Abstract
The Cn molecular symmetry implicated by the schemes with which cyclodextrins (CDs), the well-known cyclic oligosaccharides, are introduced in the literature, is not valid. Numerous studies have shown that CDs are rather flexible with their macrocycle adopting various conformations that enable the inclusion complexation of guest molecules of various shapes. In this work, the loss and gain of the C7 symmetry of the heptakis (2, 3, 6-tri-O-methyl)-β-CD (TM-β-CD) is investigated by means of its conformation geometrical features in its hydrated form and upon complexation with molecules of different shapes. For this, the crystal structure of the inclusion complex of a bulky guest molecule (giberellic acid) in TM-β-CD is presented for the first time and compared with the previously determined crystal structures of monohydrated TM-β-CD and the inclusion complex of a linear monoterpenoid (geraniol) in TM-β-CD. The structural investigation was complemented by molecular dynamics simulations in an explicit solvent, based on the crystallographically determined models. The crucial role of the guest, in the symmetry gain of the host, reveals a pronounced induced-fit complexation mechanism for permethylated CDs.
1. Introduction
Cyclodextrins (CDs) are cyclic oligomers of 1→4 linked α-D-glucopyranose monomers, obtained, in the standard way, by enzymatic degradation of starch. The well-known smallest members consist of 6, 7 or 8 units bearing the names α-, β- and γ-CDs, respectively. These three main native CDs possess a hollow, truncated conical shape, their narrower side formed by primary O6 hydroxyl groups (primary face) and the wider side by secondary O2 and O3 hydroxyl groups (secondary face) [1]. Due to their conical shape, with a hydrophilic exterior and a hydrophobic cavity, native CDs and their derivatives have the ability to encapsulate a wide variety of appropriately sized hydrophobic molecules or parts of molecules, inside their cavity, forming noncovalent dynamic inclusion complexes (i.cs.). As a result, CDs exert a profound effect on the physicochemical properties of the guest molecules, leading to a large number of chemical and biological applications [2,3].
CDs have been studied as symmetry-breaking hosts in mechanomolecules where even symmetrical guests are rendered asymmetric by the presence of an encircling CD host [4]. On the other hand, it has been early noted that CDs are introduced in the literature with schemes presenting oversimplified models of round structures and symmetric truncated cones aiming to illustrate their general characteristics and their ability for the inclusion complexation of suitable guests [5]. X-ray crystallography studies have indicated rigid truncated cone structures of high C6, C7 or C8 symmetry for α-, β- or γ-CDs, respectively, with a planar ring of glycosidic oxygen atoms [6]. However, numerous experimental and theoretical studies have strongly questioned the concept of rigid CDs [7,8,9]. In addition, the model of rigid structures cannot justify the ability of CDs to form inclusion complexes with guest molecules of various shapes and even more to separate enantiomers in chromatography [10], since this ability implies an effective host–guest fitting.
The rigidity and “roundness” of the native CDs, indicated by the X-ray crystallographically determined structures, is due mainly to the formation of intramolecular O2(n)…O3(n−1) hydrogen bonds at the CDs wide (secondary) rim. However, it should be noted that the determined crystal structures offer a positional and time-averaged view, whereas NMR and theoretical (Molecular Mechanics and Dynamics) studies have shown that these hydrogen bonds rapidly interchange at room temperature (a mechanism called “flip–flop”), elucidating the amazing flexibility of the CD structures in solution, which is of particular importance since most CD applications take place in this state [11]. Theoretical gas-phase calculations have also shown that symmetry breaking lowers the energy of these molecules [5].
In order to investigate the deviation of uncomplexed and complexed CDs from the Cn molecular symmetry by means of X-ray crystallography and complementary theoretical (molecular dynamics) studies, we chose to examine the conformation geometrical features of heptakis (2, 3, 6-tri-O-methyl)-β-CD (permethylated β-CD or TM-β-CD) in its hydrate form and upon complexation with molecules of different shapes. The permethylation of β-CD results in greater conformational flexibility, owing to the loss of its intramolecular O2(n)…O3(n−1) hydrogen bonding capability and consequently accounts for the distortion of its macrocycle. TM-β-CD compared to its unmethylated counterpart, presents an elongated cavity, which improves its complexation properties, higher aqueous solubility and greater protection from hydrolysis both in solution and in the solid state. These characteristics make TM-β-CD suitable for various applications, in particular to increase the bioavailability of a drug; by the appropriate choice of host and guest, one can achieve a very high selectivity [12,13,14].
The crystal structures of monohydrate TM-β-CD (1), the inclusion complex (i.c.) of a linear monoterpenoid (geraniol) in TM-β-CD (2) and the i.c. of a bulky guest molecule (gibberellic acid) in TM-β-CD (3) are used to compare the geometrical features of the host macrocycle.
The crystal structure of (3), is presented here for the first time in the literature. Gibberellins are known as the most powerful of the growth promotors because they induce and promote flowering in many plants and modify the flower sex expression in some plants [15,16]. Gibberellic acid (GA3) is a tetracyclic diterpenoid compound of the gibberellin family, which regulates plant growth and development (Figure 1a). It is a naturally occurring plant growth regulator (PGR) and is responsible for a variety of effects including the stimulation of seed germination in many cases. GA3 has been widely used in agriculture, horticulture and forestry [17]. One of the most important applications of GA3 is in wine growing in order to make seedless grape varieties [18]. However, it is also used in floriculture to improve the phenotypic characteristics of plants [19]. However, due to the poor water solubility of the drug, dissolving GA3 in organic solvents firstly and then diluting in water is necessary for further application [17]. To this end, the inclusion complex formation of GA3 in CDs could improve its bioavailability, enhance the stability and offer a controlled release sustaining the PGR action.
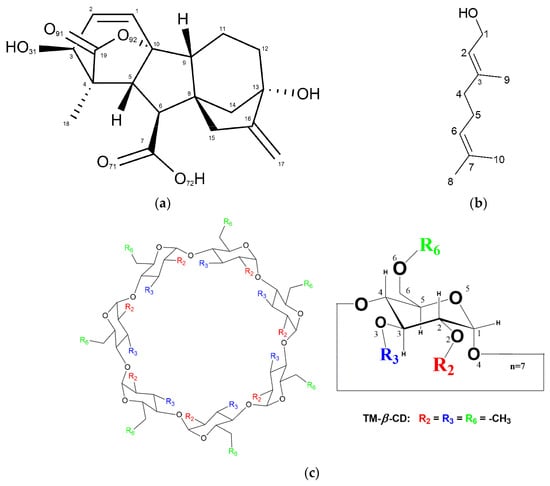
Figure 1.
Chemical representation of (a) gibberellic acid (GA3), (b) geraniol and (c) heptakis (2, 3, 6-tri-O-methyl)-β-cyclodextrin (TM-β-CD).
The monohydrate TM-β-CD crystal structure (1) has been also presented in early reports [20,21], whereas the crystal structure of (2) has been presented in a previous work by our group [22]. The choice of (2) was based on the linear and relatively rigid guest molecule (Figure 1b) due to its limited torsional degrees of freedom.
