
Two New Organic Co-Crystals Based on Acetamidophenol Molecules

 ,
,  , and
, and
Abstract
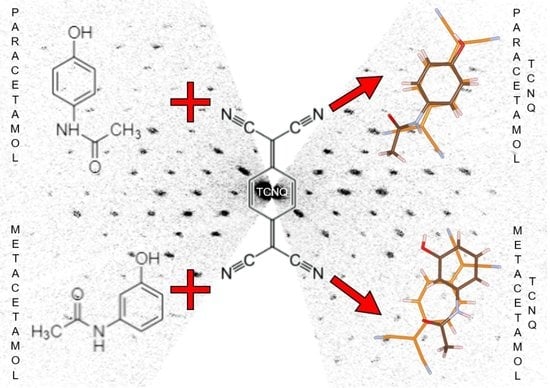
:
1. Introduction
2. Materials and Methods
2.1. Crystallisation
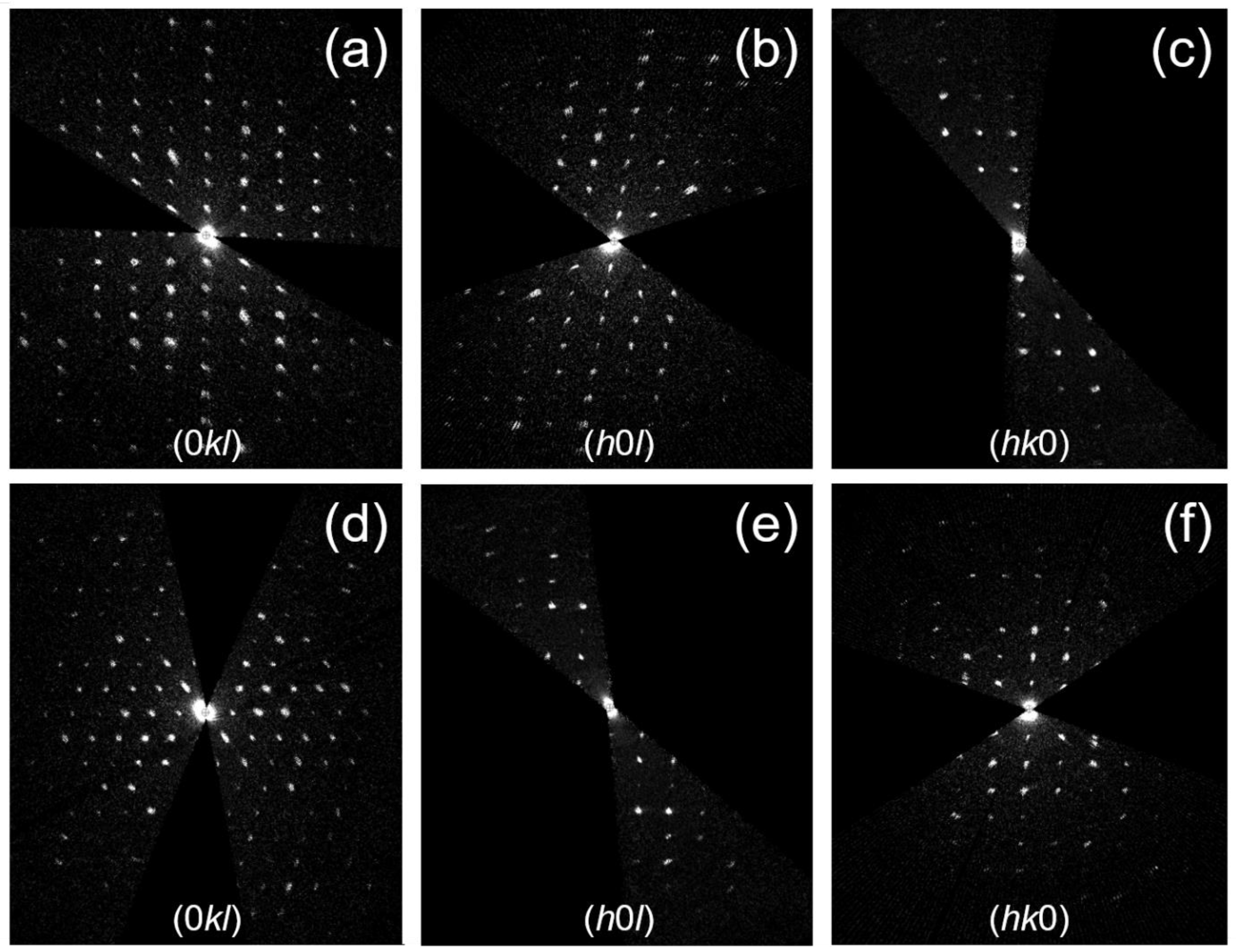
2.2. TEM and 3D ED Structure Analysis
2.3. Theoretical Calculations
3. Results and Discussion
3.1. Structure Determination of Paracetamol-TCNQ Co-Crystal
3.2. Structure Determination of Metacetamol-TCNQ Co-Crystal
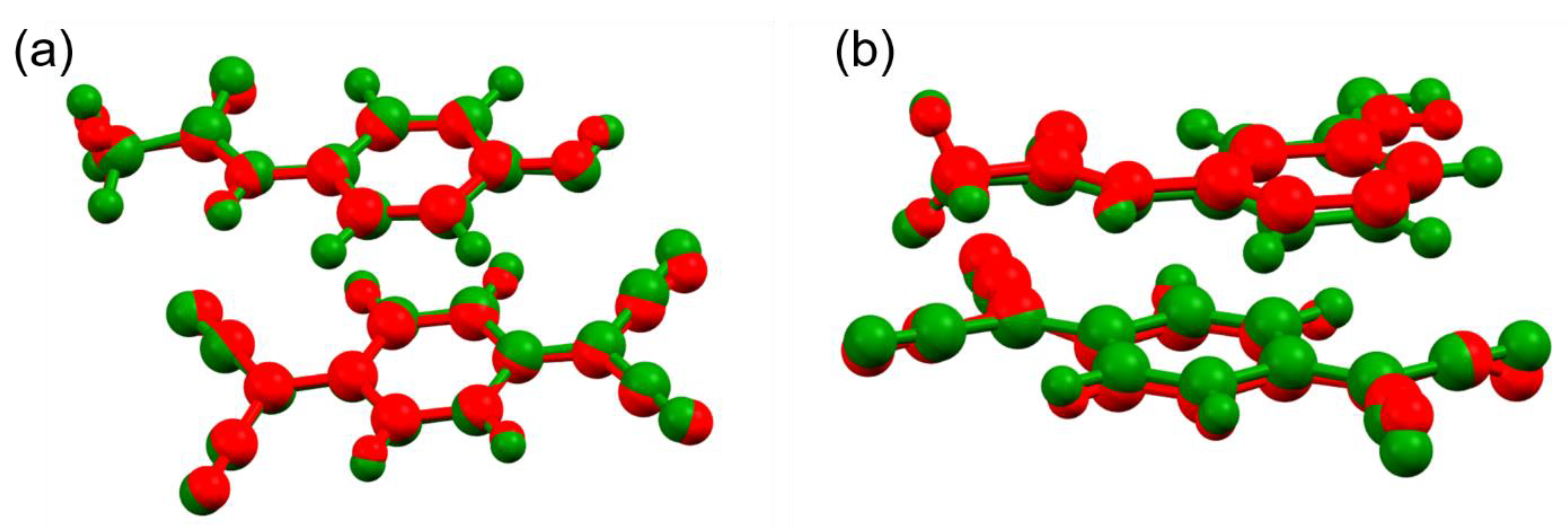
3.3. Structure Refinement and Energy Minimisation
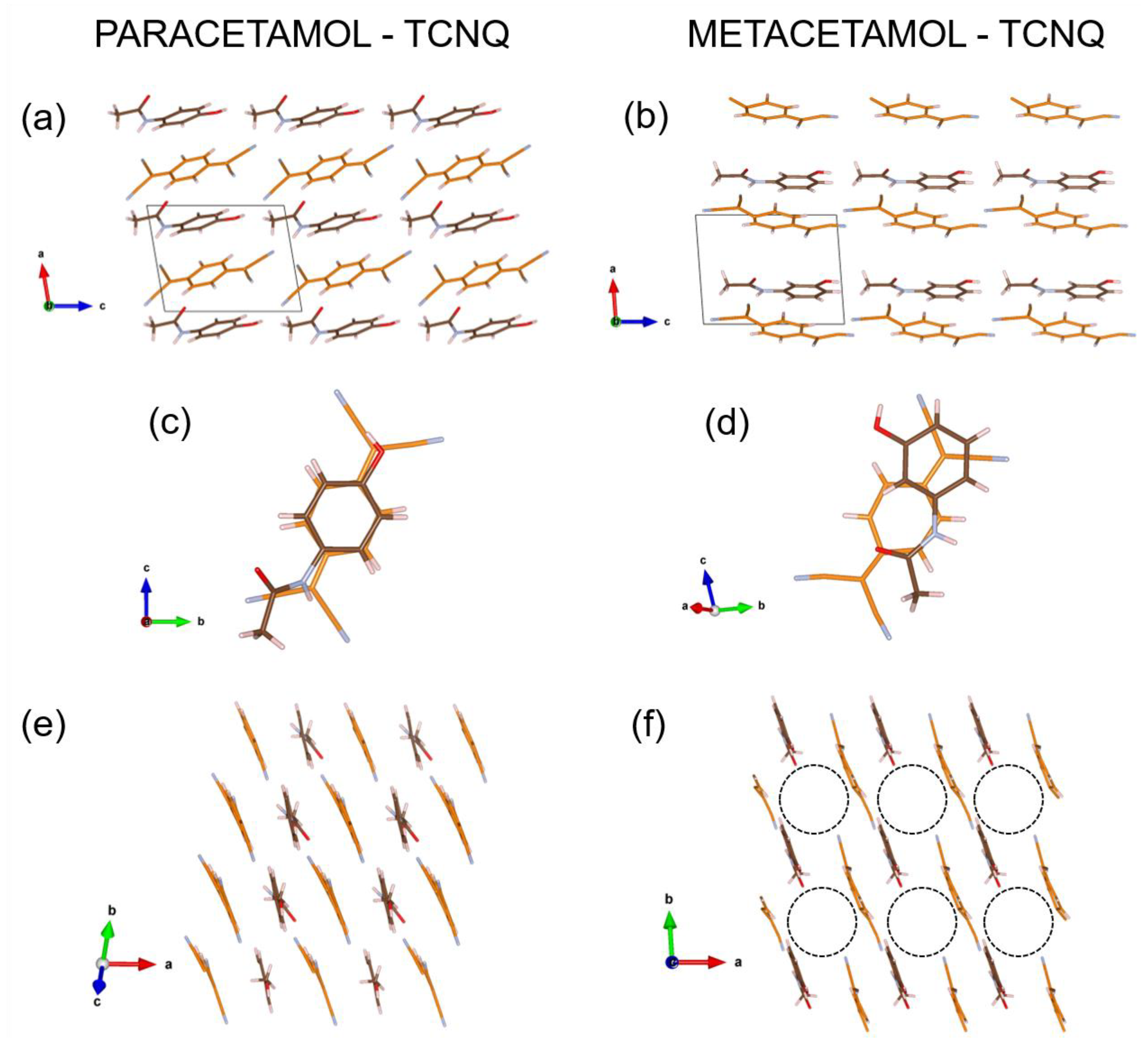
3.4. Structure Description
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kolb, U.; Gorelik, T.; Kübel, C.; Otten, M.T.; Hubert, D. Towards automated diffraction tomography: Part I—Data acquisition. Ultramicroscopy 2007, 107, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Nannenga, B.L.; Iadanza, M.G.; Gonen, T. Threedimensional electron crystallography of protein microcrystals. eLife 2013, 2, e01345. [Google Scholar] [CrossRef]
- Gemmi, M.; Mugnaioli, E.; Gorelik, T.E.; Kolb, U.; Palatinus, L.; Boullay, P.; Hovmöller, S.; Abrahams, J.P. 3D electron diffraction: The nanocrystallography revolution. ACS Cent. Sci. 2019, 5, 1315–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorset, D.L. Structural Electron Crystallography; Plenum Press: New York, NY, USA, 1995. [Google Scholar]
