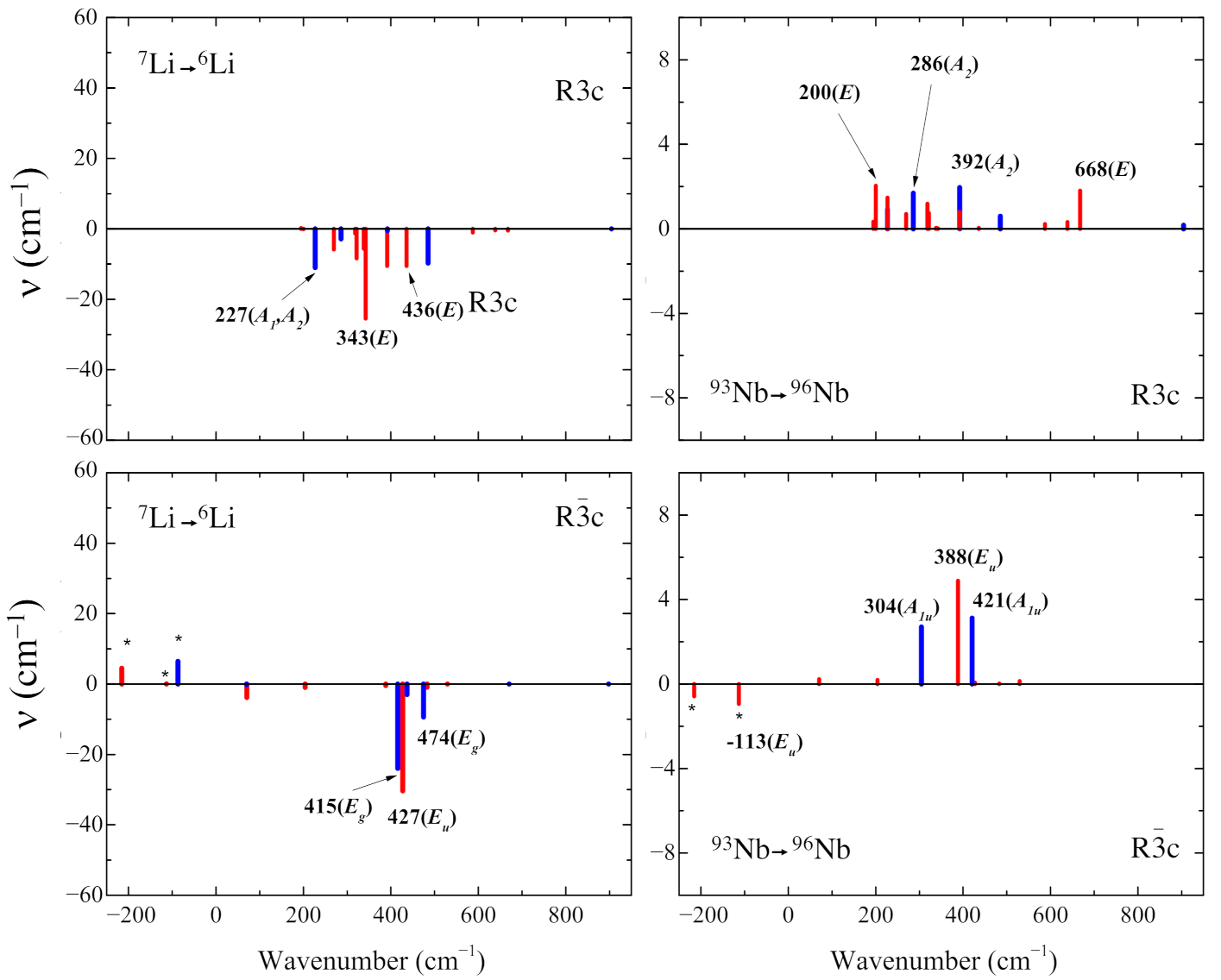
3.4. Isotopic Substitutions
4. Conclusions
- The global hybrid B3LYP calculations reproduce accurately the crystallographic data proposed in the literature, with a small underestimation of the cell parameters and the vibrational features [48]. The R3c system is a non-centro-symmetric structure with a large octahedral distortion due to the Jahn–Teller effect. The Rc centro-symmetric form has a perfect octahedral structure, in which the niobium atoms coordinate six equivalent oxygen atoms.
