
Microwave-Assisted Synthesis of 2-Methyl-1H-indole-3-carboxylate Derivatives via Pd-Catalyzed Heterocyclization

 , , , and
, , , and
Abstract
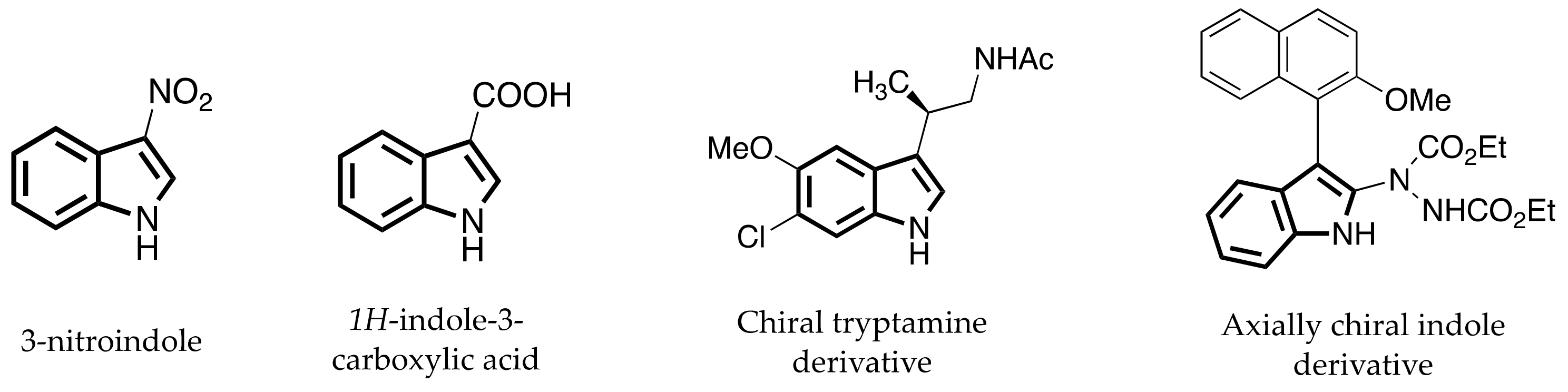
:1. Introduction
2. Materials and Methods
2.1. Materials and General Procedures
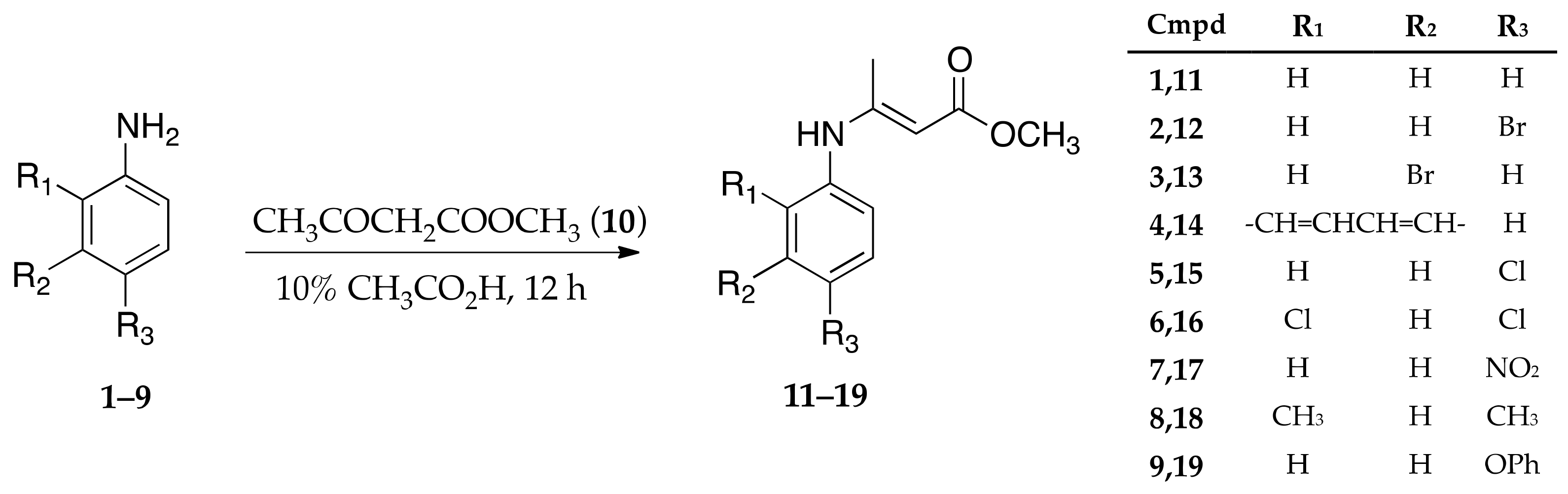
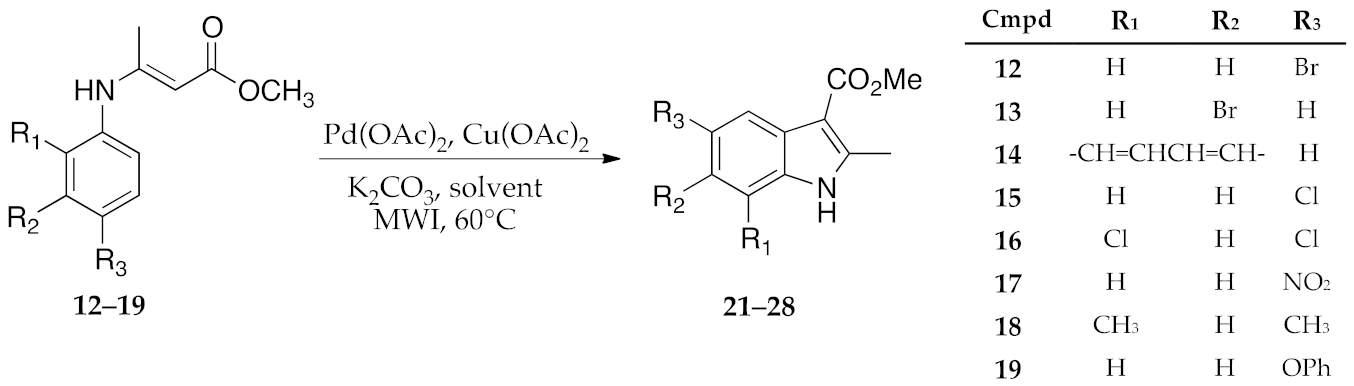
2.2. Synthesis of Enamine Derived Compounds 11–19
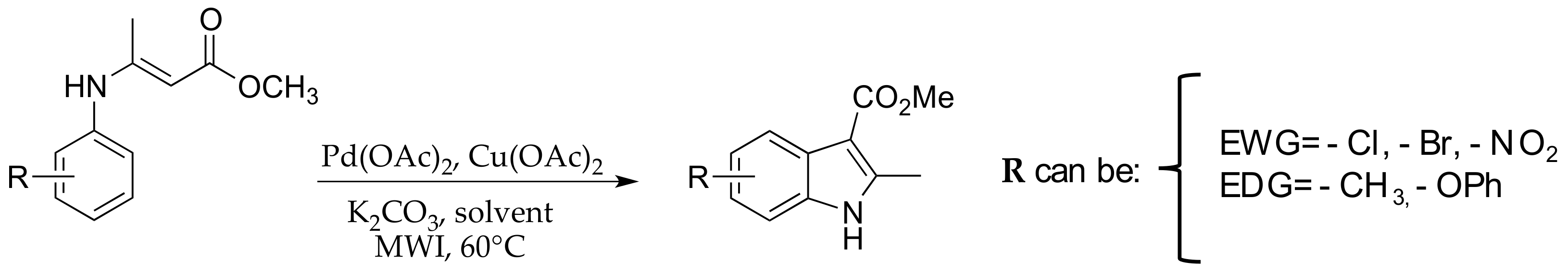
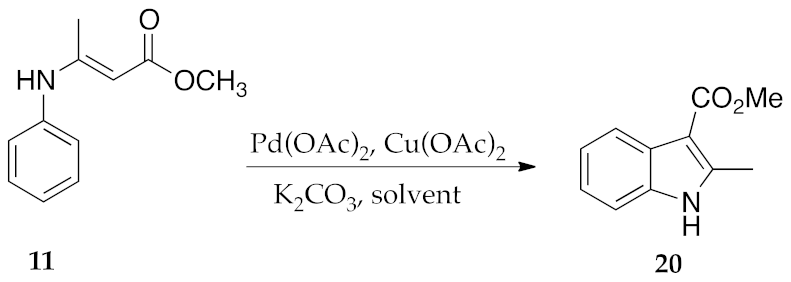
2.3. μW-Assisted Palladium-Catalyzed Oxidative Cyclization Yielding Indole Derivatives 20–28
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kaushik, N.K.; Kaushik, N.; Attri, P.; Kumar, N.; Kim, C.H.; Verma, A.K.; Choi, E.H. Biomedical Importance of Indoles. Molecules 2013, 18, 6620–6662. [Google Scholar] [CrossRef] [PubMed]
- Chadha, N.; Silakari, O. Indoles as therapeutics of interest in medicinal chemistry: Bird’s eye view. Eur. J. Med. Chem. 2017, 134, 159–184. [Google Scholar] [CrossRef] [PubMed]
- Chilton, W.S.; Bigwood, J.; Jensen, R.E. Psilocin, Bufotenine and Serotonin: Historical and Biosynthetic Observations. J. Psychedelic Drugs 1979, 11, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.S. Tryptamine: A neuromodulator or neurotransmitter in mammalian brain? Progress. Neurobiol. 1982, 19, 117–139. [Google Scholar] [CrossRef]
- Negård, M.; Uhlig, S.; Kauserud, H.; Andersen, T.; Høiland, K.; Vrålstad, T. Links between genetic groups, indole alkaloid pro-files and ecology within the grass-parasitic Claviceps purpurea species complex. Toxins 2015, 7, 1431–1456. [Google Scholar] [CrossRef] [Green Version]
- Moudi, M.; Go, R.; Yien, C.Y.; Nazre, M. Vinca alkaloids. Int. J. Prev. Med. 2013, 4, 1231–1235. [Google Scholar]