As the crystallographic studies provide structures that are affected by crystal contacts and constitute averages of possible conformations, complementary studies of the molecular dynamics (MD) simulations of TM-β-CD (Figure 1c) in (1), (2) and (3) in explicit water solvent were conducted. The initial models of the MD simulations were based on the crystallographically determined atomic coordinates. By monitoring the conformational alterations of the macrocycle in each case during the time frame of the simulations, we gain some insight into the dynamic behavior and symmetry aspects of the host molecule. In addition, the binding affinities of (2) and (3) inclusion complexes were calculated by the Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) method in order to evaluate and compare the stability of the complexes.
The findings of the present work elucidate the flexibility of permethylated β-CDs and the pronounced conformational changes caused to their macrocycle upon complexation with guest molecules of different shapes and sizes. The crucial role of the guest, in the molecular symmetry gain of the host, highlights a well-known induced-fit complexation mechanism for permethylated CDs.
2. Materials and Methods
2.1. X-ray Crystallography
2.1.1. Crystallization
Gibberellic acid (GA3, ≥90% pure) and TM-β-CD of pharmaceutical-grade quality were received as white powders from Sigma-Aldrich (St. Louis, MO, USA) and used without further purification. Crystallization of monohydrate TM-β-CD was achieved by using the common slow evaporation method. Briefly, a 0.02 M aqueous solution of the molecule was stirred for 1 h at 40 °C until it was limpid and then remained at 48 °C for one week until colorless prismatic crystals were formed. The crystallization procedure of geraniol/TM-β-CD i.c. is explicitly described in [22]. Crystals of the GA3/TM-β-CD complex were obtained by mixing a 0.02 M aqueous solution of TM-β-CD with solid GA3 in a host: guest molar ratio of 1:1. The final mixture was stirred for 1 h at 40 °C and was subsequently maintained at 48 °C for one week until colorless transparent prismatic crystals suitable for X-ray data collection were formed.
2.1.2. Data Collection, Structure Solution and Refinement
X-ray data collection for monohydrate TM-β-CD (1) and GA3/TM-β-CD (3) i.c. were undertaken using a Bruker D8-VENTURE crystallography system equipped with a Cu source (λ = 1.54 Å) and an Oxford Cryosystems low-temperature device (Oxford Cryosystems Ltd., Long Handorough, UK). In the case of geraniol/TM-β-CD i.c. (2), data were collected using synchrotron radiation light (λ = 0.81 Å) at the EMBL Hamburg (X11 beamline) at the DORIS stage ring, DESY [22]. Paraffin oil was used as cryoprotectant agent for cases (2) and (3).
In the case of monohydrate TM-β-CD (1), a total of 2006 frames were collected. The total exposure time was 9.19 h. The frames were integrated with the Bruker SAINT software package [23] using a wide-frame algorithm. The integration of the data using an orthorhombic unit cell yielded a total of 71,972 reflections to a maximum θ angle of 59.16° (0.90 Å resolution), of which 11,002 were independent (average redundancy 6.542, completeness = 99.5%, Rint = 4.07%, Rsig = 2.56%) and 10,232 (93.00%) were greater than 2σ(F2). The final cell constants of a = 14.847(2) Å, b = 19.391(5) Å, c = 26.556(5) Å and volume = 7645(3) Å3 are based upon the refinement of the XYZ-centroids of 9675 reflections above 20 σ(I) with 5.649° < 2θ < 117.8°. Data were corrected for absorption effects using the Multi-Scan method with SADABS [24]. The ratio of minimum to maximum apparent transmission was 0.699.
The crystal structure of geraniol/TM-β-CD (2) i.c. is described in detail by Christoforides et al. [22].
In the case of GA3/TM-β-CD (3), a total of 606 frames were collected. The total exposure time was 11.78 h. The frames were integrated with SAINT using a wide-frame algorithm. The integration of the data using an orthorhombic unit cell yielded a total of 97,761 reflections to a maximum θ angle of 50.61° (1.00 Å resolution). The final cell constants of a = 14.7487(8) Å, b = 22.0113(13) Å, c = 27.6009(15) Å and volume = 8960.3(11) Å3 are based upon the refinement of the XYZ-centroids of 9567 reflections above 20 σ(I) with 5.135° < 2θ < 98.02°. Data were corrected for absorption effects using the Multi-Scan method. The ratio of minimum to maximum apparent transmission was 0.864. Data were corrected for absorption effects using the Multi-Scan method with SADABS as in case (1). Further analysis of the scattering data with the program XPREP [25] indicated that the complex crystallizes in an orthorhombic system; the space group is P212121, and the unit cell dimensions are a = 14.784, b = 22.0113 and c = 27.6001 Å (Table 1).

Table 1.
Crystal data, data collection and refinement statistics.
The above crystal structures were solved by the Patterson-seeded dual-space recycling utility of the SHELXD program [26] and subsequently refined by full-matrix least squares against F2 using SHELXL-2014/7 [27] with the assistance of the SHELXLE GUI [28]. In the case of the i.c. (3), due to the limited resolution of the collected data and structural complexity of the guest (the tetracyclic diterpenoid compound of GA3), soft restraints on bond lengths and angles of the guest were applied using the PRODRG2 webserver [29]. All hydrogen atoms were placed in geometric positions and treated as riding on their parent atoms with dC-H = 0.95–1.00 Å (depending on the hybridization of carbon atom), and dO-H = 0.84 Å. Uiso(H) values were assigned in the range 1.2–1.5 times Ueq of the parent atom. Hydrogen atoms of water molecules were not included in the final models. In an effort to avoid a low data/parameters ratio, anisotropic thermal parameters were imposed to selected, nonH atoms of the host molecules. Extinction corrections were applied to the final solutions, and reflections that illustrated high divergence between the Fo and Fc values were omitted at the last stages of refinement.
The graphics programs Mercury 4.3.1 [30], PyMoL 2.0.6 [31] and Olex2 1.3 [32] were used to explore the crystal packing, analyze the structure geometry and visualize the asymmetric unit, the intermolecular interactions and the crystal packing of the complex. Crystallographic data are given in Table 1. Crystallographic information files with embedded structure factors were checked and validated for the consistency and integrity of crystal structure determination according to IUCr standards and consequently were deposited into the Cambridge Structural Database (CSD) under the deposition numbers CCDC: 2160066 and 2160065.
2.2. Molecular Dynamics (MD) Study
2.2.1. System Preparation
The 3D models that were employed for molecular dynamics simulations were based on the crystal structures of (1), (3) determined by the present crystallographic analysis and (2) determined in our previous work [22]. For MD simulations, the AMBER 12 (Assisted Model Building with Energy Refinement) software [33] was chosen along with the q4md-CD force field [34] to generate the correct permethylated CD topology. The GAFF force field was implemented to produce the topology for the two guest molecules (geraniol and GA3) with AM1-Mulliken charges using Antechamber [35]. The analyzed structures were solvated in a 10 Å truncated octahedron box consisting of the TIP3P water molecules [36]. MD calculations were performed with SANDER using a time step of 1 fs. The Particle Mesh Ewald (PME) method was applied to maintain the long-range electrostatic interactions with the nonbonded cutoff distance set to 10 Å in order to create the periodic boundaries. Temperature and pressure controls were performed using a Berendsen-type algorithm with coupling constants of 0.5 ps (equilibration) or 1.0 ps (production).