- The R3c polymorph is more stable than Rc by about 0.32 eV.
- The infrared spectra characterizing the two systems are quite different. The R3c one is dominated by four main peaks at 200, 319, 588 and 639 cm. The mode at 904 cm is not visible in the spectrum because it is IR-inactive for symmetry reasons (A symmetry).In addition, the R spectrum presents four main peaks, spanning from 388 and 529 cm, with other minor features. As for the R3c system, the highest vibration at 898 cm is not visible in the IR spectrum (A symmetry).When comparing the set of vibrational modes of the two structures, the only clearly common mode is the highest one, at 904 cm for Rc and just 6 cm redshifted in Rc.
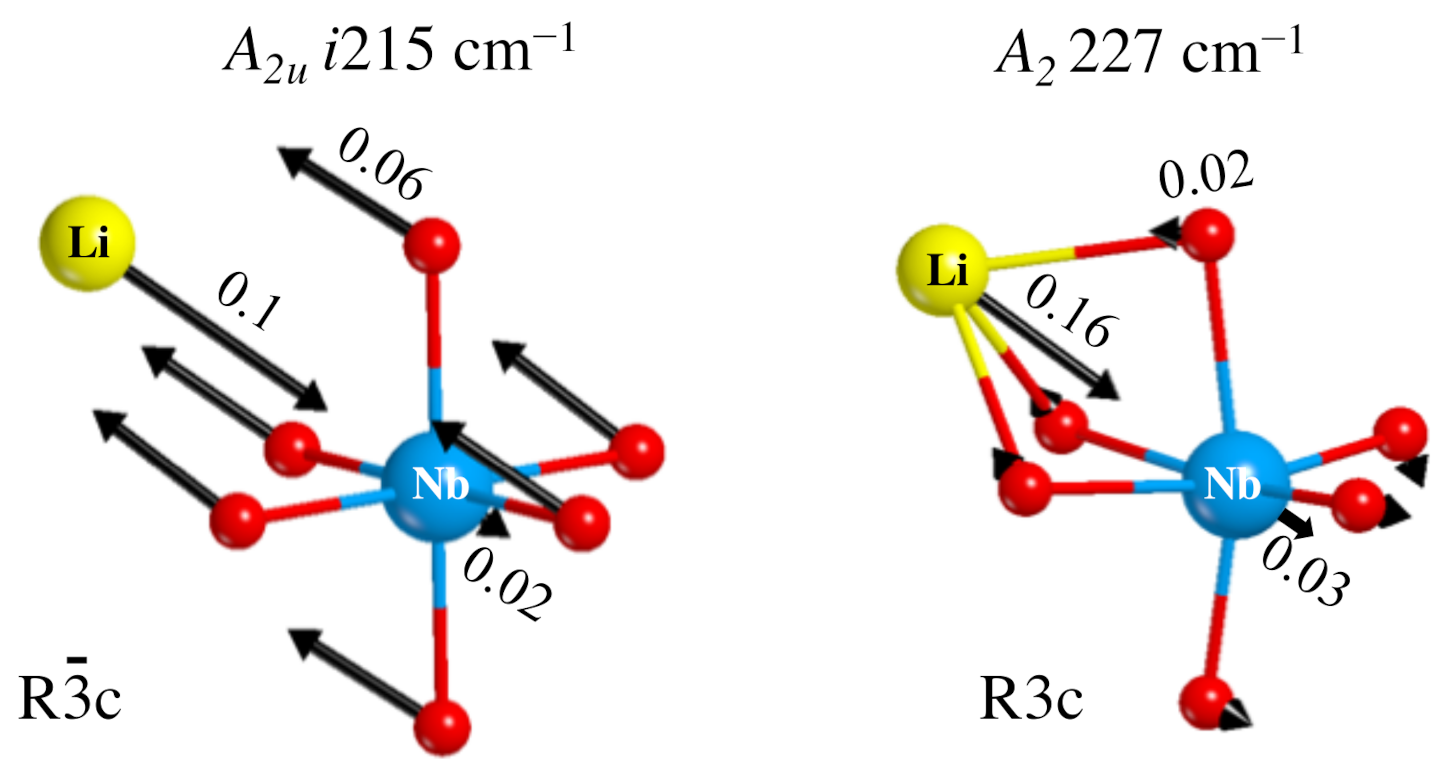
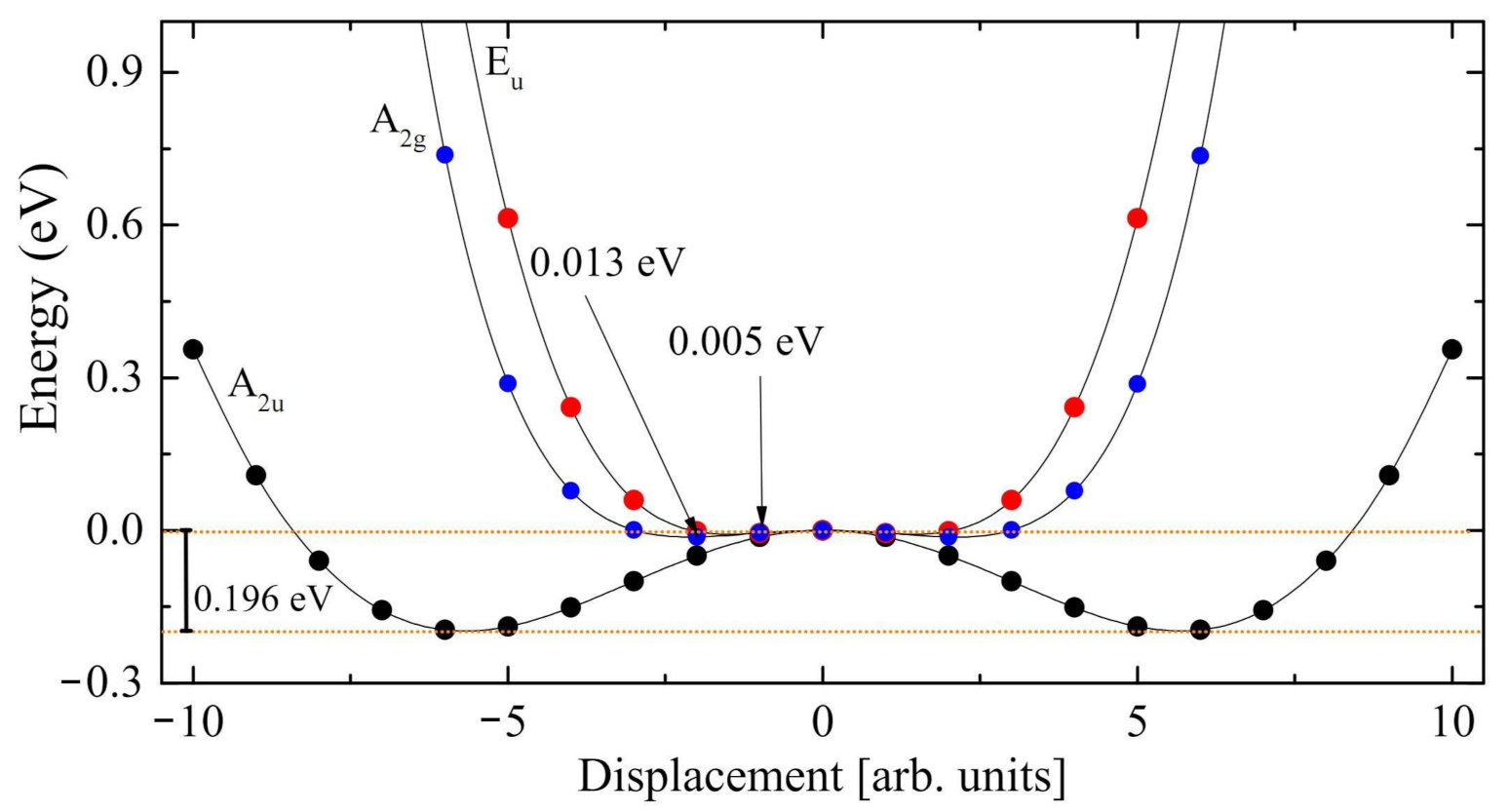
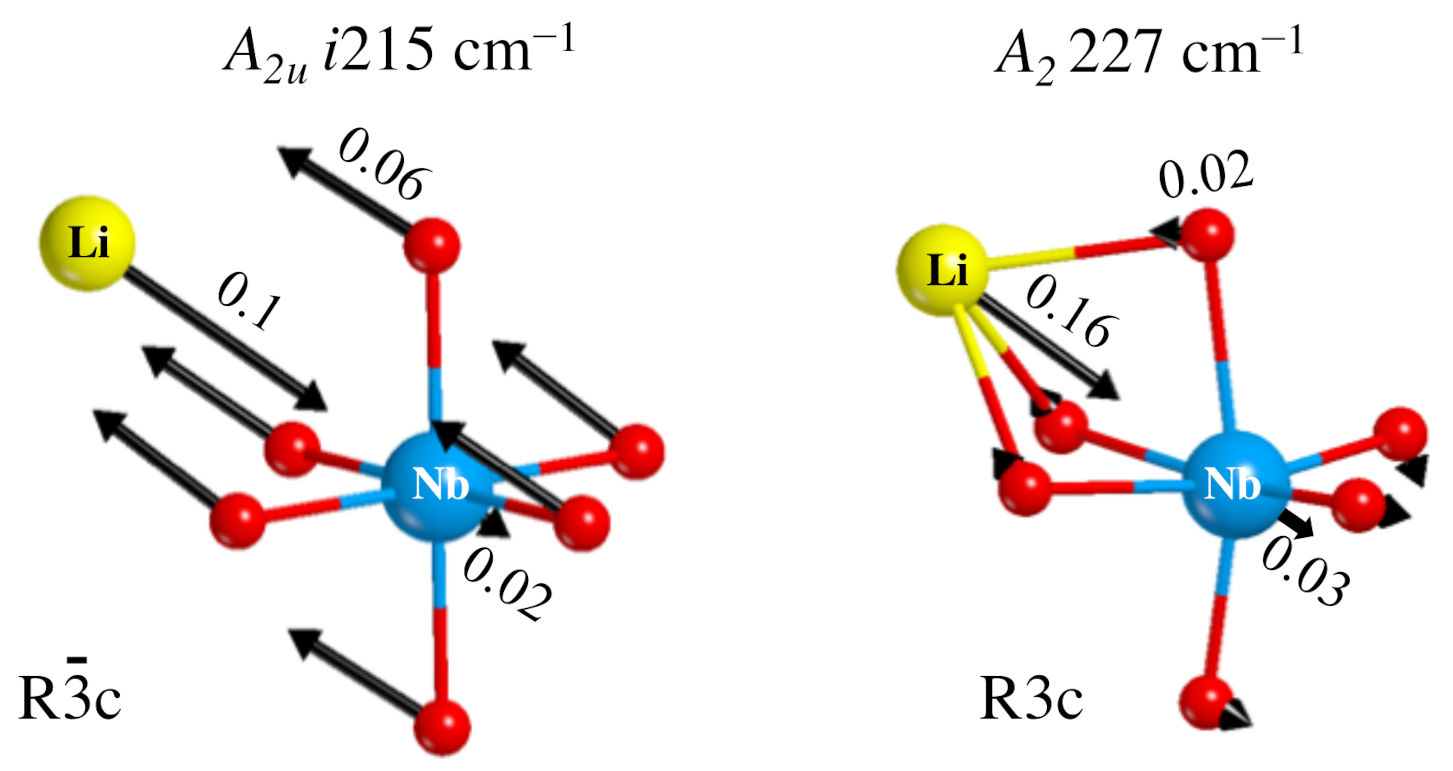
- The Rc spectrum is characterized by four imaginary eigenvalues, indicating a geometrical instability. At 0 K this structure is then unstable with respect to many Cartesian coordinates. Scanning the PES along the vibrational modes with imaginary wavenumbers it turns out that all of them present a double minimum around the common maximum of the PE structure. However, the minima of the mode with the i215 cm wavenumber are much deeper than the minima of the three other modes. This A mode is the channel for the PE to FE transition.