- Desai, N.; Somani, H.; Trivedi, A.; Bhatt, K.; Nawale, L.; Khedkar, V.M.; Jha, P.C.; Sarkar, D. Synthesis, biological evaluation and molecular docking study of some novel indole and pyridine based 1,3,4-oxadiazole derivatives as potential antitubercular agents. Bioorg. Med. Chem. Lett. 2016, 26, 1776–1783. [Google Scholar] [CrossRef]
- Li, T.; Liu, S.; Tan, W.; Shi, F. Catalytic Asymmetric Construction of Axially Chiral Indole-Based Frameworks: An Emerging Area. Chem.–A Eur. J. 2020, 26, 15779–15792. [Google Scholar] [CrossRef]
- Luz, J.G.; Carson, M.W.; Condon, B.; Clawson, D.; Pustilnik, A.; Kohlman, D.T.; Barr, R.J.; Bean, J.S.; Dill, M.J.; Sindelar, D.K.; et al. Indole Glucocorticoid Receptor Antagonists Active in a Model of Dyslipidemia Act via a Unique Association with an Agonist Binding Site. J. Med. Chem. 2015, 58, 6607–6618. [Google Scholar] [CrossRef]
- Rios, I.G.; Rosas-Hernández, A.; Martin, E. Recent Advances in the Application of Chiral Phosphine Ligands in Pd-Catalysed Asymmetric Allylic Alkylation. Molecules 2011, 16, 970–1010. [Google Scholar] [CrossRef]
- Bringmann, G.; Tasler, S.; Endress, H.; Kraus, J.; Messer, K.; Wohlfarth, M.; Lobin, W. Murrastifoline-F: First Total Synthesis, Atropo-Enantiomer Resolution, and Stereoanalysis of an Axially Chiral N,C-Coupled Biaryl Alkaloid. J. Am. Chem. Soc. 2001, 123, 2703–2711. [Google Scholar] [CrossRef] [PubMed]
- Dedashpour, S.; Emami, S. Indole in the target-based design of anticancer agents: A versatile scaffold with diverse mechanisms. Eur. J. Med. Chem. 2018, 150, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Robinson, B. The Fischer Indole Synthesis; John Wiley and Sons: New York, NY, USA, 1982; pp. 48–59. [Google Scholar]
- Baudin, J.-B.; Julia, S.A. Synthesis of indoles from N-aryl-1-alkenylsulphinamides. Tetrahedron Lett. 1986, 27, 837–840. [Google Scholar] [CrossRef]