- Steeds, J.W. Convergent beam electron diffraction. In Introduction to Analytical Electron Microscopy; Hren, J.J., Goldstein, J.I., Joy, D.C., Eds.; Springer: Boston, MA, USA, 1979. [Google Scholar]
- Zuo, J.M.; Spence, J.C.H. Automated structure factor refinement from convergent-beam patterns. Ultramicroscopy 1991, 35, 185–186. [Google Scholar] [CrossRef]
- Mugnaioli, E.; Gorelik, T.; Kolb, U. “Ab Initio” structure solution from electron diffraction data obtained by a combination of automated diffraction tomography and precession technique. Ultramicroscopy 2009, 109, 758–765. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Oleynikov, P.; Hovmöller, S.; Zou, X. Collecting 3D electron diffraction data by the rotation method. Z. Kristallogr. 2010, 225, 94–102. [Google Scholar] [CrossRef]
- Nederlof, I.; van Genderen, E.; Li, Y.-W.; Abrahams, J.P. A Medipix quantum area detector allows rotation electron diffraction data collection from submicrometre three-dimensional protein crystals. Acta Crystallogr. D 2013, 69, 1223–1230. [Google Scholar] [CrossRef] [Green Version]
- Nannenga, B.L.; Shi, D.; Leslie, A.G.W.; Gonen, T. High-resolution structure determination by continuous-rotation data collection in MicroED. Nat. Methods 2014, 11, 927–930. [Google Scholar] [CrossRef] [Green Version]