2.2.2. MD Simulations
The MD simulations were performed according to standard MD protocols for CD complexes available in the literature [13]. Initially, each system was equilibrated prior to the production runs using the following protocol: (i) energy minimization with the steepest descent algorithm for the first 500 cycles and the conjugate gradient algorithm for rest of the cycles with positional restrains of 50 Kcal·mol−1·Å−2 on the heavy atoms of the complexes, (ii) heating equilibration of the solvent in the canonical (NVT) ensemble to keep the system’s volume and temperature constant using positional restraints, (iii) 1000-step unrestrained energy minimization, (iv) gradual temperature increase from 0 K to 300 K in six 15-ps steps with 10 Kcal mol−1 Å−2 restraints on the complex and gradual release of the restraints in six 15-ps steps at 300 K and (v) density equilibration in the NPT ensemble for 250 ps and additional 400 ps NPT simulation at 1 atm, 300 K. To monitor the system’s equilibrium convergence, the density, temperature and energy parameters were examined. Finally, a 12 ns MD production run with constant pressure and temperature (NPT), was carried out under physiological conditions (1 atm, 300 K) combined with the Generalized Born (MM-GBSA) explicit solvation models, as described below. For analysis, the CPPTRAJ module [37] of AMBER 12 was used to monitor the root mean square displacement (RMSD) of the molecules, the distances between the centers of masses (CoMs) of each guest and the respective host, the radius of gyration (RoG) of the systems and the value distribution for some geometrical features that are associated with the host macrocycle’s flexibility. Visualization and further structural analysis were conducted with VMD [38].
2.2.3. Free Energy Prediction
A well-known postprocessing approach (MM/GBSA method) was used for investigating the binding free energy of geraniol and GA3 guests to TM-β-CD in solution simulation environment [39]. The approach combines the molecular mechanical (MM) energies with the continuum solvent approaches (Generalized Born model and Surface Area Continuum Solvation method (GBSA)).
The enthalpic term ΔH or ΔG(GB) was calculated by:
where ΔGgas and ΔGsolv represent the energy differences in gas phase and upon solvation during the simulation time, respectively.
ΔH = ΔGgas + ΔGsolv,
The entropic penalty (ΔS) incurred upon guest binding was calculated by extracting snapshots from the MD trajectories every 100 frames using the NMODE module of AMBER 12. The binding free energy was derived by Equation (2):
ΔGbind = ΔH − T∙ΔS,
3. Results
3.1. X-ray Analysis
Crystal Structures
The crystal structure of monohydrate TM-β-CD (1) was determined by X-ray crystallography at room temperature. This form of uncomplexed TM-β-CD (monohydrate) crystallizes in the P212121 space group, its asymmetric unit containing one severely distorted TM-β-CD and a highly disordered water molecule, located at the wide rim and hydrogen bonded with an O2 atom of the CD (Figure 2a). The structural analysis verified previously published results about the singular conformation of monohydrate TM-β-CD in the structural chemistry of CDs [20,21]. The uncomplexed TM-β-CD molecule is somewhat collapsed in an attempt to minimize the cavity volume in the absence of a hydrophobic guest. The values of the geometrical features of the CD macrocycle, given in Table 2 and presented in radar plots in Figure 3, span a wide range, revealing a large deviation from a C7 molecular symmetry. In particular, the heptagon formed by the glucosidic O4n atoms is not planar as the O4(n) atoms deviate noticeably from their mean plane (d = distances of O4n from their mean plane range from −1.089(2) to 0.629(3) Å). In addition, the spanning values of D (O4(n)…O4(n+1) distances) from 4.082(4) to 4.703(4), DK (K…O4(n) distances between the approximate center K of the O4(n) heptagon and the O4n atoms) from 3.414(3) to 5.940(3) and Φn (O4(n−1)…O4(n)…O4(n+1) angles) from 92.27(8) to 161.28(10) indicate the disruption of the “round” annular structure and the formation of an “irregular”, roughly elliptical shape. Five methylglucose residues have a positive tilt angle (τ = tilt angle between the optimum O4(n) plane and the mean plane atoms O4(n−1), C1(n), C4(n), O4(n)), showing that their primary sides incline toward the host cavity, whereas two methylglucose residues (G4 and G7) have negative tilt angles inclining out of the cavity. Especially the primary methoxy group of G6, which has the largest positive tilt angle (τ angle: O4(n−1)–C1(n)–C4(n)–O4(n)), forms a ‘lid’, closing off the primary side of TM-β-CD as has been previously reported [20]. The torsion angles t (t = O5(n)–C5(n)–C6(n)–O6(n)), given in Table 2, indicate that the primary groups of residues G1, G5, G6 and G7 have always the gg conformation pointing out of the cavity, whereas with the residues G2 and G3, the gt conformation points in to the cavity. TM-β-CD molecules are arranged in the crystal lattice in a ‘cage’ mode with the remaining shallow cavity of each molecule filled only by the O6 methyl group of a glucose residue (G4) of an adjacent molecule (Figure 2b), which is inverted from the normal 4C1 to the 1C4 chair conformation, a dramatic structural change, which is not observed in any other CD crystal structure [20,21].
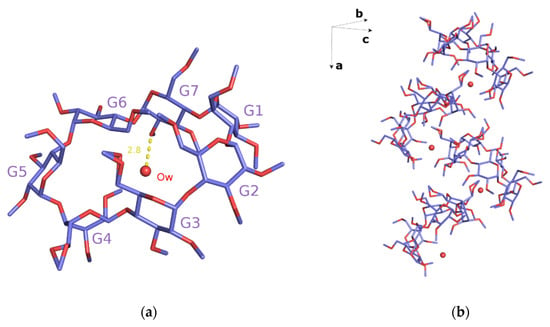
Figure 2.
(a) The asymmetric unit of the monohydrate TM-β-CD (1). G4 glucose unit is inverted from the normal 4C1 to the 1C4 chair conformation; G6 illustrates the largest positive tilt angle forming a ‘lid’, and Ow water is hydrogen bonded with an O2 atom of G7 and (b) the crystal packing of (1). Hydrogen atoms are omitted for clarity. Images were rendered with PyMoL.
Table 2.
Conformational characteristics of the host molecule (TΜ-β-CD) in three distinctive cases (A) monohydrate TΜ-β-CD, (B) geraniol/TΜ-β-CD and (C) GA3/TΜ-β-CD inclusion complexes.
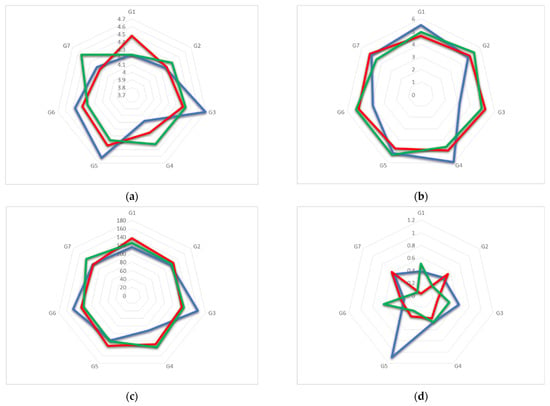
Figure 3.