- Bartoli, G.; Palmieri, G.; Bosco, M.; Dalpozzo, R. The reaction of vinyl grignard reagents with 2-substituted nitroarenes: A new approach to the synthesis of 7-substituted indoles. Tetrahedron Lett. 1989, 30, 2129–2132. [Google Scholar] [CrossRef]
- Willis, M.C.; Brace, G.N.; Holmes, I.P. Palladium-Catalyzed Tandem Alkenyl and Aryl C?N Bond Formation: A Cascade N-Annulation Route to 1-Functionalized Indoles. Angew. Chem. Int. Ed. 2005, 44, 403–406. [Google Scholar] [CrossRef]
- Zhang, Y.-C.; Jiang, F.; Shi, F. Organocatalytic Asymmetric Synthesis of Indole-Based Chiral Heterocycles: Strategies, Reactions, and Outreach. Accounts Chem. Res. 2019, 53, 425–446. [Google Scholar] [CrossRef]
- Kumari, A.; Singh, R.K. Medicinal chemistry of indole derivatives: Current to future therapeutic prospectives. Bioorg. Chem. 2019, 89, 103021. [Google Scholar] [CrossRef]
- Bahekar, R.H.; Jain, M.R.; Goel, A.; Patel, D.N.; Prajapati, V.M.; Gupta, A.A.; Jadav, P.A.; Patel, P.R. Design, synthesis, and biological evaluation of substituted-N-(thieno[2,3-b]pyridin-3-yl)-guanidines, N-(1H-pyrrolo[2,3-b]pyridin-3-yl)-guanidines, and N-(1H-indol-3-yl)-guanidines. Bioorg. Med. Chem. 2007, 15, 3248–3265. [Google Scholar] [CrossRef]
- Eleftheriadis, N.; Neochoritis, C.G.; Leus, N.G.J.; Van Der Wouden, P.E.; Dömling, A.; Dekker, F.J. Rational Development of a Potent 15-Lipoxygenase-1 Inhibitor with in Vitro and ex Vivo Anti-inflammatory Properties. J. Med. Chem. 2015, 58, 7850–7862. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, C.R.; Czekaj, M.; Kaye, S.S.; Gao, Z.; Pribish, J.; Pauls, H.; Liang, G.; Sides, K.; Cramer, D.; Cairns, J.; et al. Design, synthesis, and biological activity of potent and selective inhibitors of mast cell tryptase. Bioorg. Med. Chem. Lett. 2005, 15, 2734–2737. [Google Scholar] [CrossRef]