- Gruene, T.; Wennmacher, J.T.C.; Zaubitzer, C.; Holstein, J.J.; Heidler, J.; Fecteau-Lefebvre, A.; De Carlo, S.; Müller, E.; Goldie, K.N.; Regeni, I.; et al. Rapid structure determination of microcrystalline molecular compounds using electron diffraction. Angew. Chem. Int. Ed. 2018, 57, 16313–16317. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.G.; Martynowycz, M.W.; Hattne, J.; Fulton, T.J.; Stoltz, B.M.; Rodriguez, J.A.; Nelson, H.M.; Gonen, T. The cryoEM method microED as a powerful tool for small molecule structure determination. ACS Cent. Sci. 2018, 4, 1587–1592. [Google Scholar] [CrossRef]
- Huang, Z.; Grape, S.E.; Li, J.; Inge, A.K.; Zou, X. 3D electron diffraction as an important technique for structure elucidation of metal-organic frameworks and covalent organic frameworks. Coordin. Chem. Rev. 2021, 427, 213583. [Google Scholar] [CrossRef]
- Gemmi, M.; Lanza, A.E. 3D electron diffraction techniques. Acta Crystallogr. B 2019, 75, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Ge, M.; Yang, T.; Wang, Y.; Carraro, F.; Liang, W.; Doonan, C.; Falcaro, P.; Zheng, H.; Zou, X.; Huang, Z. On the completeness of three-dimensional electron diffraction data for structural analysis of metal–organic frameworks. Faraday Discuss. 2021, 231, 66–80. [Google Scholar] [CrossRef] [PubMed]