- The isotopic substitution shows that the i215 cm is mainly contributed by oxygen, as the graphical animation of the mode confirms.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Belik, A.A.; Furubayashi, T.; Yusa, H.; Takayama-Muromachi, E. Perovskite, LiNbO3, Corundum, and Hexagonal Polymorphs of (In1–x M x) MO3. J. Am. Chem. Soc. 2011, 133, 9405–9412. [Google Scholar] [CrossRef] [PubMed]
- Xue, D.; He, X. Dopant Occupancy and Structural Stability of Doped Lithium Niobate Crystals. Phys. Rev. B 2006, 73, 064113. [Google Scholar] [CrossRef]
- Choubey, R.; Sen, P.; Sen, P.; Bhatt, R.; Kar, S.; Shukla, V.; Bartwal, K. Optical Properties of MgO Doped LiNbO3 Single Crystals. Opt. Mater. 2006, 28, 467–472. [Google Scholar] [CrossRef]
- Beyer, O.; Breunig, I.; Kalkum, F.; Buse, K. Photorefractive Effect in Iron-Doped Lithium Niobate Crystals Induced by Femtosecond Pulses of 1.5 μm Wavelength. Appl. Phys. Lett. 2006, 88, 051120. [Google Scholar] [CrossRef]
- Yang, G.; Kong, Y.; Hou, W.; Yan, Q. Heating Behavior and Crystal Growth Mechanism in Microwave Field. J. Phys. Chem. B 2005, 109, 1371–1379. [Google Scholar] [CrossRef]
- Yu, B.; Cao, B.; Cao, H.; Zhang, X.; Chen, D.; Qu, J.; Niu, H. Synthesis and Nonlinear Optical Properties of Single-Crystalline KNb3O8 Nanowires. Nanotechnology 2013, 24, 085704. [Google Scholar] [CrossRef]
- Oikawa, S.; Kawanishi, T.; Higuma, K.; Matsuo, Y.; Izutsu, M. Double-Stub Structure for Resonant-Type Optical Modulators Using 20-μm-Thick Electrode. IEEE Photonics Technol. Lett. 2003, 15, 221–223. [Google Scholar] [CrossRef]
- Ali, R.F.; Gates, B.D. Synthesis of Lithium Niobate Nanocrystals with Size Focusing Through an Ostwald Ripening Process. Chem. Mater. 2018, 30, 2028–2035. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Molina, R.; Hernández-Márquez, J.; Enríquez-Carrejo, J.; Farias-Mancilla, J.; Mani-González, P.; Vigueras Santiago, E.; Rodríguez-Aranda, M.; Vargas-Ortíz, A.; Yáñez-Limón, J. Synthesis by Wet Chemistry and Characterization of LiNbO3 Nanoparticles. Superficies y Vacío 2015, 28, 115–118. [Google Scholar]
- Dutto, F.; Raillon, C.; Schenk, K.; Radenovic, A. Nonlinear Optical Response in Single Alkaline Niobate Nanowires. Nano Lett. 2011, 11, 2517–2521. [Google Scholar] [CrossRef] [Green Version]
- Araujo, R.M.; Valerio, M.E.; Jackson, R.A. Computer Modelling of Hafnium Doping in Lithium Niobate. Crystals 2018, 8, 123. [Google Scholar] [CrossRef] [Green Version]
- Mamoun, S.; Merad, A.; Guilbert, L. Energy Band Gap and Optical Properties of Lithium Niobate from Ab Initio Calculations. Comput. Mater. Sci. 2013, 79, 125–131. [Google Scholar] [CrossRef]
- Young, S.M.; Zheng, F.; Rappe, A.M. First-Principles Materials Design of High-Performing Bulk Photovoltaics with the LiNbO3 Structure. Phys. Rev. Appl. 2015, 4, 054004. [Google Scholar] [CrossRef] [Green Version]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Re´rat, M.; Casassa, S.; Baima, J.; Salustro, S.; et al. Quantum-Mechanical Condensed Matter Simulations with CRYSTAL. Wires 2018, 8, e1360. [Google Scholar] [CrossRef]
- Dovesi, R.; Pascale, F.; Civalleri, B.; Doll, K.; Harrison, N.M.; Bush, I.; D’arco, P.; Noël, Y.; Rérat, M.; Carbonniere, P.; et al. The CRYSTAL Code, 1976–2020 and Beyond, a Long Story. J. Chem. Phys. 2020, 152, 204111. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R. Development of the Colle-Salvetti Correlation-Energy Formula Into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Dovesi, R.; Saunders, V.; Roetti, C.; Orlando, R.; Zicovich-Wilson, C.; Pascale, F.; Civalleri, B.; Doll, K.; Harrison, N.; Bush, I.; et al. CRYSTAL17 User’s Manual. Manuals/crystal17.pdf. 2017. Available online: http://www.crystal.unito.it (accessed on 1 August 2021).