- Wolfard, J.; Xu, J.; Zhang, H.; Chung, C.K. Synthesis of chiral tryptamines via a regioselective indole alkylation. Org. Lett. 2018, 20, 5431–5434. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Chen, K.; Wu, P.; Zhang, Y.; Jiao, Y.; Shi, F. A Strategy for Synthesizing Axially Chiral Naphthyl-Indoles: Catalytic Asymmetric Addition Reactions of Racemic Substrates. Angew. Chem. Int. Ed. 2019, 58, 15104–15110. [Google Scholar] [CrossRef] [PubMed]
- Würtz, S.; Rakshit, S.; Neumann, J.J.; Dröge, T.; Glorius, F. Palladium-Catalyzed Oxidative Cyclization ofN-Aryl Enamines: From Anilines to Indoles. Angew. Chem. Int. Ed. 2008, 47, 7230–7233. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.A.; Miller, D.D. Microwave-Assisted Synthesis of Medicinally Relevant Indoles. Curr. Med. Chem. 2011, 18, 615–637. [Google Scholar] [CrossRef] [PubMed]
- Wilson, N.S.; Roth, G.P. Recent trends in microwave-assisted synthesis. Curr. Opin. Drug Discov. Dev. 2002, 5, 620–629. [Google Scholar] [CrossRef]
- Wathey, B.; Tierney, J.; Lidström, P.; Westman, J. The impact of microwave-assisted organic chemistry on drug discovery. Drug Discov. Today 2002, 7, 373–380. [Google Scholar] [CrossRef]
- Berrino, E.; Supuran, C.T. Advances in microwave-assisted synthesis and the impact of novel drug discovery. Expert Opin. Drug Discov. 2018, 13, 861–873. [Google Scholar] [CrossRef]
- Franco, L.H.; Palermo, J.A. Synthesis of 2-(Pyrimidin-4-yl)indoles. Chem. Pharm. Bull. 2003, 5, 975–977. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zhang, S.; Shi, G.; Chen, Z. Electrochemical synthesis of 1,2,4,5-tetrasubstituted imidazoles from enamines and benzylamines. Org. Biomol. Chem. 2021, 19, 6682–6686. [Google Scholar] [CrossRef]
- Huang, W.; Zhu, C.; Li, M.; Yu, Y.; Wu, W.; Tu, Z.; Jiang, H. TBAI or KI-Promoted Oxidative Coupling of Enamines and N -Tosylhydrazine: An Unconventional Method toward 1,5- and 1,4,5-Substituted 1,2,3-Triazoles. Adv. Synth. Catal. 2018, 360, 3117–3123. [Google Scholar] [CrossRef]
