
One for All, All for One: The Peculiar Dynamics of TNF-Receptor-Associated Factor (TRAF2) Subunits

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Spectroscopic Assays
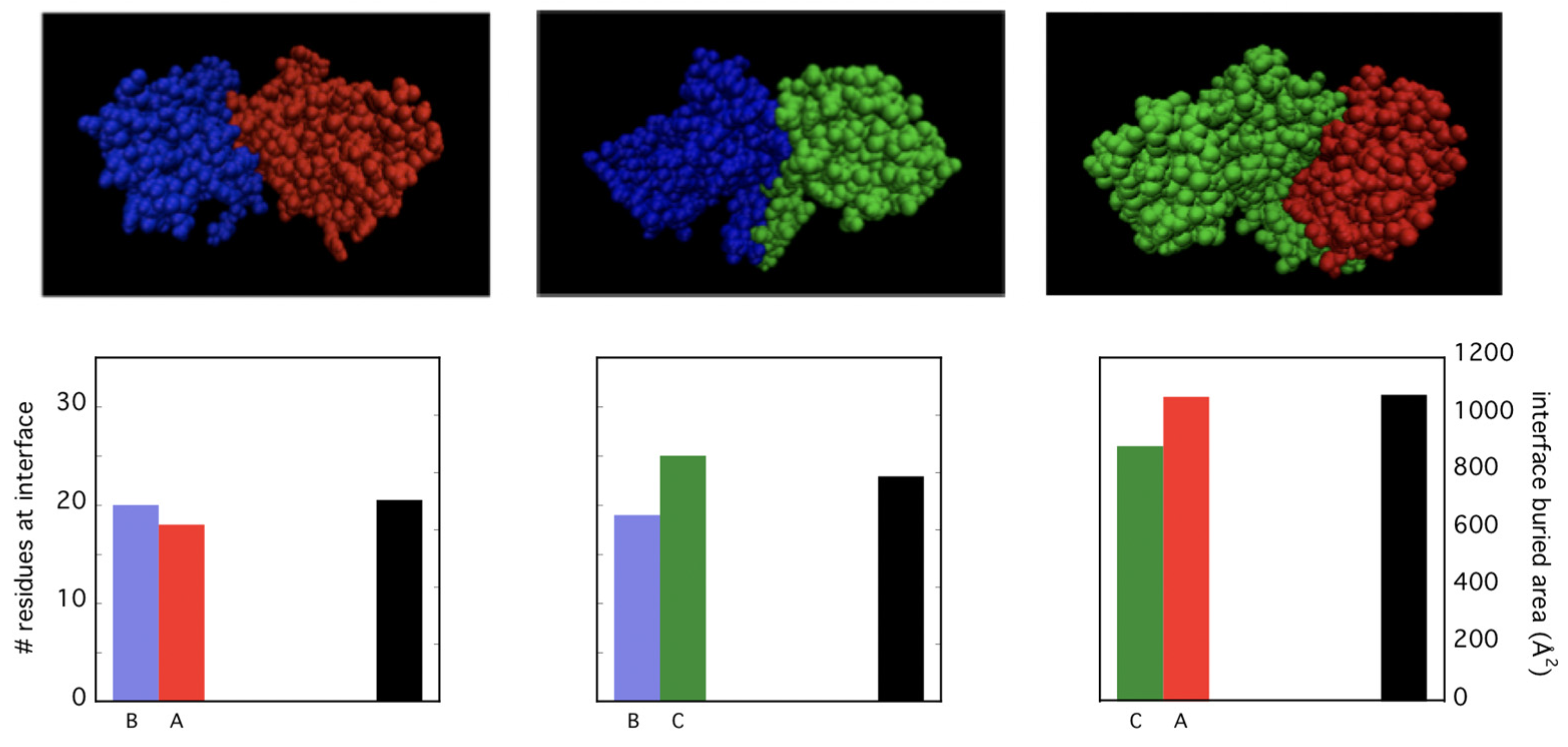
2.3. Molecular Graphics and Analysis of Subunits Interfaces
2.4. Molecular Dynamics Simulation
2.5. Dynamic Cross Correlation and Canonical Correlation Analysis
3. Results and Discussion
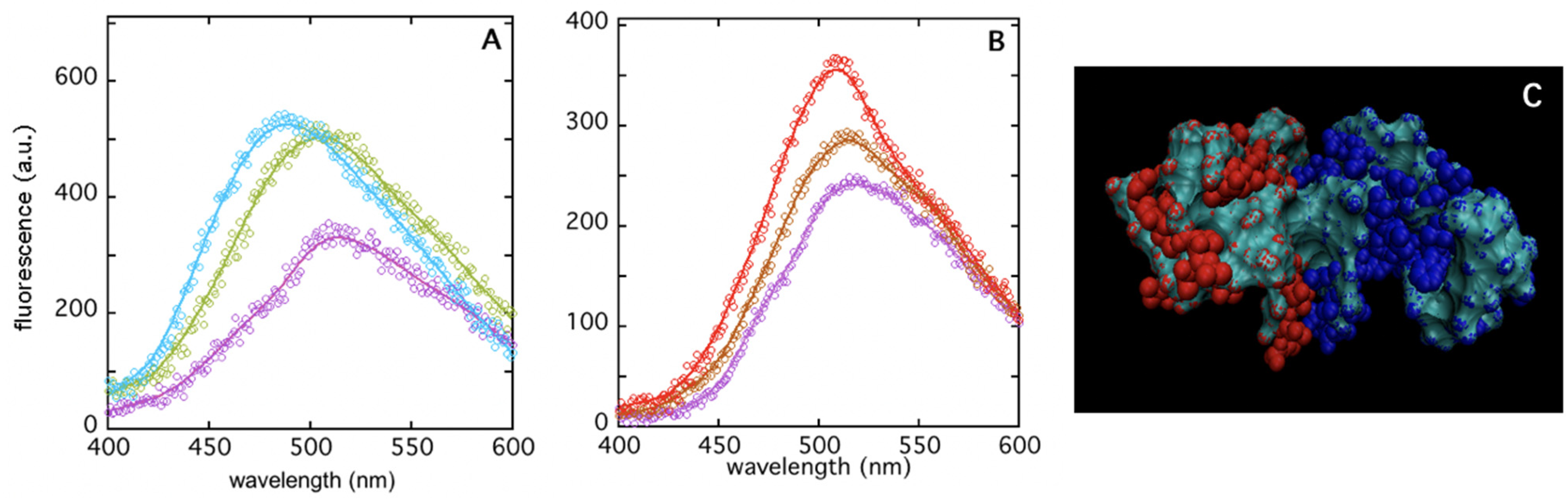
3.1. Insights on Inter-Subunits Interface by Dissociation Measurements
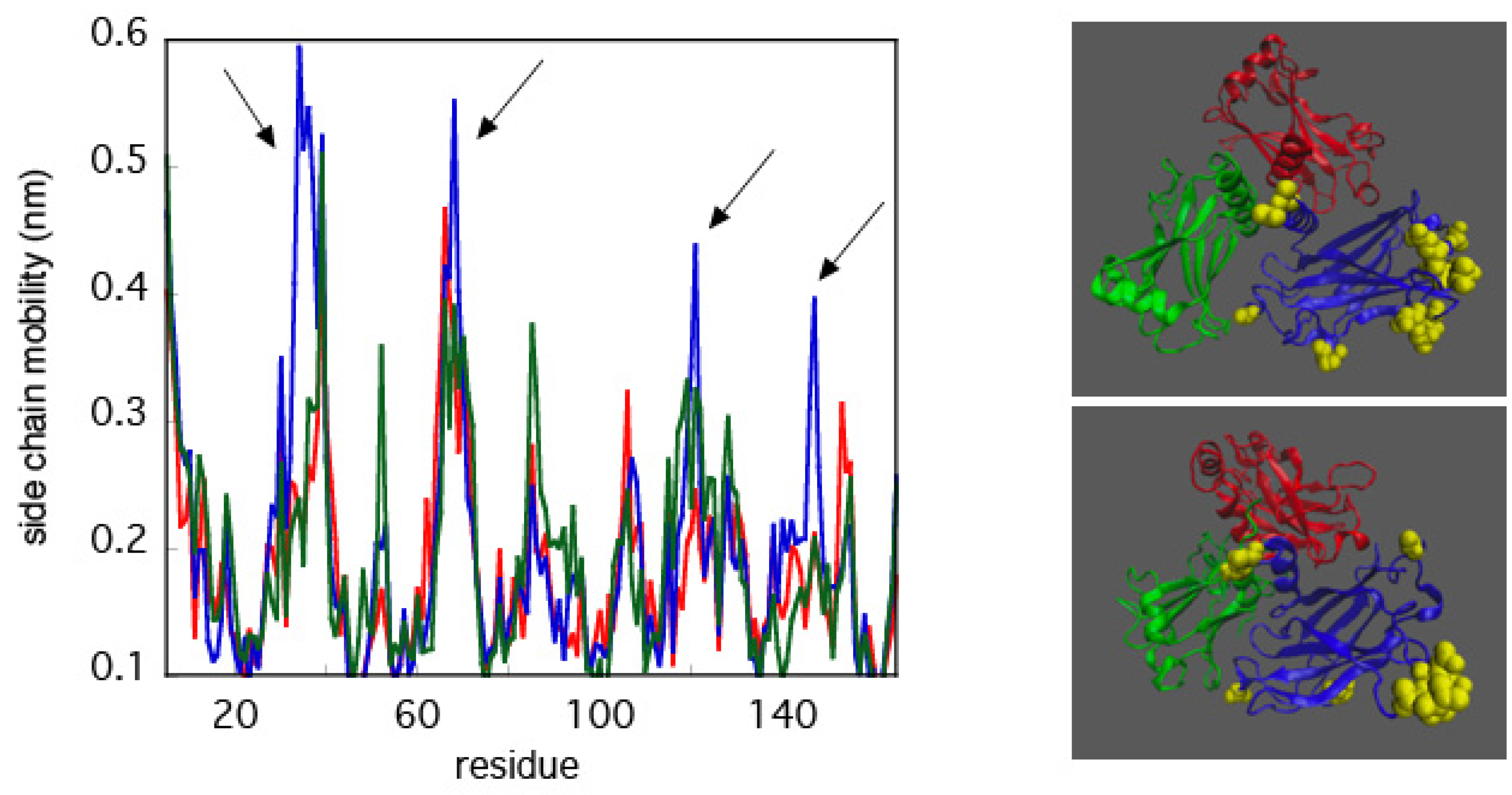
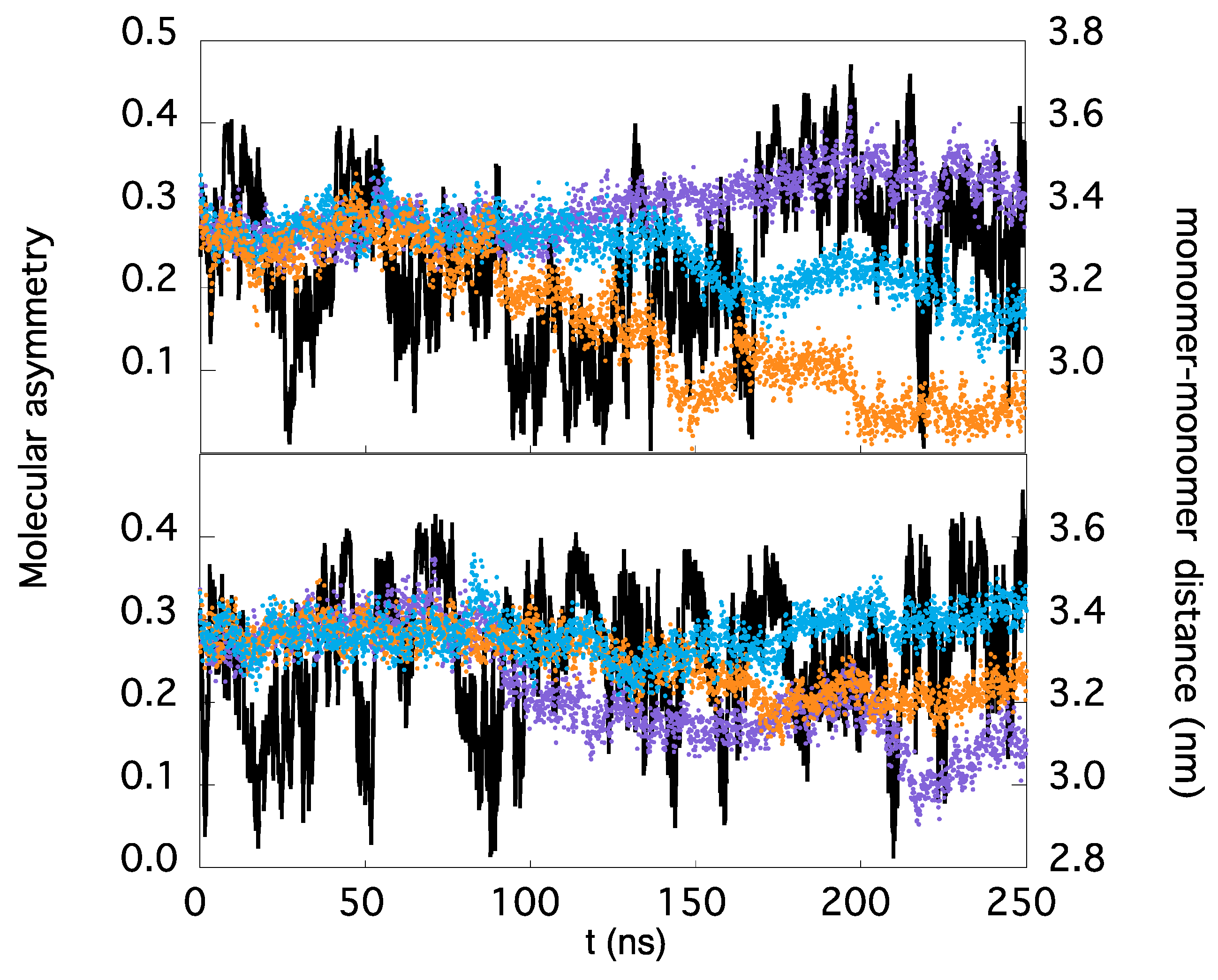
3.2. Asymmetric Mobility of TRAF2-C Monomers
3.3. Asymmetric and Correlated Motions in TRAF2-C Dynamics
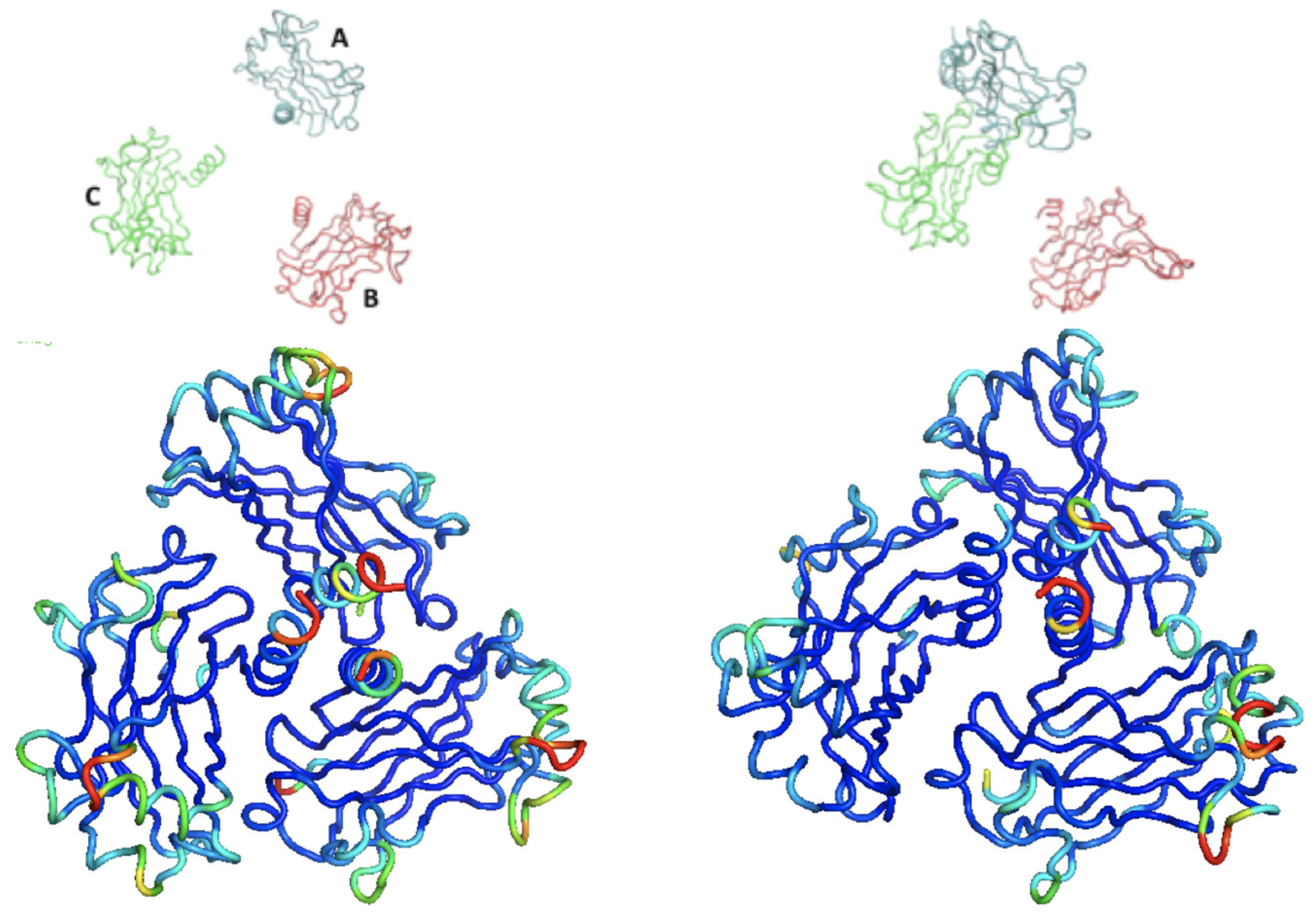
3.4. Dynamic Clusters Analysis of TRAF2-C
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Doyle, C.M.; Rumfeldt, J.A.; Broom, H.R.; Broom, A.; Stathopulos, P.B.; Vassall, K.A.; Almey, J.J.; Meiering, E.M. Energetics of oligomeric protein folding and association. Arch. Biochem. Biophys. 2013, 531, 44–64. [Google Scholar] [CrossRef]