- Andrusenko, I.; Hamilton, V.; Mugnaioli, E.; Lanza, A.; Hall, C.; Potticary, J.; Hall, S.R.; Gemmi, M. The crystal structure of orthocetamol solved by 3D electron diffraction. Angew. Chem. Int. Ed. 2019, 58, 10919–10922. [Google Scholar] [CrossRef]
- Andrusenko, I.; Hamilton, V.; Lanza, A.E.; Hall, C.L.; Mugnaioli, E.; Potticary, J.; Buanz, A.; Gaisford, S.; Piras, A.M.; Zambito, Y.; et al. Structure determination, thermal stability and dissolution rate of δ-indomethacin. Int. J. Pharm. 2021, 608, 121067. [Google Scholar] [CrossRef]
- Brázda, P.; Palatinus, L.; Babor, M. Electron diffraction determines molecular absolute configuration in a pharmaceutical nanocrystal. Science 2019, 364, 667–669. [Google Scholar] [CrossRef]
- Andrusenko, I.; Potticary, J.; Hall, S.R.; Gemmi, M. A new olanzapine cocrystal obtained from volatile deep eutectic solvents and determined by 3D electron diffraction. Acta Crystallogr. B 2020, 76, 1036–1044. [Google Scholar] [CrossRef]
- Hitchen, J.; Andrusenko, I.; Hall, C.L.; Mugnaioli, E.; Potticary, J.; Gemmi, M.; Hall, S.R. Organic cocrystals of TCNQ and TCNB based on an orthocetamol backbone solved by three-dimensional electron diffraction. Cryst. Growth Des. 2022, 22, 1155–1163. [Google Scholar] [CrossRef]