- Neumann, J.J.; Rakshit, S.; Dröge, T.; Würtz, S.; Glorius, F. Exploring the Oxidative Cyclization of Substituted N -Aryl Enamines: Pd-Catalyzed Formation of Indoles from Anilines. Chem.–A Eur. J. 2011, 17, 7298–7303. [Google Scholar] [CrossRef] [PubMed]
- Zoller, J.; Fabry, D.C.; Ronge, M.A.; Rueping, M. Synthesis of Indoles Using Visible Light: Photoredox Catalysis for Palla-dium-Catalyzed CH Activation. Angew. Chem. Int. Ed. 2014, 53, 13264–13268. [Google Scholar] [CrossRef] [PubMed]
- Beletskaya, I.; Cheprakov, A.V. The Heck Reaction as a Sharpening Stone of Palladium Catalysis. Chem. Rev. 2000, 100, 3009–3066. [Google Scholar] [CrossRef] [PubMed]
- Beccalli, E.M.; Broggini, G.; Martinelli, M.; Sottocornola, S. C-C, C-O, C-N bond formation on sp2 Carbon by Pd(II)-Catalyzed reactions involving on oxidant agents. Chem. Rev. 2007, 107, 5318–5365. [Google Scholar] [CrossRef] [PubMed]
- Newman, S.G.; Lautens, M. The Role of Reversible Oxidative Addition in Selective Palladium(0)-Catalyzed Intramolecular Cross-Couplings of Polyhalogenated Substrates: Synthesis of Brominated Indoles. J. Am. Chem. Soc. 2010, 132, 11416–11417. [Google Scholar] [CrossRef]
- Dallinger, D.; Kappe, C.O. Microwave-Assisted Synthesis in Water as Solvent. Chem. Rev. 2007, 107, 2563–2591. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Entry | Solvent | Energy Input | Time (h) | Isolated Yields (%) |
|---|---|---|---|---|
| 1 | DMF | a | 16 | 76 |
| 2 | DMF | b | 0.5 | 93 |
| 3 | DMF | b | 0.25 | 67 |
| 4 | DMF | c | 0.5 | 19 |
| Compound | Microwave | Conventional Heating | ||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Solvent | Time (h) | Isolated Yield (%) | Entry | Solvent | Time (h) | Isolated Yield (%) | |
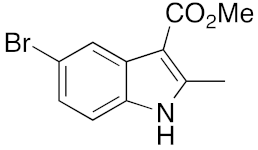 21 | 1 | DMF | 3 | 94 | 3 | DMF | 16 | 89 |
| 2 | DMF | 1 | 55 | 4 | DMF | 6 | 51 | |
