
Structural and Biochemical Basis of Etoposide-Resistant Mutations in Topoisomerase IIα


Abstract
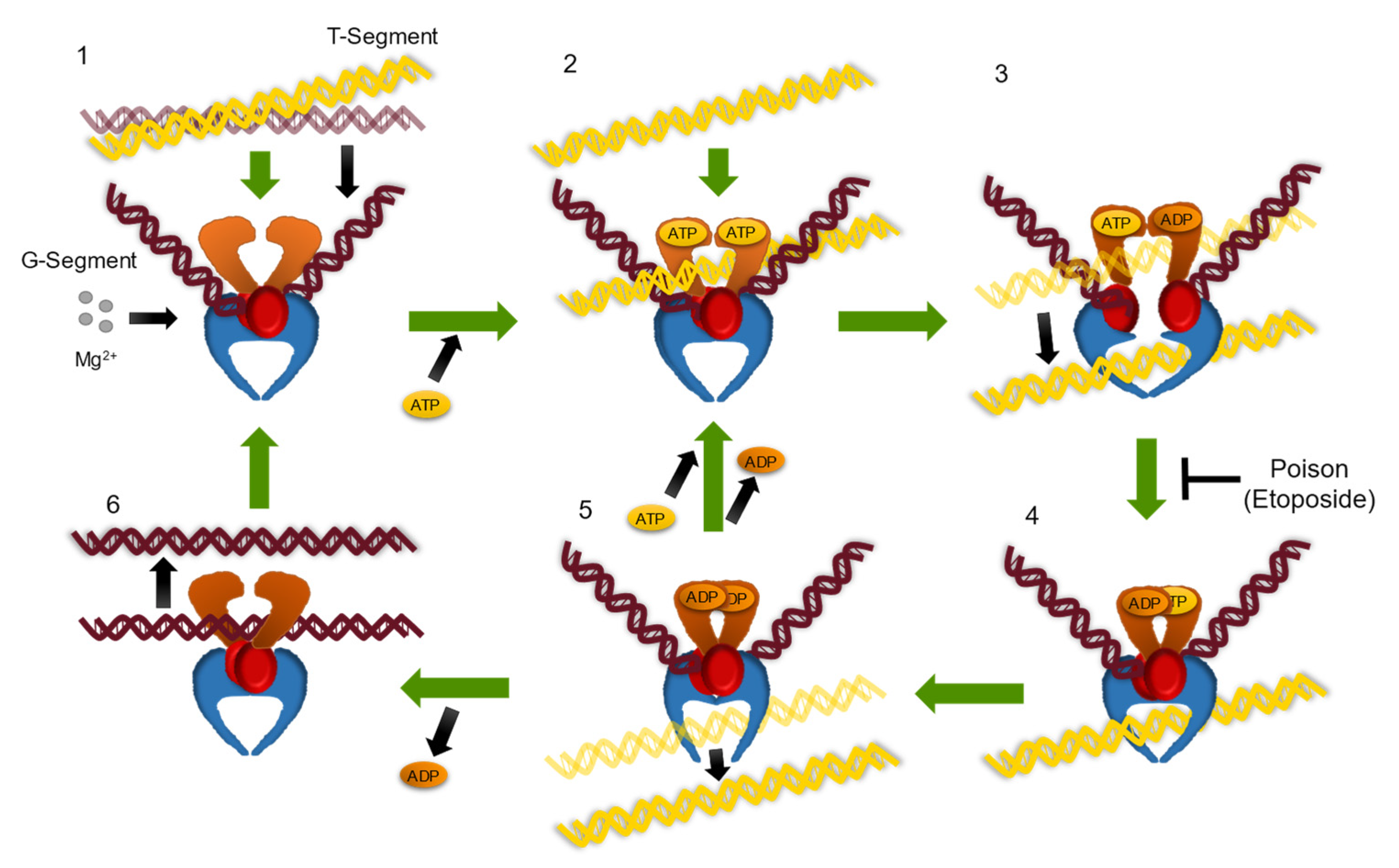
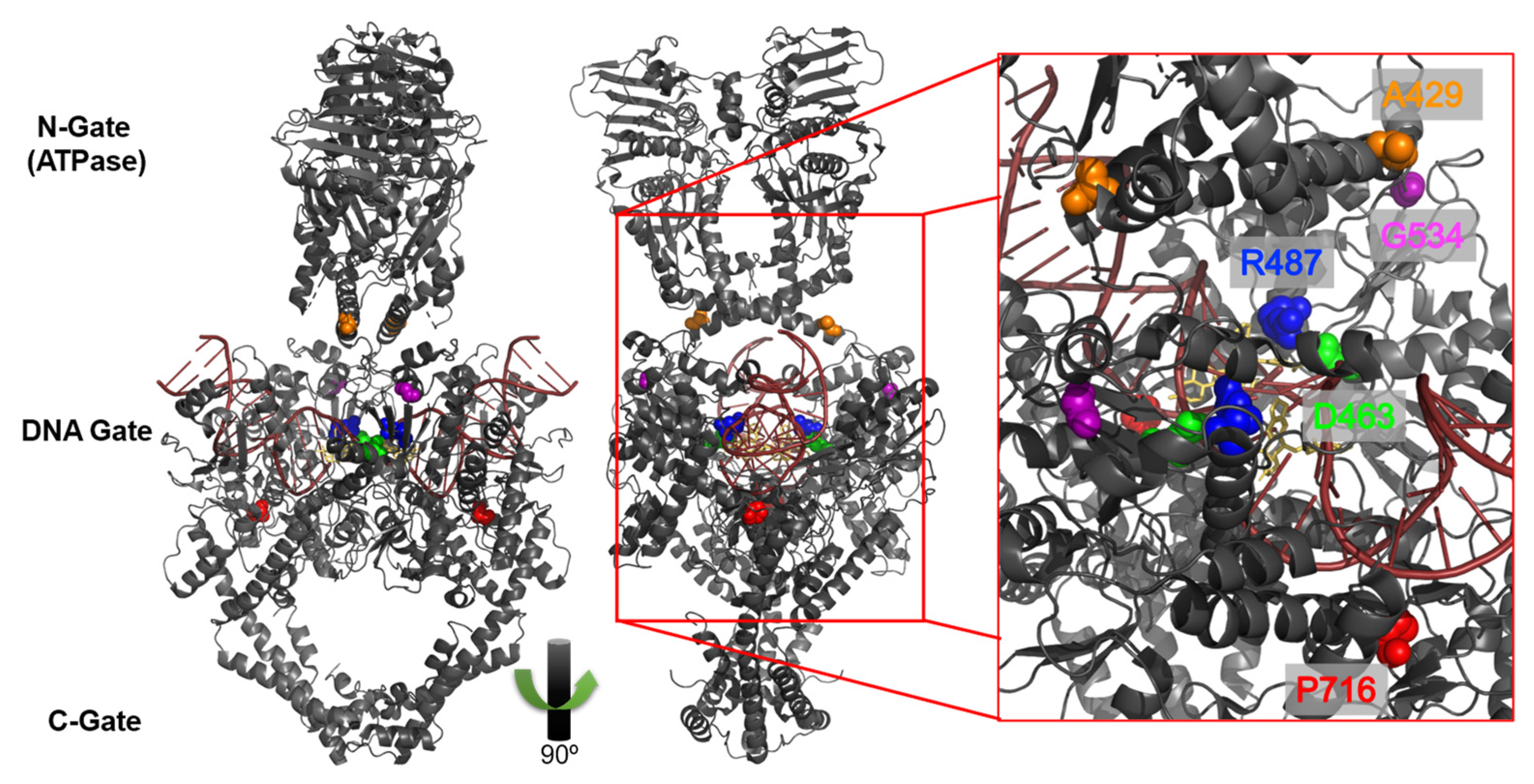
:1. Introduction
2. Materials and Methods
2.1. Enzymes and Materials
2.2. Topoisomerase II-Mediated Plasmid DNA Relaxation
2.3. Topoisomerase II-Mediated Cleavage of Plasmid DNA
3. Results
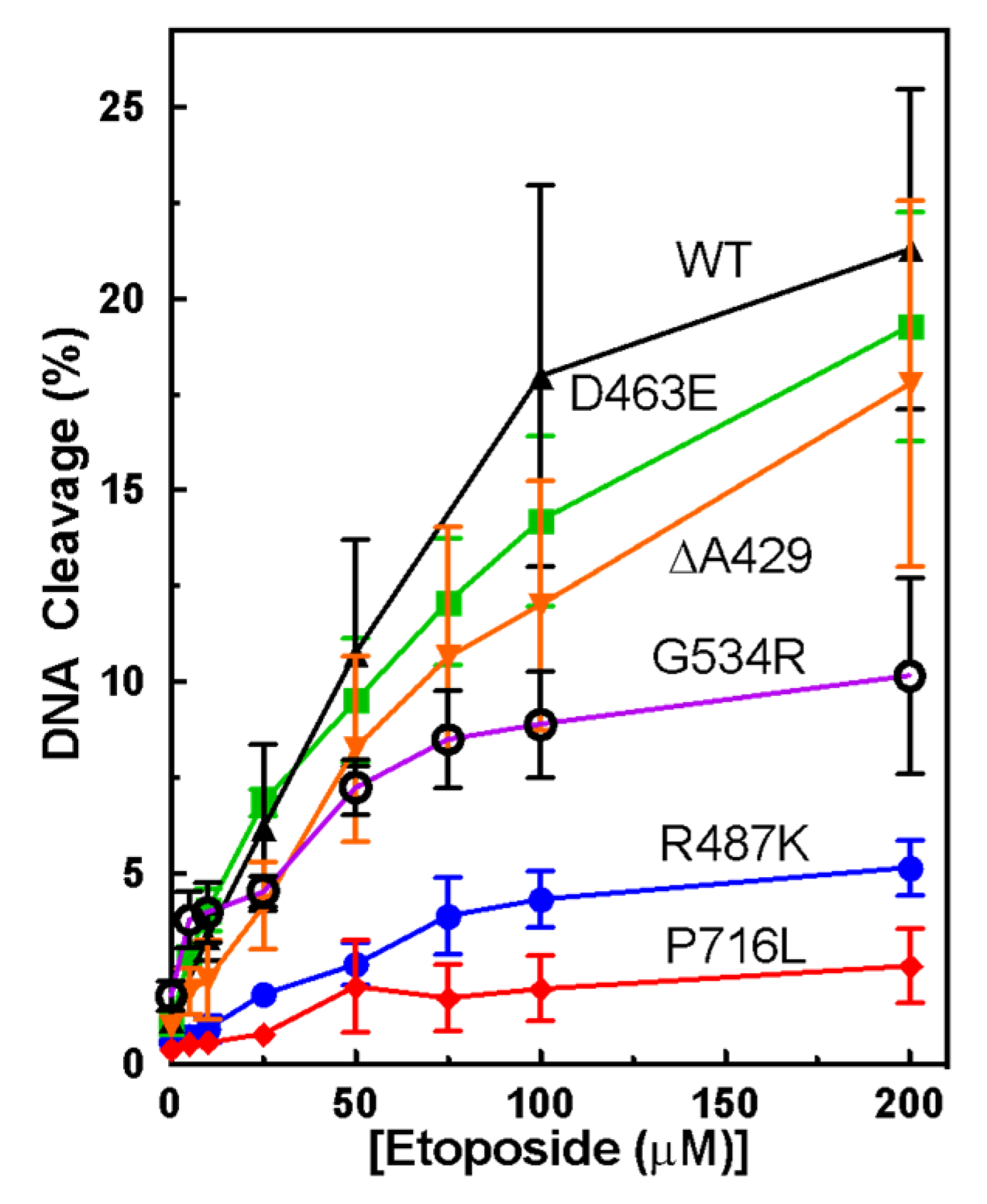
3.1. Plasmid DNA Cleavage Demonstrates Varying Levels of Etoposide-Induced Cleavage among Point Mutants
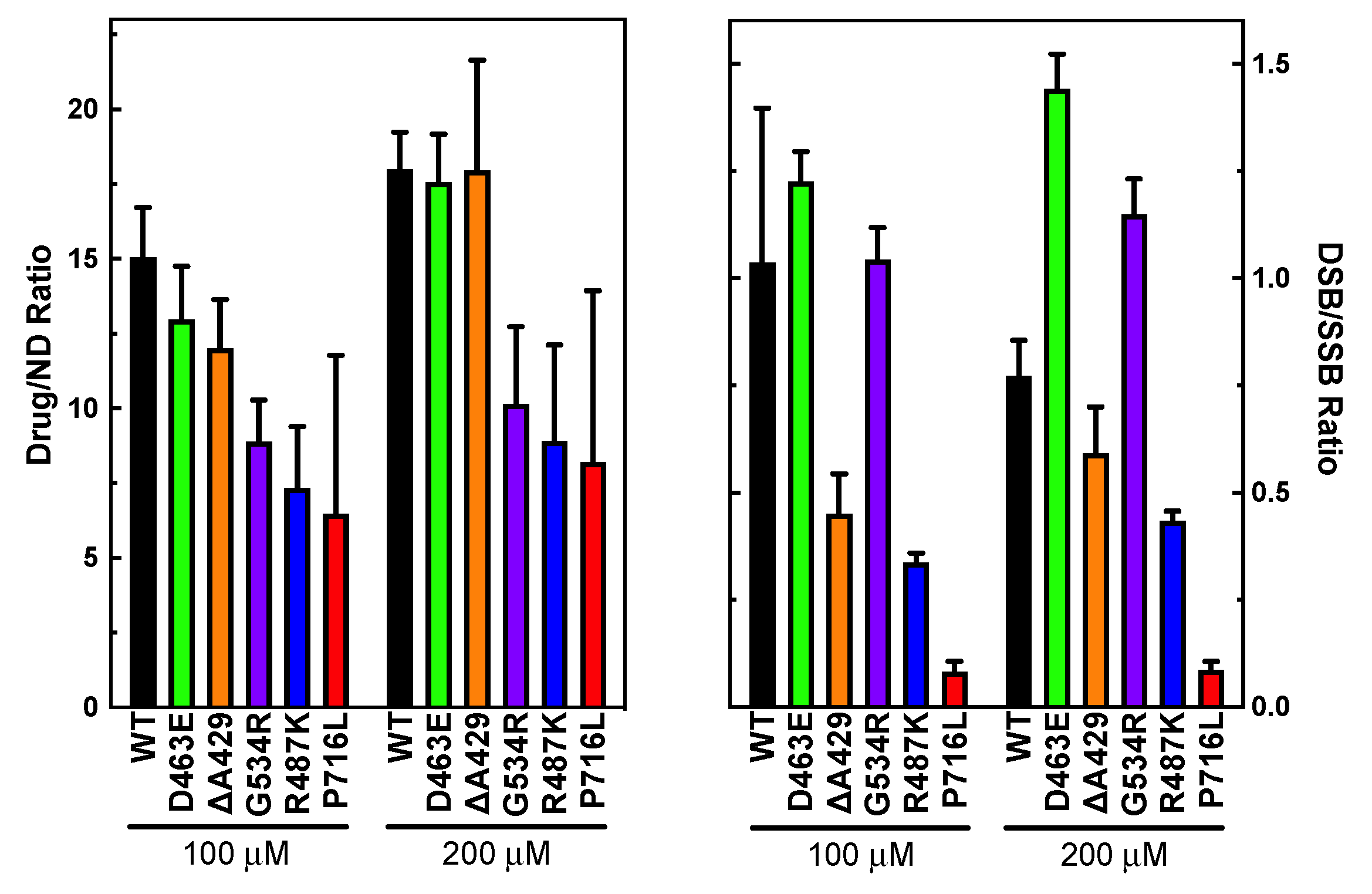
3.2. Measuring Etoposide-Induced Cleavage Enhancement
3.3. Examining the Level of Coordination during DNA Cleavage
3.4. Plasmid DNA Relaxation with Selected Mutants of TOP2A
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pommier, Y.; Sun, Y.; Huang, S.N.; Nitiss, J.L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol. 2016, 17, 703–721. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer 2009, 9, 327–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deweese, J.E.; Osheroff, N. The Use of Divalent Metal Ions by Type II Topoisomerases. Metallomics 