- Lanza, A.; Margheritis, E.; Mugnaioli, E.; Cappello, V.; Garau, G.; Gemmi, M. Nanobeam precession-assisted 3D electron diffraction reveals a new polymorph of hen egg-white lysozyme. IUCrJ 2019, 6, 178–188. [Google Scholar] [CrossRef]
- Vincent, R.; Midgley, P.A. Double conical beam-rocking system for measurement of integrated electron diffraction intensities. Ultramicroscopy 1994, 53, 271–282. [Google Scholar] [CrossRef]
- Palatinus, L.; Brázda, P.; Jelínek, M.; Hrdá, J.; Steciuk, G.; Klementová, M. Specifics of the data processing of precession electron diffraction tomography data and their implementation in the program PETS2.0. Acta Crystallogr. B 2019, 75, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Burla, M.C.; Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; Cuocci, C.; Giacovazzo, C.; Mallamo, M.; Mazzone, A.; Polidori, G. Crystal structure determination and refinement via SIR2014. J. Appl. Crystallogr. 2015, 48, 306–309. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge structural database. Acta Crystallogr. B 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Momma, K.; Izumi, F.J. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S.; et al. Quantum-mechanical condensed matter simulations with CRYSTAL. WIREs Comput. Mol. Sci. 2018, 8, e1360. [Google Scholar] [CrossRef]