- Vilela Oliveira, D.; Laun, J.; Peintinger, M.F.; Bredow, T. BSSE-Correction Scheme for Consistent Gaussian Basis Sets of Double-and Triple-Zeta Valence with Polarization Quality for Solid-State Calculations. J. Comput. Chem. 2019, 40, 2364–2376. [Google Scholar] [CrossRef]
- Gentile, F.S.; Pannico, M.; Causà, M.; Mensitieri, G.; Di Palma, G.; Scherillo, G.; Musto, P. Metal Defects in HKUST-1 MOF Revealed by Vibrational Spectroscopy: A Combined Quantum Mechanical and Experimental Study. J. Mater. Chem. A 2020, 8, 10796–10812. [Google Scholar] [CrossRef]
- Gage, S.H.; Ngo, C.; Molinari, V.; Causa, M.; Richards, R.M.; Gentile, F.S.; Pylypenko, S.; Esposito, D. Strong Metal–Support Interactions of TiN–and TiO2–Nickel Nanocomposite Catalysts. J. Phys. Chem. C 2018, 122, 339–348. [Google Scholar] [CrossRef]
- Gentile, F.S.; Mackrodt, W.C.; Allan, N.L.; Dovesi, R. Predicted Strong Spin-Phonon Interactions in Li-Doped Diamond. Phys. Chem. Chem. Phys. 2020, 22, 20612–20617. [Google Scholar] [CrossRef]
- Mackrodt, W.C.; Rérat, M.; Gentile, F.S.; Dovesi, R. An All-Electron Study of the Low-Lying Excited States and Optical Constants of Al2O3 in the Range 5–80 eV. J. Phys. Condens. Matter 2019, 32, 085901. [Google Scholar] [CrossRef] [PubMed]
- Gentile, F.S.; Salustro, S.; Desmarais, J.K.; Ferrari, A.M.; D’Arco, P.; Dovesi, R. Vibrational Spectroscopy of Hydrogens in Diamond: A Quantum Mechanical Treatment. Phys. Chem. Chem. Phys. 2018, 20, 11930–11940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Palma, G.; Kirtman, B.; Gentile, F.S.; Platonenko, A.; Ferrari, A.M.; Dovesi, R. The VN2 Negatively Charged Defect in Diamond. A Quantum Mechanical Investigation of the EPR Response. Carbon 2020, 159, 443–450. [Google Scholar] [CrossRef]
- Causa, M.; D’Amore, M.; Garzillo, C.; Gentile, F.; Savin, A. The Bond Analysis Techniques (ELF and Maximum Probability Domains) Application to a Family of Models Relevant to Bio-Inorganic Chemistry. Struct. Bond 2013, 150, 119–142. [Google Scholar]
- Salustro, S.; Gentile, F.S.; D’Arco, P.; Civalleri, B.; Rérat, M.; Dovesi, R. Hydrogen Atoms in the Diamond Vacancy Defect. A Quantum Mechanical Vibrational Analysis. Carbon 2018, 129, 349–356. [Google Scholar] [CrossRef]
- Salustro, S.; Gentile, F.S.; Erba, A.; Carbonniére, P.; El-Kelany, K.; Dovesi, R. The Characterization of the VNxHy Defects in Diamond through the Infrared Vibrational Spectrum. A Quantum Mechanical Investigation. Carbon 2018, 132, 210–219. [Google Scholar] [CrossRef]
- Ferrari, A.; Salustro, S.; Gentile, F.; Mackrodt, W.; Dovesi, R. Substitutional Nitrogen Atom in Diamond. A Quantum Mechanical Investigation of the Electronic and Spectroscopic Properties. Carbon 2018, 134, 354–365. [Google Scholar] [CrossRef] [Green Version]