2010, 2, 450–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wendorff, T.J.; Schmidt, B.H.; Heslop, P.; Austin, C.A.; Berger, J.M. The structure of DNA-bound human topoisomerase IIα: Conformational mechanisms for coordinating inter-subunit interactions with DNA cleavage. J. Mol. Biol. 2012, 424, 109–124. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.B.; Mercer, S.L.; Deweese, J.E. Inhibitors and Poisons of Mammalian Type II Topoisomerases. In Advances in Molecular Toxicology; Fishbein, J.C., Heilman, J., Eds.; Academic Press: Cambridge, MA, USA, 2017; Volume 11, pp. 203–240. [Google Scholar] [CrossRef]
- Campain, J.A.; Gottesman, M.M.; Pastan, I. A novel mutant topoisomerase II alpha present in VP-16-resistant human melanoma cell lines has a deletion of alanine 429. Biochemistry 1994, 33, 11327–11332. [Google Scholar] [CrossRef]
- Patel, S.; Keller, B.A.; Fisher, L.M. Mutations at arg486 and glu571 in human topoisomerase IIalpha confer resistance to amsacrine: Relevance for antitumor drug resistance in human cells. Mol. Pharmacol. 2000, 57, 784–791. [Google Scholar] [CrossRef] [Green Version]