- Suplatov, D.; Švedas, V. Study of Functional and Allosteric Sites in Protein Superfamilies. Acta Nat. 2015, 7, 34–45. [Google Scholar] [CrossRef] [Green Version]
- Levy, E.D.; Boeri Erba, E.; Robinson, C.V.; Teichmann, S.A. Assembly reflects evolution of protein complexes. Nature 2008, 453, 1262–1265. [Google Scholar] [CrossRef] [Green Version]
- Venkatakrishnan, A.J.; Levy, E.D.; Teichmann, S.A. Homomeric protein complexes: Evolution and assembly. Biochem. Soc. Trans. 2010, 38, 879–882. [Google Scholar] [CrossRef]
- Goodsell, D.S.; Olson, A.J. Structural symmetry and protein function. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 105–153. [Google Scholar] [CrossRef]
- Swapna, L.S.; Srikeerthana, K.; Srinivasan, N. Extent of structural asymmetry in homodimeric proteins: Prevalence and relevance. PLoS ONE 2012, 7, e36688. [Google Scholar] [CrossRef] [Green Version]
- Minicozzi, V.; Di Venere, A.; Nicolai, E.; Giuliani, A.; Caccuri, A.M.; Di Paola, L.; Mei, G. Non-Symmetrical Structural Behavior of a Symmetric Protein: The Case of Homo-Trimeric TRAF2 (Tumor Necrosis Factor-Receptor Associated Factor 2). J. Biomol. Struct. Dyn. 2021, 39, 319–329. [Google Scholar] [CrossRef]