- Gentile, F.; Salustro, S.; Di Palma, G.; Causá, M.; D’arco, P.; Dovesi, R. Hydrogen, Boron and Nitrogen Atoms in Diamond: A Quantum Mechanical Vibrational Analysis. Theor. Chem. Acc. 2018, 137, 154. [Google Scholar] [CrossRef] [Green Version]
- Di Palma, G.; Gentile, F.S.; Lacivita, V.; Mackrodt, W.; Dovesi, R. N 2 positively charged defects in diamond. A Quantum Mechanical investigation of the structural, electronic, EPR and vibrational properties. J. Mater. Chem. C 2020, 8, 5239–5247. [Google Scholar] [CrossRef]
- Becke, A.D. A Multicenter Numerical Integration Scheme for Polyatomic Molecules. J. Chem. Phys. 1988, 88, 2547–2553. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Pascale, F.; Zicovich-Wilson, C.M.; Gejo, F.L.; Civalleri, B.; Orlando, R.; Dovesi, R. The Calculation of the Vibrational Frequencies of the Crystalline Compounds and Its Implementation in the CRYSTAL Code. J. Comput. Chem. 2004, 25, 888–897. [Google Scholar] [CrossRef]
- Dall’Olio, S.; Dovesi, R.; Resta, R. Spontaneous Polarization as a Berry Phase of the Hartree-Fock Wavefunction: The Case of KNbO3. Phys. Rev. B 1997, 56, 10105–10114. [Google Scholar] [CrossRef]
- King-Smith, R.; Vanderbilt, D. First-Principles Investigation of Ferroelectricity in Perovskite Compounds. Phys. Rev. B 1994, 49, 5828. [Google Scholar] [CrossRef]
- Kumada, N.; Kinomura, N.; Muto, F. Crystal Structures of Ilmenite Type LiNbO3 and NaNbO3. J. Ceram. Soc. Jpn. 1990, 98, 384–388. [Google Scholar] [CrossRef] [Green Version]
- Boysen, H.; Altorfer, F. A Neutron Powder Investigation of the High-Temperature Structure and Phase Transition in LiNbO3. Acta Crystallogr. Sect. B Struct. Sci. 1994, 50, 405–414. [Google Scholar] [CrossRef]
- Lehnert, H.; Boysen, H.; Frey, F.; Hewat, A.; Radaelli, P. A Neutron Powder Investigation of the High-Temperature Structure and Phase Transition in Stoichiometric LiNbO3. Z.-Krist. Cryst. Mater. 1997, 212, 712–719. [Google Scholar] [CrossRef] [Green Version]
- Persson, K. Materials Data on LiNbO3 (SG:161) by Materials Project. 2014. Available online: https://materialsproject.org/materials/mp-3731/ (accessed on 7 September 2021). [CrossRef]
- Persson, K. Materials Data on LiNbO3 (SG:167) by Materials Project. 2014. Available online: https://materialsproject.org/materials/mp-552588/ (accessed on 7 September 2021). [CrossRef]
- Toyoura, K.; Ohta, M.; Nakamura, A.; Matsunaga, K. First-Principles Study on Phase Transition and Ferroelectricity in Lithium Niobate and Tantalate. J. Appl. Phys. 2015, 118, 064103. [Google Scholar] [CrossRef]
- Zhang, Z.G.; Abe, T.; Moriyoshi, C.; Tanaka, H.; Kuroiwa, Y. Study of Materials Structure Physics of Isomorphic LiNbO3 and LiTaO3 Ferroelectrics by Synchrotron Radiation X-Ray Diffraction. Jpn. J. Appl. Phys. 2018, 57, 11UB04. [Google Scholar] [CrossRef]