- Leontiou, C.; Lakey, J.H.; Lightowlers, R.; Turnbull, R.M.; Austin, C.A. Mutation P732L in human DNA topoisomerase IIβ abolishes DNA cleavage in the presence of calcium and confers drug resistance. Mol. Pharmacol. 2006, 69, 130–139. [Google Scholar] [CrossRef]
- Leontiou, C.; Watters, G.P.; Gilroy, K.L.; Heslop, P.; Cowell, I.G.; Craig, K.; Lightowlers, R.N.; Lakey, J.H.; Austin, C.A. Differential selection of acridine resistance mutations in human DNA topoisomerase IIbeta is dependent on the acridine structure. Mol. Pharmacol. 2007, 71, 1006–1014. [Google Scholar] [CrossRef] [Green Version]
- Oestergaard, V.H.; Bjergbaek, L.; Skouboe, C.; Giangiacomo, L.; Knudsen, B.R.; Andersen, A.H. The transducer domain is important for clamp operation in human DNA topoisomerase IIalpha. J. Biol. Chem. 2004, 279, 1684–1691. [Google Scholar] [CrossRef] [Green Version]
- Vanden Broeck, A.; Lotz, C.; Drillien, R.; Haas, L.; Bedez, C.; Lamour, V. Structural basis for allosteric regulation of Human Topoisomerase IIα. Nat. Commun. 2021, 12, 2962. [Google Scholar] [CrossRef]