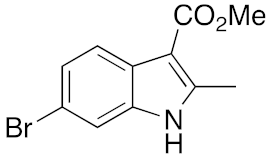 22 | 5 | DMF | 3 | 29 | 8 | DMF | 16 | 17 |
| 6 | DMSO | 3 | 74 | 9 | DMSO | 16 | 33 | |
| 7 | ACN a | 3 | 8 | 10 | ACN b | 16 | ND | |
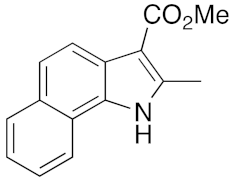 23 | 11 | DMF | 1 | 95 | 12 | DMF | 6 | 91 |
| 13 | DMF | 6 | 78 | |||||
 24 | 14 | DMF | 1 | 90 | 16 | DMF | 16 | 73 |
| 15 | DMF | 0.5 | 82 | |||||
 25 | 17 | DMF | 1 | 95 | 19 | DMF | 3 | 91 |
| 18 | DMF | 0.5 | 82 | 20 | DMF | 1 | 23 | |
 26 | 21 | DMF | 3 | 18 | 23 | DMF | 16 | 12 |
| 22 | DMSO | 3 | ND | 24 | DMSO | 16 | ND | |
| 25 | ACN a | 3 | 91 | 27 | ACN b | 16 | 24 | |
| 26 | ACN a | 1 | 12 | |||||
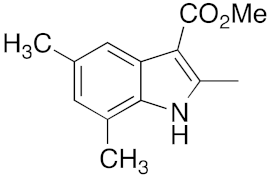 27 | 28 | DMF | 3 | 94 | 30 | DMF | 6 | 89 |
| 29 | DMF | 1 | 38 | 31 | DMF | 3 | 58 | |
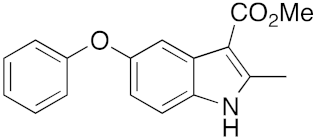 28 | 32 | DMF | 1 | 95 | 34 | DMF | 3 | 90 |
| 33 | DMF | 0.5 | 62 | 35 | DMF | 1 | 43 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bellavita, R.; Casertano, M.; Grasso, N.; Gillick-Healy, M.; Kelly, B.G.; Adamo, M.F.A.; Menna, M.; Merlino, F.; Grieco, P. Microwave-Assisted Synthesis of 2-Methyl-1H-indole-3-carboxylate Derivatives via Pd-Catalyzed Heterocyclization. Symmetry 2022, 14, 435. https://doi.org/10.3390/sym14030435
Bellavita R, Casertano M, Grasso N, Gillick-Healy M, Kelly BG, Adamo MFA, Menna M, Merlino F, Grieco P. Microwave-Assisted Synthesis of 2-Methyl-1H-indole-3-carboxylate Derivatives via Pd-Catalyzed Heterocyclization. Symmetry. 2022; 14(3):435. https://doi.org/10.3390/sym14030435
Chicago/Turabian StyleBellavita, Rosa, Marcello Casertano, Nicola Grasso, Malachi Gillick-Healy, Brian G. Kelly, Mauro F. A. Adamo, Marialuisa Menna, Francesco Merlino, and Paolo Grieco. 2022. "Microwave-Assisted Synthesis of 2-Methyl-1H-indole-3-carboxylate Derivatives via Pd-Catalyzed Heterocyclization" Symmetry 14, no. 3: 435. https://doi.org/10.3390/sym14030435