- Servoin, J.; Gervais, F. Soft Vibrational Mode in LiNbO3 and LiTaO3. Solid State Commun. 1979, 31, 387–391. [Google Scholar] [CrossRef]
- Inbar, I.; Cohen, R. Origin of Ferroelectricity in LiTaO3 and LiNbO3; LAPW Total energy Calculations. Ferroelectrics 1995, 164, 45–55. [Google Scholar] [CrossRef]
- Inbar, I.; Cohen, R. Comparison of the Electronic Structures and Energetics of Ferroelectric LiNbO3 and LiTaO3. Phys. Rev. B 1996, 53, 1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caciuc, V.; Postnikov, A.; Borstel, G. Ab Initio Structure and Zone-Center Phonons in LiNbO3. Phys. Rev. B 2000, 61, 8806. [Google Scholar] [CrossRef]
- Nogueira, B.A.; Milani, A.; Castiglioni, C.; Fausto, R. The Correlation Between Experimental Polarized Raman Spectra and Their Density Functional Theory Prediction in the LCAO Framework: The R3c LiNbO3 Crystal as a Test Case. J. Raman Spectrosc. 2021, 52, 995–1010. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| a = b | c | = | (3) | Refs. | ||
|---|---|---|---|---|---|---|
| Exp R3c | 5.212 | 14.356 | 90 | 120 | 337.733 | [37] |
| Exp RC | 5.285 | 13.848 | 90 | 120 | 334.971 | [38,39] |
| Theory R3c | 5.223 | 14.129 | 90 | 120 | 333.801 | [40] |
| Theory Rc | 5.269 | 13.903 | 90 | 120 | 334.326 | [41] |
| Theory R3c | 5.194 | 13.962 | 90 | 120 | 326.191 | this work |
| Theory Rc | 5.227 | 13.757 | 90 | 120 | 325.484 | this work |
| R | R | q | q | q | OV | OV | |
|---|---|---|---|---|---|---|---|
| R3c | 2.094–2.260 | 1.889–2.141 | +0.513 | +2.682 | −1.065 | 0.062–0.051 | 0.158–0.072 |
| RC | 1.998 | 1.992 | +0.556 | +2.704 | −1.086 | 0.073 | 0.119 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gentile, F.S.; Diana, R.; Panunzi, B.; Caruso, U.; Platonenko, A.; Pascale, F.; Dovesi, R. Vibrational Analysis of Paraelectric–Ferroelectric Transition of LiNbO3: An Ab-Initio Quantum Mechanical Treatment. Symmetry 2021, 13, 1650. https://doi.org/10.3390/sym13091650
Gentile FS, Diana R, Panunzi B, Caruso U, Platonenko A, Pascale F, Dovesi R. Vibrational Analysis of Paraelectric–Ferroelectric Transition of LiNbO3: An Ab-Initio Quantum Mechanical Treatment. Symmetry. 2021; 13(9):1650. https://doi.org/10.3390/sym13091650
Chicago/Turabian StyleGentile, Francesco Silvio, Rosita Diana, Barbara Panunzi, Ugo Caruso, Alexander Platonenko, Fabien Pascale, and Roberto Dovesi. 2021. "Vibrational Analysis of Paraelectric–Ferroelectric Transition of LiNbO3: An Ab-Initio Quantum Mechanical Treatment" Symmetry 13, no. 9: 1650. https://doi.org/10.3390/sym13091650
APA StyleGentile, F. S., Diana, R., Panunzi, B., Caruso, U., Platonenko, A., Pascale, F., & Dovesi, R. (2021). Vibrational Analysis of Paraelectric–Ferroelectric Transition of LiNbO3: An Ab-Initio Quantum Mechanical Treatment. Symmetry, 13(9), 1650. https://doi.org/10.3390/sym13091650