- Bradley, J.R.; Pober, J.S. Tumor necrosis factor receptor-associated factors (TRAFs). Oncogene 2001, 20, 6482–6491. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.C.; Burkitt, V.; Villa, A.R.; Tong, L.; Wu, H. Structural basis for self-association and receptor recognition of human TRAF2. Nature 1999, 398, 533–538. [Google Scholar] [CrossRef]
- Ceccarelli, A.; Di Venere, A.; Nicolai, E.; De Luca, A.; Minicozzi, V.; Rosato, N.; Caccuri, A.M.; Mei, G. TNFR-Associated Factor-2 (TRAF2): Not Only a Trimer. Biochemistry 2015, 54, 6153–6161. [Google Scholar] [CrossRef]
- Di Venere, A.; Nicolai, E.; Sinibaldi, F.; Di Pierro, D.; Caccuri, A.M.; Mei, G. Studying the TRAF2 binding to model membranes: The role of subunits dissociation. Biotechnol. Appl. Biochem. 2018, 65, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, A.; Di Venere, A.; Nicolai, E.; De Luca, A.; Rosato, N.; Gratton, E.; Mei, G.; Caccuri, A.M. New insight into the interaction of TRAF2 C-terminal domain with lipid raft microdomains. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 813–822. [Google Scholar] [CrossRef] [Green Version]