- Das, P.P.; Mugnaioli, E.; Nicolopoulos, S.; Tossi, C.; Gemmi, M.; Galanis, A.; Borodi, G.; Pop, M.M. Crystal structures of two important pharmaceuticals solved by 3D precession electron diffraction tomography. Org. Process Res. Dev. 2018, 22, 1365–1372. [Google Scholar] [CrossRef]
- Feyand, M.; Mugnaioli, E.; Vermoortele, F.; Bueken, B.; Dieterich, J.M.; Reimer, T.; Kolb, U.; de Vos, D.; Stock, N. Automated diffraction tomography for the structure elucidation of twinned, sub-micrometer crystals of a highly porous, catalytically active bismuth metal–organic framework. Angew. Chem. Int. Ed. 2012, 51, 10373–10376. [Google Scholar] [CrossRef]
- Yu, W.; Wang, X.-Y.; Li, J.; Li, Z.-T.; Yan, Y.-K.; Wang, W.; Pei, J. A photoconductive charge-transfer crystal with mixed-stacking donor–acceptor heterojunctions within the lattice. Chem. Commun. 2013, 49, 54–56. [Google Scholar] [CrossRef]
- Zhu, L.; Yi, Y.; Fonari, A.; Corbin, N.S.; Coropceanu, V.; Brédas, J.-L. Electronic properties of mixed-stack organic charge-transfer crystals. J. Phys. Chem. C 2014, 118, 14150–14156. [Google Scholar] [CrossRef]






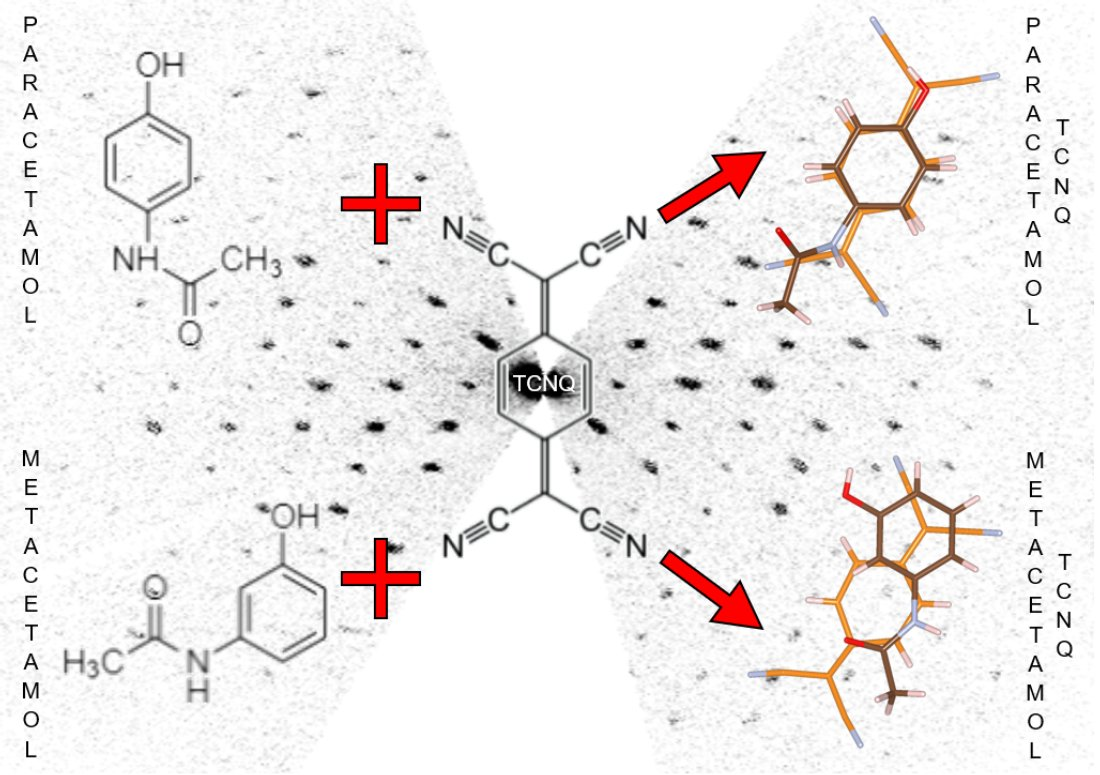{kind=link}
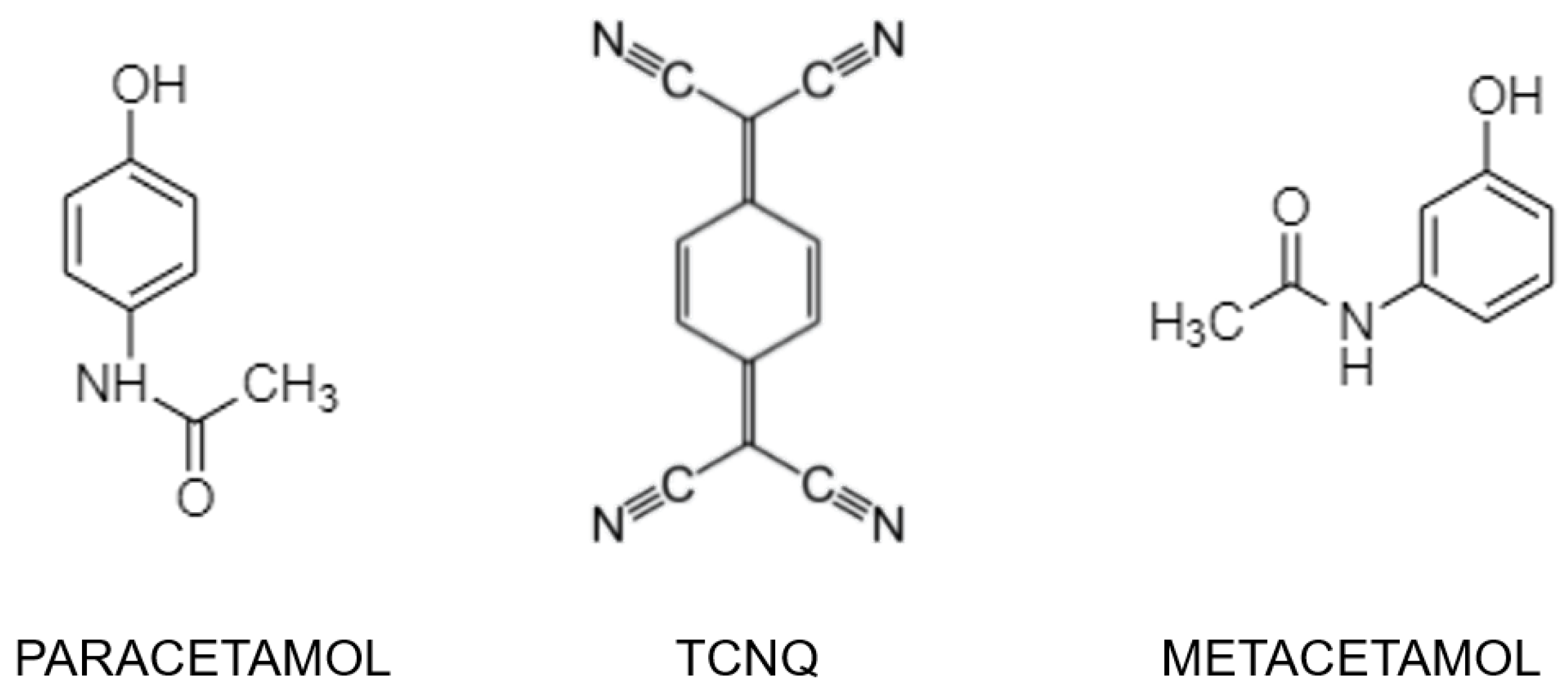{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Paracetamol-TCNQ | Metacetamol-TCNQ | |
|---|---|---|
| ab initio structure determination by SIR2014 | ||
| Data resolution (Å) | 0.8 | 0.9 |
| Sampled reflections (No.) | 2296 | 2763 |
| Independent reflections (No.) | 1251 | 1403 |