- Regal, K.M.; Mercer, S.L.; Deweese, J.E. HU-331 is a catalytic inhibitor of topoisomerase IIα. Chem. Res. Toxicol. 2014, 27, 2044–2051. [Google Scholar] [CrossRef] [PubMed]
- Fortune, J.M.; Osheroff, N. Merbarone inhibits the catalytic activity of human topoisomerase IIα by blocking DNA cleavage. J. Biol. Chem. 1998, 273, 17643–17650. [Google Scholar] [CrossRef] [Green Version]
- Bromberg, K.D.; Burgin, A.B.; Osheroff, N. A two-drug model for etoposide action against human topoisomerase IIα. J. Biol. Chem. 2003, 278, 7406–7412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deweese, J.E.; Osheroff, M.A.; Osheroff, N. DNA topology and topoisomerases: Teaching a “knotty” subject. Biochem. Mol. Biol. Educ. 2009, 37, 2–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClendon, A.K.; Dickey, J.S.; Osheroff, N. Ability of viral topoisomerase II to discern the handedness of supercoiled DNA: Bimodal recognition of DNA geometry by type II enzymes. Biochemistry 2006, 45, 11674–11680. [Google Scholar] [CrossRef] [Green Version]
- Dickey, J.S.; Osheroff, N. Impact of the C-terminal domain of topoisomerase IIα on the DNA cleavage activity of the human enzyme. Biochemistry 2005, 44, 11546–11554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClendon, A.K.; Osheroff, N. The geometry of DNA supercoils modulates topoisomerase-mediated DNA cleavage and enzyme response to anticancer drugs. Biochemistry 2006, 45, 3040–3050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClendon, A.K.; Gentry, A.C.; Dickey, J.S.; Brinch, M.; Bendsen, S.; Andersen, A.H.; Osheroff, N. Bimodal recognition of DNA geometry by human topoisomerase II alpha: Preferential relaxation of positively supercoiled DNA requires elements in the C-terminal domain. Biochemistry 2008, 47, 13169–13178. [Google Scholar] [CrossRef] [Green Version]
- Gilroy, K.L.; Austin, C.A. The impact of the C-terminal domain on the interaction of human DNA topoisomerase II alpha and beta with DNA. PLoS ONE 2011, 6, e14693. [Google Scholar] [CrossRef] [Green Version]
- Meczes, E.L.; Gilroy, K.L.; West, K.L.; Austin, C.A. The impact of the human DNA topoisomerase II C-terminal domain on activity. PLoS ONE 2008, 3, e1754. [Google Scholar] [CrossRef] [Green Version]