- Di Venere, A.; Nicolai, E.; Minicozzi, V.; Caccuri, A.M.; Di Paola, L.; Mei, G. The Odd Faces of Oligomers: The Case of TRAF2-C, A Trimeric C-Terminal Domain of TNF Receptor-Associated Factor. Int. J. Mol. Sci. 2021, 22, 5871. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Mehrabi, P.; Ren, Z.; Sljoka, A.; Ing, C.; Bezginov, A.; Ye, L.; Pomès, R.; Prosser, R.S.; Pai, E.F. The role of dimer asymmetry and protomer dynamics in enzyme catalysis. Science 2017, 355, eaag2355. [Google Scholar] [CrossRef]
- Bonjack, M.; Avnir, D. The near-symmetry of protein oligomers: NMR-derived structures. Sci. Rep. 2020, 10, 8367. [Google Scholar] [CrossRef] [PubMed]
- Di Venere, A.; Horn, T.; Stehling, S.; Mei, G.; Masgrau, L.; González-Lafont, À.; Kühn, H.; Ivanov, I. Role of Arg403 for thermostability and catalytic activity of rabbit 12/15-lipoxygenase. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2013, 1831, 1079–1088. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Molec. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Gunsteren, W.F.; Billeter, S.R.; Eking, A.A.; Hiinenberger, P.H.; Kriiger, P.; Mark, A.E.; Scott, W.R.P.; Tironi, I.G. Biomolecular Simulation: The GROMOS96 Manual and User Guide; VDF Hochschulverlag AG an der ETH Zürich: Zürich, Switzerland, 1996. [Google Scholar]