| Independent reflection coverage (%) | 71 | 76 |
| Global thermal factor U iso (Å2) | 0.02820 | 0.03214 |
| Rint (%) | 20.19 | 16.01 |
| RSIR (%) | 27.60 | 29.82 |
| structure refinement by SHELXL | ||
| Data resolution (Å) | 0.9 | 0.9 |
| Reflections total (No.) | 1492 | 2763 |
| Reflections > 4σ (No.) | 648 | 1449 |
| R14σ (%) | 37.01 | 34.60 |
| R1all (%) | 39.46 | 46.28 |
| GooF | 2.161 | 3.850 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andrusenko, I.; Hitchen, J.; Mugnaioli, E.; Potticary, J.; Hall, S.R.; Gemmi, M. Two New Organic Co-Crystals Based on Acetamidophenol Molecules. Symmetry 2022, 14, 431. https://doi.org/10.3390/sym14030431
Andrusenko I, Hitchen J, Mugnaioli E, Potticary J, Hall SR, Gemmi M. Two New Organic Co-Crystals Based on Acetamidophenol Molecules. Symmetry. 2022; 14(3):431. https://doi.org/10.3390/sym14030431
Chicago/Turabian StyleAndrusenko, Iryna, Joseph Hitchen, Enrico Mugnaioli, Jason Potticary, Simon R. Hall, and Mauro Gemmi. 2022. "Two New Organic Co-Crystals Based on Acetamidophenol Molecules" Symmetry 14, no. 3: 431. https://doi.org/10.3390/sym14030431
APA StyleAndrusenko, I., Hitchen, J., Mugnaioli, E., Potticary, J., Hall, S. R., & Gemmi, M. (2022). Two New Organic Co-Crystals Based on Acetamidophenol Molecules. Symmetry, 14(3), 431. https://doi.org/10.3390/sym14030431