- Zechiedrich, E.L.; Osheroff, N. Eukaryotic topoisomerases recognize nucleic acid topology by preferentially interacting with DNA crossovers. EMBO J. 1990, 9, 4555–4562. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme |
Relaxation Activity No Drug |
Relaxation Activity +Etoposide |
Processive (P) or Distributive (D) |
|---|---|---|---|
| WT | -- | -- | P |
| D463E | = | = | P |
| ΔA429 | = | Stimulated | P |
| G534R | = | Less sensitive | P |
| R487K | Slower | Less sensitive | D |
| P716L | Slower | Stimulated | D |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gibson, E.G.; Deweese, J.E. Structural and Biochemical Basis of Etoposide-Resistant Mutations in Topoisomerase IIα. Symmetry 2022, 14, 1309. https://doi.org/10.3390/sym14071309
Gibson EG, Deweese JE. Structural and Biochemical Basis of Etoposide-Resistant Mutations in Topoisomerase IIα. Symmetry. 2022; 14(7):1309. https://doi.org/10.3390/sym14071309
Chicago/Turabian StyleGibson, Elizabeth G., and Joseph E. Deweese. 2022. "Structural and Biochemical Basis of Etoposide-Resistant Mutations in Topoisomerase IIα" Symmetry 14, no. 7: 1309. https://doi.org/10.3390/sym14071309