Radar plots of the values of the geometric features (a) D, (b) DK, (c) Φn and (d) d of TM-β-CD molecule in monohydrate TM-β-CD case (blue), geraniol/TM-β-CD case (red) and GA3/TM-β-CD case (green).
The crystal structure of geraniol/TM-β-CD (2) has been presented in detail in a previous work [22]. Briefly, the (2) i.c. crystallizes in the P212121 space group, its asymmetric unit consisting of one TM-β-CD molecule, one encapsulated geraniol molecule, disordered over three sites (sites S1, S2 and S3), and a water molecule occupying interstices in the crystal lattice of the inclusion complexes (Figure 4a). Geometrical characteristics and parameters regarding the host molecule conformation are given in (Table 2) and presented in the radar plots of Figure 3. As indicated by the D, DK and Φn values, the glucosidic O4n atoms form a regular heptagon. However, a deviation from planarity is observed as the d values vary from 0.556(3) Å to −0.609(3) Å. The methylglucose ring tilt angles span in a range from −13.90(2) to 43.3(2)°, which is noticeably shorter compared to those of (1) and (3). The primary methoxy groups of three residues (G1, G5 and G7) form the characteristic ‘lid’ in the primary region of the host, preventing the deep penetration of the guest. The O5(n)–C5(n)–C6(n)–O6(n) torsion angles indicate that the primary methoxy groups (two of them were found disordered) have the usual gauche–gauche (gg) and gauche–trans (gt) conformations. The complex units are arranged in a head-to-tail mode along the b axis forming screw channels (Figure 4b) stabilized by many intermolecular C-H…O bonds between the adjacent hosts. This crystal packing mode characterizes several isostructural TM-β-CD complexes not only with other linear guests, such as β-citronellol (DEWMIE) [40] or citral (EMIWAB) [41], but also with more bulky guest molecules, such as pesticide fenitrothion (CIJVEY) [42] and the cyclic 3-cyclooctyl-1,1-dimethylurea (OYAPIO) [42].
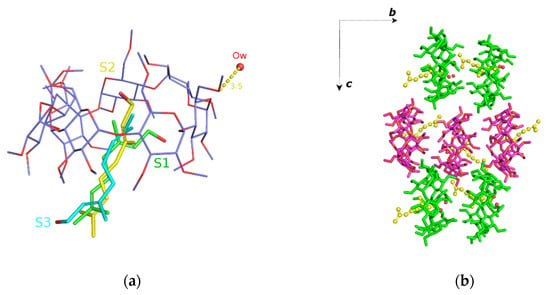
Figure 4.
(a) The asymmetric unit of geraniol/TM-β-CD (2). The guest is disordered over three sites: S1 (green with s.o.f. 60%), S2 (yellow with s.o.f. 20%) and S3 (cyan with s.o.f. 20%) and (b) the complex units arranged in a head-to-tail mode forming antiparallel (green and magenta sticks) screw channels along the b axis. For clarity, only the S2 site of geraniol (as yellow spheres) is shown.
The GA3/TΜ-β-CD (3) i.c. crystallizes in the orthorhombic P212121 space group, and its asymmetric unit contains one TΜ-β-CD encapsulating a highly disordered GA3 molecule and one water molecule bridging via hydrogen bonds adjacent complex units (Figure 5a,b and Table 3). The inclusion complex is stabilized only by van der Waals intermolecular forces and contacts between the hydrophobic sites of the guest and host molecules. The lack of directional interactions explains the high disorder of the guest. Four prominent guest-binding modes were determined (sites S1, S2, S3 and S4 with the occupancy factors of 0.20, 0.20, 0.20 and 0.40, respectively, Figure 5a). The guest occupying these sites is partially encapsulated inside the hydrophobic cavity of the TΜ-β-CD, its main part protruding from the wide rim of the host to the hydrophobic interspace formed by adjacent hosts. The geometric features of the TΜ-β-CD host in its complex with GA3 are quoted in Table 2. The O4(n) atoms that interconnect the host’s glucose units form an elliptical heptagon, as the range of their D values is noticeably wider than that observed in the case of the inclusion complex (2) (Figure 5a). A significant deviation from planarity was also observed as the d values span from −0.621(7) to 0.513(7) Å (Table 2 and Figure 3). Five methylglucose residues have positive tilt angles indicating that their primary sides incline toward the approximate sevenfold axis of the macrocycle, whereas two methylglucose residues (G1 and G5) have negative tilt angles inclining out of the host cavity (Table 2). The net result is the partial capping of the primary side that gives to the host TM-β-CD molecule the (commonly observed in the majority of its inclusion complexes) ‘closed’ cup-shaped conformation that prevents a deep immersion of the guest inside the cavity [14]. The primary methoxy groups of the host, participating in crystal contacts (intermolecular C-H..O bonds with adjacent host molecules and a H-bond with the bridging water molecule) are found disordered in five out of the seven methylglucose residues, adopting the usual gg and gt conformations (Table 2).
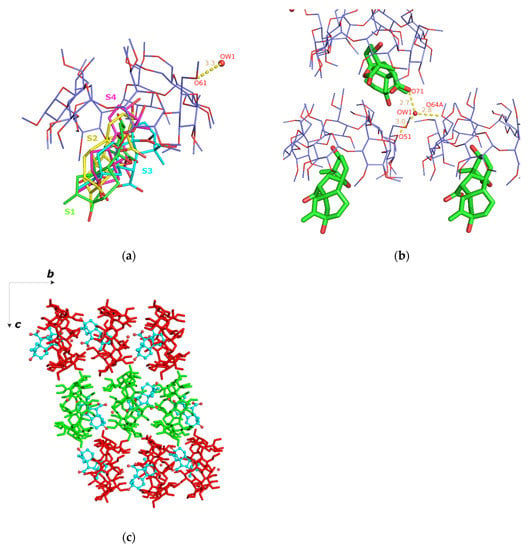
Figure 5.
(a) The asymmetric unit of the GA3/TΜ-β-CD (3) crystal structure. The guest is disordered over four sites: S1 (green with s.o.f. 20%), S2 (yellow with s.o.f. 20%), S3 (cyan with s.o.f. 20%) and S4 (magenta with s.o.f. 40%); (b) one water molecule interconnects via H-bonds adjacent hosts and (c) the crystal packing consists of antiparallel columns (green and red sticks) along b axis. For clarity, only the S3 site of GA3 (as cyan spheres) is shown.
Table 3.
H-bonds and their lengths (in Å) between the sole water Ow1 and both host and guest molecules in GA3/TM-β-CD crystal structure. Structural analysis was performed with Olex2.