- Carbonaro, M.; Di Venere, A.; Filabozzi, A.; Maselli, P.; Minicozzi, V.; Morante, S.; Nicolai, E.; Nucara, A.; Placidi, E.; Stellato, F. Role of dietary antioxidant (-)-epicatechin in the development of β-lactoglobulin fibrils. Biochim. Biophys. Acta Proteins Proteom. 2016, 1864, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, K.; Fukuda, I.; Nakamura, H. A Novel Approach of Dynamic Cross Correlation Analysis on Molecular Dynamics Simulations and Its Application to Ets1 Dimer-DNA Complex. PLoS ONE 2014, 9, e112419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cumbo, F.; Paci, P.; Santoni, D.; Di Paola, L.; Giuliani, A. GIANT: A Cytoscape Plugin for Modular Networks. PLoS ONE 2014, 9, e105001. [Google Scholar] [CrossRef] [PubMed]
- Guimerà, R.; Amaral, L.A.N. Functional Cartography of Complex Metabolic Networks. Nature 2005, 433, 895–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Nardo, G.; Di Venere, A.; Zhang, C.; Nicolai, E.; Castrignanò, S.; Di Paola, L.; Gilardi, G.; Mei, G. Polymorphism on Human Aromatase Affects Protein Dynamics and Substrate Binding: Spectroscopic Evidence. Biol. Direct 2021, 16, 1–12. [Google Scholar] [CrossRef]