The inclusion complex units stack along the b axis forming columns, in which the host molecules are arranged coaxially so that the wide rim (head) of the one faces the narrow rim (tail) of the other (head-to-tail mode) (Figure 5c). Based on the orientation of individual inclusion complexes, the crystal packing consists of antiparallel columns related by the c twofold screw axis, which is commonly observed for the TM-β-CD inclusion complexes crystallizing in the space group P212121 [14]. A thorough search in the Cambridge Structural Database (CSD) [43], using cell check v 1.0, resulted in two isostructural entries that share similar cell dimensions and same space group. In both cases, (4-hydroxyazobenzene/TΜ-β-CD (CCDC code: DORLEC) [44] and 2-naphthylacetic acid/TΜ-β-CD inclusion complexes (CCDC code: BEZCIU) [45]), the complexes form antiparallel columns along the b axis and are arranged in a head-to-tail manner.
3.2. Trajectory Analysis
The crystallographically determined atomic coordinates of the uncomplexed TM-β-CD molecule (1), the geraniol/TM-β-CD i.c. (2) considering a host:guest ratio of 1:1 and the GA3/TM-β-CD i.c. with the same host:guest ratio were used as the initial structures for three distinctive MD simulations of 12 ns as described in Section 2.2.2. All structural and energy analyses of the three produced single trajectories, including the root mean square displacement (RMSD), root mean square fluctuation (RMSF), radius of gyration (RoG), center of mass (CoM) distance monitoring and Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) free energy calculations, were performed with the aid of CPPTRAJ implemented in AMBER 12 as explained before and presented here.
Starting with the case of (1), the distortion of the macrocycle is eminent from the first frames (Figure 6a,b). As indicated by the glycosidic O4(n) atoms, the macrocycle deviates far from planarity and the sevenfold symmetry of a regular heptagon (Figure 6a,b), adopting an elongated form during the time frame of the simulation. Its G6 and G3 glucose units are also characterized by high torsion angles, building the characteristic “lid” at the narrow rim of the molecule.
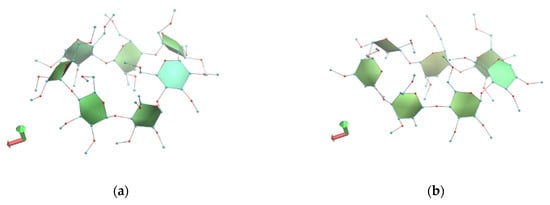
Figure 6.
Representative snapshots of monohydrate TM-β-CD system at 0 (a) and 10th ns (b) of the simulation time. Images were rendered with VMD.
For the (2) i.c. (geraniol/TΜ-β-CD) case, the guest occupying the site of the highest occupancy (S1) was chosen for the starting model. By monitoring the frames during the time interval of the simulation, we observed that the guest molecule remains constantly encapsulated inside the hydrophobic TΜ-β-CD cavity and frequently changes its initial equatorial orientation to an axial one and vice versa. The axial accommodation of the guest comes along with the opening of the host’s characteristic lid, allowing the protrusion of the linear guest molecule from the narrow rim of the host (Figure 7a,b). The RMSD plot (Figure 7c) reveals the high mobility of the guest. Moreover, the geraniol’s hydroxyl group forms alternate hydrogen bonds with TΜ-β-CD methoxy groups of both the primary and secondary rims during the time frame of the simulation (see [22]). Thus, the inclusion mode is affected by the guest’s size, shape and characteristic group.
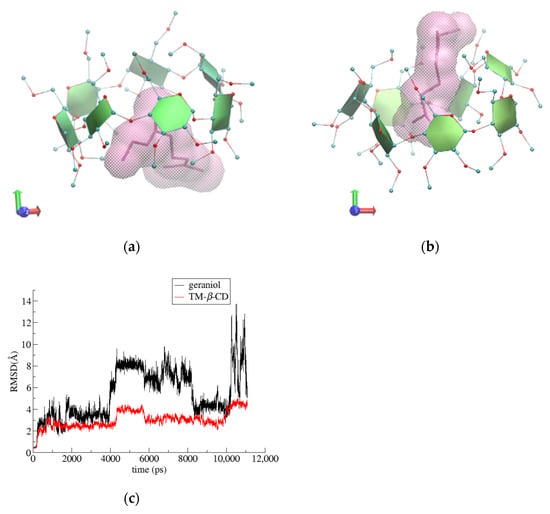
Figure 7.
Representative snapshots of (2) i.c. (geraniol/TΜ-β-CD) at 0 (a) and the 8th ns (b) of the simulation time and (c) RMSD vs. time for host and guest molecules in the formed complex.
Finally, in the case of (3) i.c. (GA3/TM-β-CD), once again, the crystallographically determined site of the guest molecule with the highest occupancy (S4) was used. The bulky guest molecule is always partially encapsulated in the CD’s cavity, protruding from the wide rim of the host’s macrosycle as its narrow rim is closed by the formation of the characteristic ‘lid’ (Figure 8a,b). A hydroxyl group of GA3 (O13-H) is alternately (but always) hydrogen-bonded with two particular etheric oxygens of the TΜ-β-CD secondary methoxy groups (Figure 8c), contributing to the limited mobility of the guest and the overall stability of the i.c. as is indicated by the RMSD plot given in Figure 8d.
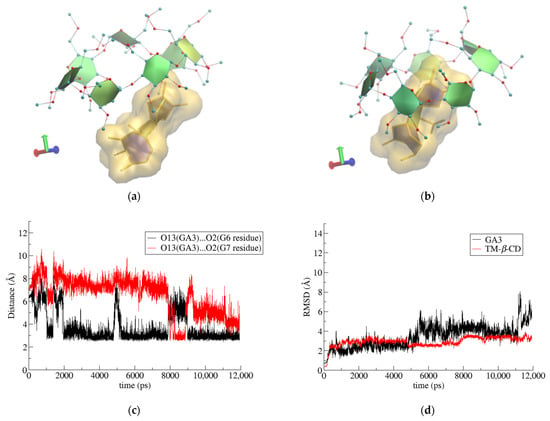
Figure 8.
Representative snapshots of GA3/TΜ-β-CD inclusion complex at (a) 0 and (b) 10 ns of the simulation time, (c) hydrogen-bond distance between O13 atom of GA3 and O2 atoms of G6 and G7 glucose units at host’s secondary rim as a function of simulation time and (d) RMSD vs. time for host and guest molecules in the formed complex.
Once again, the size, shape and characteristic group of the guest define its inclusion mode in the particular host. However, the guest’s limited ability to intrude into the host’s cavity also affects the host’s conformation. The macrocycle of TΜ-β-CD remains severely distorted, deviating significantly from a molecular C7 symmetry.
3.3. Binding Affinity Calculations
Binding affinities of (2) and (3) i.c., estimated by the Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) method, and the values of their energetic components are listed in Table 4. Van der Waals interactions seem to be predominant in both cases. It is also clear that the calculated absolute value of ΔG(GB) for (2) i.c. is higher than that of (3) i.c., indicating a more stable inclusion complex. These results are in accordance with the observations made by monitoring the MD simulation trajectories; in the case of (2), the guest is found immersed deeper inside the CD cavity, participating in more van der Waals interactions with it, whereas in the case of (3), the guest is only partially included inside the CD cavity, and the available surface for nonpolar interactions is decreased. The plot of the host–guest Center of Mass (CoM) distance vs. time for (2) and (3) (Figure 9) indicates that for a large fraction of the simulation time, the geraniol is found fully entrapped inside the CD cavity and it is accommodated close to the O4n plane area, whereas the GA3 is always partially encapsulated in the CD’s cavity, located near the host’s secondary rim.
Table 4.
Binding free energies and their standard deviations (kcal/mole) resulting from MM/GBSA analysis of the inclusion compounds of geraniol and GA3 with TM-β-CD and with host: guest ratios of 1:1.
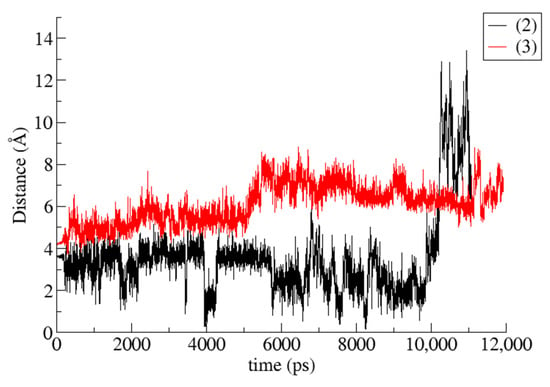
Figure 9.
CoM (Center of Mass) distance between host and guest during the time frame of the simulations for geraniol/TM-β-CD (2) and GA3/TM-β-CD (3) cases.
3.4. Comparison
The RMSD and RMSF plots depicted for the TM-β-CD in the three examined cases (Figure 10a,b) do not give any valuable information about the gain or loss of the molecular symmetry. On the other hand, by monitoring the radius of gyration (RoG) of TM-β-CD in cases (1), (2) and (3) during the simulation time, we obtained the mean values summarized in Table 5. The considerably lower value of TM-β-CD RoG in case (2) indicates that the “roundness” of the host molecule is better achieved upon its complexation with the particular linear guest (geraniol).
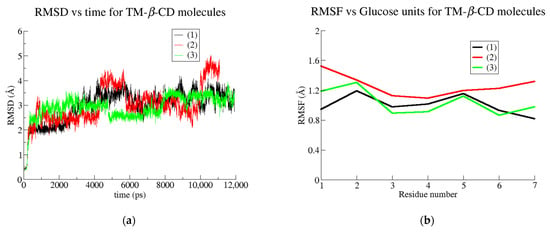
Figure 10.
(a) RMSD plots for the simulated systems of monohydrated TM-β-CD (black line), geraniol/TM-β-CD complex (red) and GA3/TM-β-CD (green) and (b) RMSF vs. glucose units for the simulated systems of monohydrated TM-β-CD (black), geraniol/TM-β-CD complex (red) and GA3/TM-β-CD (green).
Table 5.
Mean values of radius of gyration (RoG) and their standard deviations (Å) of TM-β-CD in (1) monohydrate TM-β-CD, (2) geraniol/TM-β-CD i.c. and (3) GA3/TM-β-CD i.c.
The gain or loss of C7 molecular symmetry of the host macrocycle is better shown by the distribution values over the MD simulation time of the following geometric features: D: Distance between the adjacent glycosidic oxygens O4n and O4(n+1). (n = 1, 2, … 7); DK: Distance between the centroid of the glycosidic oxygens (KA) and O4(n); and Φn: O4(n−1)…O4n…O4(n+1) angle. These features have also been described in the crystallographic analysis (Table 2 and Figure 3), which offers a positional and time average of the structures. In Figure 11, the distribution of the values of these characteristic features during the time frame of the MD simulations for each one examined of the examined cases (1), (2) and (3) is presented. It is clear by the dispersion of the values of these geometrical features that the conformation of the macrocycle of TM-β-CD is not stable, deviating far from the form of a regular heptagon in the case of (1) (Figure 11a,d,g). A similar dynamic behavior is observed in the case of (3), where the bulky guest is found always partially encapsulated in the host’s cavity (Figure 11c,f,i). On the other hand, in the case of (2), where the linear guest intrudes inside the host’s cavity and opens its closed ‘lid’, the dispersion of the values of these geometrical features is significantly limited (Figure 11b,e,h), revealing a tendency of the host to adopt a stable conformation, which approaches better the sevenfold molecular symmetry.
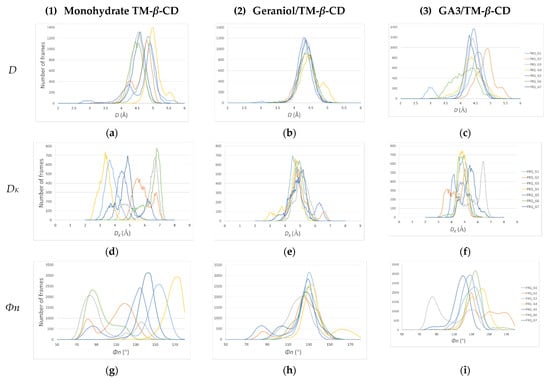
Figure 11.
Distribution of the values of the host’s macrocycle geometrical features over the MD simulations time frame. (a–c) D for monohydrate TM-β-CD (1), geraniol/TM-β-CD (2) and GA3/TM-β-CD (3) inclusion complexes; (d–f) DK for (1), (2) and (3); (g–i) Φn for (1), (2) and (3). Each color line corresponds to the distribution of the three characteristic features for each one of the seven glucose units (G1, G2, … G7).
4. Discussion
Permethylated β-CDs lacking the ability to form intramolecular O(2)…O(3′) hydrogen bonds like their parent β-CDs, present high conformational flexibility and thus a great ability to host suitable guests of different shapes and sizes. In this work, the distortion of the TM-β-CD macrocycle and its deviation from a C7 molecular symmetry, was examined in its uncomplexed form and upon complexation with guest molecules of different shapes and sizes. The study was based on the analysis of the TM-β-CD geometrical features, obtained initially by X-ray crystallography, which offers a positional and time average of the structures, and further by MD studies of the crystallographically determined structures, in an aqueous environment and in the absence of crystal contacts.
In particular, three structures were used in this work for the comparison analysis of the host’s geometrical features: (1) The crystal structure of monohydrate TM-β-CD determined at room temperature. As reported previously [20,21], this form of uncomplexed TM-β-CD (monohydrate) crystallizes in the P212121 space group, its asymmetric unit containing one TM-β-CD molecule, which is somewhat collapsed in an attempt to minimize the cavity volume in the absence of a hydrophobic guest. (2) The crystal structure of the inclusion complex of a linear monoterpenoid (geraniol) in TM-β-CD, which has been presented in detail in a previous work of our group [22], and (3) the crystal structure of the inclusion complex of a bulky molecule, gibberellic acid (GA3) in TM-β-CD, which is presented here for the first time in the literature. The GA3/TM-β-CD inclusion complex crystallizes in the P212121 space group, and its asymmetric unit contains one host and one partially entrapped GA3 guest molecule revealing a 1:1 host-to-guest stoichiometry in the crystalline state. The guest molecule is found disordered over four sites, entering the TM-β-CD cavity from its secondary rim, while the rest of the molecule is located mainly in the interspace formed by three adjacent neighboring hosts. The complex units form antiparallel channels deployed along the crystallographic b axis in a “head-to-tail” manner.