- Arrigo, N.; Paci, P.; Di Paola, L.; Santoni, D.; De Ruvo, M.; Giuliani, A.; Castiglione, F. Characterizing Protein Shape by a Volume Distribution Asymmetry Index. Open Bioinform. J. 2012, 6, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Di Paola, L.; Paci, P.; Santoni, D.; De Ruvo, M.; Giuliani, A. Proteins as sponges: A statistical journey along protein structure organization principles. J. Chem. Inf. Model. 2012, 52, 474–482. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy; Kluwer Academic/Plenum Publishers: New York, NY, USA, 1999. [Google Scholar] [CrossRef]
- Matsuzawa, A.; Tseng, P.H.; Vallabhapurapu, S.; Luo, J.L.; Zhang, W.; Wang, H.; Vignali, D.A.; Gallagher, E.; Karin, M. Essential cytoplasmic translocation of a cytokine receptor- assembled signaling complex. Science 2008, 321, 663–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.C.; Ye, H.; Hsia, C.; Segal, D.; Rich, R.L.; Liou, H.-C.; Myszka, D.G.; Wu, H. A Novel Mechanism of TRAF Signaling Revealed by Structural and Functional Analyses of the TRADD-TRAF2 Interaction. Cell 2000, 101, 777–787. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Minicozzi, V.; Di Venere, A.; Caccuri, A.M.; Mei, G.; Di Paola, L. One for All, All for One: The Peculiar Dynamics of TNF-Receptor-Associated Factor (TRAF2) Subunits. Symmetry 2022, 14, 720. https://doi.org/10.3390/sym14040720
Minicozzi V, Di Venere A, Caccuri AM, Mei G, Di Paola L. One for All, All for One: The Peculiar Dynamics of TNF-Receptor-Associated Factor (TRAF2) Subunits. Symmetry. 2022; 14(4):720. https://doi.org/10.3390/sym14040720
Chicago/Turabian StyleMinicozzi, Velia, Almerinda Di Venere, Anna Maria Caccuri, Giampiero Mei, and Luisa Di Paola. 2022. "One for All, All for One: The Peculiar Dynamics of TNF-Receptor-Associated Factor (TRAF2) Subunits" Symmetry 14, no. 4: 720. https://doi.org/10.3390/sym14040720
APA StyleMinicozzi, V., Di Venere, A., Caccuri, A. M., Mei, G., & Di Paola, L. (2022). One for All, All for One: The Peculiar Dynamics of TNF-Receptor-Associated Factor (TRAF2) Subunits. Symmetry, 14(4), 720. https://doi.org/10.3390/sym14040720