The comparative structural analysis of the TM-β-CD conformation in the three aforementioned cases was based on the values of geometrical features of the CD macrocycle, such as the distance between the adjacent glycosidic oxygens O4(n) and O4(n+1) (D), the distance between the centroid of the glycosidic oxygens (KA) and O4(n) (DK), the O4(n−1)…O4n…O4(n+1) angle (Φn) and the deviation of the O4(n) atom from the least squares optimum plane of the seven glycosidic oxygens (d). The crystallographically determined values of these geometrical features reveal a severely distorted macrocycle in the case of (1), deviating far from a C7 molecular symmetry. However, in the case of (2), where the linear and relatively rigid (due to its limited torsional degrees of freedom) guest molecule intrudes more effectively inside the hydrophobic host cavity than the bulky guest of the case (3), a pronounced gain of molecular symmetry for TM-β-CD is noticed (Table 2, Figure 3).
Similar results were obtained by recording the values of these geometrical features during the time frame of the MD simulations. The distribution of these values is much more dispersed in the case of (1) than in that of (3) and especially (2), indicating a significant gain of molecular symmetry for TM-β-CD in the latter case (Figure 11). By monitoring the frames of the MD trajectories, we observed that in the case of (1) (no guest) and (3) (with the bulky guest being always partially encapsulated in the host’s cavity), the narrow rim of the host is always closed by the formation of a characteristic ‘lid’ by its primary methoxy groups that has a significant role in the distortion of the macrocycle. On the other hand, in the case of (2), the linear guest is found protruding the host’s primary rim, and thus it opens the host’s ‘lid’, forming a stable inclusion complex, as indicated by the MM/GBSA-calculated binding affinities ∆G(GB) and endowing TM-β-CD with a noticeable gain of molecular symmetry
5. Conclusions
Although permethylated β-CDs are flexible molecules adapting conformations that deviate far from a C7 molecular symmetry, they have the inclusion capacity and the ability to adjust the topological features of their host cavity to achieve by an induced-fit mechanism the inclusion of a guest molecule. In the present study, it was shown that if the guest molecule has the proper size, shape and conformational rigidity to intrude fully inside the CD cavity, a particular stable inclusion complex is formed in which the host adopts the shape of an ‘open’ cone that approaches significantly the sevenfold molecular symmetry.
Author Contributions
Conceptualization, E.C. and K.B.; methodology, E.C., A.A., E.E. and K.B.; software, E.C., A.A. and K.B.; validation, E.C., A.A., E.E. and K.B.; formal analysis, E.C. and K.B.; investigation, E.C., A.A., E.E. and K.B.; resources, E.C., A.A., E.E. and K.B.; data curation, E.C., A.A., E.E. and K.B.; writing—original draft preparation, E.C. and K.B.; writing—review and editing, E.C., A.A., E.E. and K.B.; visualization, E.C., A.A., E.E. and K.B.; supervision, K.B.; project administration, E.E. and K.B.; funding acquisition, E.E. and K.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Crystallographic data have been deposited into the Cambridge Structural Database (CSD) under the deposition numbers CCDC: 2160066 and 2160065.
Acknowledgments
We acknowledge support of this work by the project “INSPIRED-The National [Research Infrastructures on Integrated Structural Biology, Drug Screening Efforts and Drug target functional characterization” (MIS 5002550), which is implemented under the Action “Reinforcement of the Research and Innovation Infrastructure”, funded by the Operational Programme “Competitiveness, Entrepreneurship and Innovation” (NSRF 2014-2020) and cofinanced by Greece and the European Union (European Regional Development Fund).
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations: Permethylated beta-cyclodextrin (TM-β-CD); gibberellic acid (GA3); molecular dynamics simulation (MD); site occupancy factor (s.o.f.)
References
- Poulson, B.G.; Alsulami, Q.A.; Sharfalddin, A.; El Agammy, E.F.; Mouffouk, F.; Emwas, A.-H.; Jaremko, L.; Jaremko, M. Cyclodextrins: Structural, Chemical, and Physical Properties, and Applications. Polysaccharides 2022, 3, 1–31. [Google Scholar] [CrossRef]
- Fourmentin, S.; Crini, G.; Lichtfouse, E. Cyclodextrin Fundamentals, Reactivity and Analysis; Environmental Chemistry for a Sustainable World; Springer: Cham, Switzerland, 2018; ISBN 978-3-030-09418-8. [Google Scholar]
- Liu, Z.; Ye, L.; Xi, J.; Wang, J.; Feng, Z. Cyclodextrin Polymers: Structure, Synthesis, and Use as Drug Carriers. Prog. Polym. Sci. 2021, 118, 101408. [Google Scholar] [CrossRef]
- Bruns, C.J. Exploring and Exploiting the Symmetry-Breaking Effect of Cyclodextrins in Mechanomolecules. Symmetry 2019, 11, 1249. [Google Scholar] [CrossRef]
- Lipkowitz, K.B. Symmetry Breaking in Cyclodextrins: A Molecular Mechanics Investigation. J. Org. Chem. 1991, 56, 6357–6367. [Google Scholar] [CrossRef]
- Saenger, W. Cyclodextrin Inclusion Compounds in Research and Industry. Angew. Chem. Int. Ed. Engl. 1980, 19, 344–362. [Google Scholar] [CrossRef]
- Dodziuk, H. Molecules with Holes–Cyclodextrins. In Cyclodextrins and Their Complexes; John Wiley & Sons, Ltd.: New York, NY, USA, 2006; pp. 1–30. ISBN 978-3-527-60898-0. [Google Scholar]
- Wang, J.; Li, S.; Ye, L.; Zhang, A.-Y.; Feng, Z.-G. Formation of a Polypseudorotaxane via Self-Assembly of γ-Cyclodextrin with Poly(N-Isopropylacrylamide). Macromol. Rapid Commun. 2012, 33, 1143–1148. [Google Scholar] [CrossRef] [PubMed]
- Miura, T.; Kida, T.; Akashi, M. Recognition of Stereoregularity of Poly(Methacrylic Acid)s with γ-Cyclodextrin. Macromolecules 2011, 44, 3723–3729. [Google Scholar] [CrossRef]
- Tang, J.; Zhang, S.; Lin, Y.; Zhou, J.; Pang, L.; Nie, X.; Zhou, B.; Tang, W. Engineering Cyclodextrin Clicked Chiral Stationary Phase for High-Efficiency Enantiomer Separation. Sci. Rep. 2015, 5, 11523. [Google Scholar] [CrossRef]
- Betzel, C.; Saenger, W.; Hingerty, B.E.; Brown, G.M. Topography of Cyclodextrin Inclusion Complexes, Part 20. Circular and Flip-Flop Hydrogen Bonding in.Beta.-Cyclodextrin Undecahydrate: A Neutron Diffraction Study. J. Am. Chem. Soc. 1984, 106, 7545–7557. [Google Scholar] [CrossRef]
- Caira, M.R.; Bourne, S.A.; Mzondo, B. Encapsulation of the Antioxidant R-(+)-α-Lipoic Acid in Permethylated α- and β-Cyclodextrins: Thermal and X-Ray Structural Characterization of the 1:1 Inclusion Complexes. Molecules 2017, 22, 866. [Google Scholar] [CrossRef]
- Papaioannou, A.; Christoforides, E.; Bethanis, K. Inclusion Complexes of Naringenin in Dimethylated and Permethylated β-Cyclodextrins: Crystal Structures and Molecular Dynamics Studies. Crystals 2019, 10, 10. [Google Scholar] [CrossRef]
- Bethanis, K.; Christoforides, E.; Tsorteki, F.; Fourtaka, K.; Mentzafos, D. Structural Studies of the Inclusion Compounds of α-Naphthaleneacetic Acid in Heptakis(2,6-Di-O-Methyl)-β-Cyclodextrin and Heptakis(2,3,6-Tri-O-Methyl)-β-Cyclodextrin by X-Ray Crystallography and Molecular Dynamics. J. Incl. Phenom. Macrocycl. Chem. 2018, 92, 157–171. [Google Scholar] [CrossRef]
- Hedden, P.; Sponsel, V. A Century of Gibberellin Research. J. Plant Growth Regul. 2015, 34, 740–760. [Google Scholar] [CrossRef] [PubMed]
- Schwechheimer, C. Gibberellin Signaling in Plants—The Extended Version. Front. Plant Sci. 2012, 2, 107. [Google Scholar] [CrossRef]
- Camara, M.C.; Vandenberghe, L.P.S.; Rodrigues, C.; de Oliveira, J.; Faulds, C.; Bertrand, E.; Soccol, C.R. Current Advances in Gibberellic Acid (GA3) Production, Patented Technologies and Potential Applications. Planta 2018, 248, 1049–1062. [Google Scholar] [CrossRef]
- Gao, X.-T.; Wu, M.-H.; Sun, D.; Li, H.-Q.; Chen, W.-K.; Yang, H.-Y.; Liu, F.-Q.; Wang, Q.-C.; Wang, Y.-Y.; Wang, J.; et al. Effects of Gibberellic Acid (GA3) Application before Anthesis on Rachis Elongation and Berry Quality and Aroma and Flavour Compounds in Vitis Vinifera L. “Cabernet Franc” and “Cabernet Sauvignon” Grapes. J. Sci. Food Agric. 2020, 100, 3729–3740. [Google Scholar] [CrossRef]
- Gao, S.; Chu, C. Gibberellin Metabolism and Signaling: Targets for Improving Agronomic Performance of Crops. Plant Cell Physiol. 2020, 61, 1902–1911. [Google Scholar] [CrossRef]
- Caira, M.R.; Griffith, V.J.; Nassimbeni, L.R.; van Oudtshoorn, B. Unusual 1C4 Conformation of a Methylglucose Residue in Crystalline Permethyl-[Small Beta]-Cyclodextrin Monohydrate. J. Chem. Soc. Perkin Trans. 2 1994, 2071–2072. [Google Scholar] [CrossRef]
- Steiner, T.; Saenger, W. Closure of the Cavity in Permethylated Cyclodextrins through Glucose Inversion, Flipping, and Kinking. Angew. Chem. Int. Ed. 1998, 37, 3404–3407. [Google Scholar] [CrossRef]
- Christoforides, E.; Fourtaka, K.; Andreou, A.; Bethanis, K. X-ray Crystallography and Molecular Dynamics Studies of the Inclusion Complexes of Geraniol in β-Cyclodextrin, Heptakis (2,6-Di-O-Methyl)-β-Cyclodextrin and Heptakis (2,3,6-Tri-O-Methyl)-β-Cyclodextrin. J. Mol. Struct. 2020, 1202, 127350. [Google Scholar] [CrossRef]
- Sheldrick G, M. SAINT, Version 8.37; Bruker AXS Inc.: Madison, WI, USA, 2013. [Google Scholar]
- Sheldrick, G.M. SADABS; Bruker AXS Inc.: Madison, WI, USA, 2012. [Google Scholar]
- Sheldrick, G.M. XPREP; Bruker AXS Inc.: Madison, WI, USA, 2008. [Google Scholar]
- Sheldrick, G.M. Experimental Phasing with It SHELXC/It D/It E: Combining Chain Tracing with Density Modification. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt graphical user interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Schüttelkopf, A.W.; van Aalten, D.M.F. PRODRG: A Tool for High-Throughput Crystallography of Protein–Ligand Complexes. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New Features for the Visualization and Investigation of Crystal Structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Schrödinger, LLC. The PyMOL Molecular Graphics System, Version 1.8; Schrodinger, LLC: New York, NY, USA, 2015. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An Overview of the Amber Biomolecular Simulation Package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 3, 198–210. [Google Scholar] [CrossRef]
- Cezard, C.; Trivelli, X.; Aubry, F.; Djedaini-Pilard, F.; Dupradeau, F.-Y. Molecular Dynamics Studies of Native and Substituted Cyclodextrins in Different Media: 1. Charge Derivation and Force Field Performances. Phys. Chem. Chem. Phys. 2011, 13, 15103–15121. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. 3rd PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.Py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Fourtaka, K.; Christoforides, E.; Mentzafos, D.; Bethanis, K. Crystal Structures and Molecular Dynamics Studies of the Inclusion Compounds of β-Citronellol in β-Cyclodextrin, Heptakis(2,6-Di-O-Methyl)-β-Cyclodextrin and Heptakis(2,3,6-Tri-O-Methyl)-β-Cyclodextrin. J. Mol. Struct. 2018, 1161, 1–8. [Google Scholar] [CrossRef]
- Fourtaka, K.; Christoforides, E.; Tzamalis, P.; Bethanis, K. Inclusion of Citral Isomers in Native and Methylated Cyclodextrins: Structural Insights by X-Ray Crystallography and Molecular Dynamics Simulation Analysis. J. Mol. Struct. 2021, 1234, 130169. [Google Scholar] [CrossRef]
- Cruickshank, D.L.; Rougier, N.M.; Maurel, V.J.; de Rossi, R.H.; Buján, E.I.; Bourne, S.A.; Caira, M.R. Permethylated β-Cyclodextrin/Pesticide Complexes: X-Ray Structures and Thermogravimetric Assessment of Kinetic Parameters for Complex Dissociation. J. Incl. Phenom. Macrocycl. Chem. 2013, 75, 47–56. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Shi, J.; Guo, D.-S.; Ding, F.; Liu, Y. Unique Regioselective Binding of Permethylated β-Cyclodextrin with Azobenzene Derivatives. Eur. J. Org. Chem. 2009, 2009, 923–931. [Google Scholar] [CrossRef]
- Triantafyllopoulou, V.; Tsorteki, F.; Mentzafos, D.; Bethanis, K. Inclusion Compounds of Plant Growth Regulators in Cyclodextrins, Part VII: Study of the Crystal Structures of 2-Naphthylacetic Acid Encapsulated in β-Cyclodextrin and Heptakis(2,3,6-Tri-O-Methyl)-β-Cyclodextrin Complexes by X-Ray Crystallography. J. Incl. Phenom. Macrocycl. Chem. 2013, 75, 303–310. [Google Scholar] [CrossRef][Green Version]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).