


Steady State Kinetics for Enzymes with Multiple Binding Sites Upstream of the Catalytic Site

 , , , ,
, , , ,
Abstract
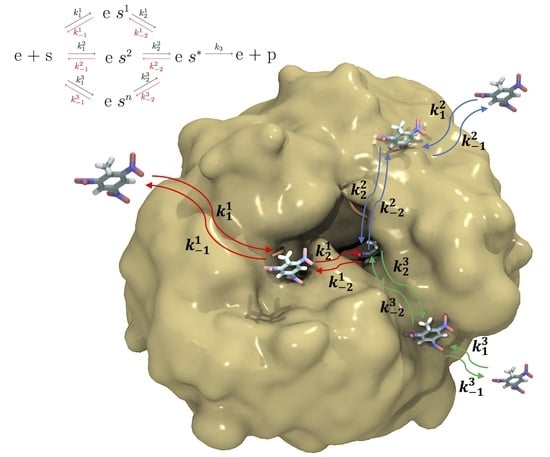
:
1. Introduction
2. Materials and Methods
3. Results
3.1. Steady-State Kinetics for an Enzyme with Several PCBSs
3.2. Numerical System Simulation with PCBS
3.3. Dependence of Catalytic Efficiency on PCBS Number
4. Discussion
5. Conclusions
6. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MM | Michaelis–Menten |
| PCBS | precatalytic binding site |
Appendix A
Appendix A.1. Model with Two PCBSs
Appendix A.2. Model with n PCBSs under Constraints for and
Appendix A.3. Algorithm Used

References
- Ma, X.; Hortelao, A.C.; Patino, T.; Sanchez, S. Enzyme catalysis to power micro/nanomachines. ACS Nano 2016, 10, 9111–9122. [Google Scholar] [CrossRef] [PubMed]
- Riziotis, I.; Ribeiro, A.J.M.; Borkakoti, N.; Thornton, J.M. Conformational Variation in Enzyme Catalysis: A Structural Study on Catalytic Residues. JMB 2022, 434, 167517. [Google Scholar]
- Osuna, S. Conformational Variation in Enzyme Catalysis: The challenge of predicting distal active site mutations in computational enzyme design. Wires Comput. Mol. Sci. 2021, 11, e1502. [Google Scholar]
- Marques, S.M.; Bednar, D.; Damborsky, J. Computational study of protein-ligand unbinding for enzyme engineering. Front. Chem. 2016, 10, 650–665. [Google Scholar] [CrossRef]
- Jagger, B.R.; Kochanek, S.E.; Haldar, S.A.; Rommie, E.; Mulholland, A.J. Multiscale simulation approaches to modeling drug–protein binding. Curr. Opin. Struct. Biol. 2020, 61, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Nerukh, D.; Okimoto, N.; Suenaga, A.; Taiji, M. Ligand diffusion on protein surface observed in molecular dynamics simulation. Curr. Opin. Struct. Biol. 2012, 3, 3476–3479. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, V.; Fischer, M.; Zhu, Y.; Pleiss, J. Modelling of substrate access and substrate binding to cephalosporin acylases. J. Phys. Chem. Lett. 2019, 9, 12402. [Google Scholar] [CrossRef] [PubMed]
- Buch, I.; Giorgino, T.; De Fabritiis, G. Complete reconstruction of an enzyme-inhibitor binding process by molecular dynamics simulations. Proc. Natl. Acad. Sci. USA 2011, 108, 10184–10189. [Google Scholar] [CrossRef]
- Lorsch, J.R. Chapter One—Practical Steady-State Enzyme Kinetics. In Laboratory Methods in Enzymology: Protein Part A; Lorsch, J., Ed.; Academic Press: Baltimore, MD, USA, 2007; pp. 3–15. [Google Scholar]
- Voet, D.; Voet, J. Chapter fourteen—Rates of Enzymatic Reactions. In Biochemistry; Lorsch, J., Ed.; Academic Press: Baltimore, MD, USA, 2007; pp. 3–15. [Google Scholar]
- Goutelle, S.; Maurin, M.; Rougier, F.; Barbaut, X.; Bourguignon, L.; Ducher, M.; Maire, P. The Hill equation: A review of its capabilities in pharmacological modelling. Fundam. Clin. Pharmacol. 2008, 22, 633–648. [Google Scholar] [CrossRef]
- Burke, M.A. Maini’s many contributions to mathematical enzyme kinetics: A review. J. Theor. Biol. 2019, 481, 24–27. [Google Scholar] [CrossRef]
- Baez, J.C.; Pollard, B.S. A compositional framework for reaction networks. Rev. Math. Phys. 2017, 29, 1750028. [Google Scholar] [CrossRef]
- Feinberg, M. Nondegeneracy of Semi-open Reaction Networks: An Enzyme Example and the Wnt Pathway. In Foundations of Chemical Reaction Network Theory; Springer: Cham, Switzerland, 2019; pp. 3–15. [Google Scholar]
- Custom Reactions. Available online: https://github.com/willfinnigan/kinetics/tree/master/docs (accessed on 14 August 2023).
- pCBS Kinetics. Available online: https://github.com/HumanOsv/pCBS_Kinetics (accessed on 14 August 2023).
- Finnigan, W.; Cutlan, R.; Snajdrova, R.; Adams, J.P.; Littlechild, J.A.; Harmer, N.J. Engineering a seven enzyme biotransformation using mathematical modelling and characterized enzyme parts. ChemCatChem 2019, 11, 3474–3489. [Google Scholar] [CrossRef]
- Wilding, M.; Hong, N.; Spence, M.; Buckle, A.M.; Jackson, C.J. Protein engineering: The potential of remote mutations. Biochem. Soc. Trans 2019, 47, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Jacquier, H.; Birgy, A.; Le Nagard, H.; Mechulam, Y.; Schmitt, E.; Glodt, J.; Bercot, B.; Petit, E.; Poulain, J.; Barnaud, G. Capturing the mutational landscape of the beta-lactamase TEM-1. Proc. Natl. Acad. Sci. USA 2013, 47, 13067–13072. [Google Scholar] [CrossRef] [PubMed]
- Mendonça, L.M.F.; Marana, S.R. Single mutations outside the active site affect the substrate specificity in a β-glycosidase. Biochim. Biophys. Acta Proteins Proteom. 2011, 1814, 13067–13072. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Kinetic Parameters | Michaelis–Menten * | |||||
|---|---|---|---|---|---|---|
| Catalytic efficiency |
| Kinetic Parameters | Michaelis–Menten * | |||||
|---|---|---|---|---|---|---|
| Catalytic efficiency |
| Kinetic Parameters | Michaelis–Menten * | |||||
|---|---|---|---|---|---|---|
| Catalytic efficiency |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osorio, M.I.; Petrache, M.; Salinas, D.G.; Valenzuela-Ibaceta, F.; González-Nilo, F.; Tiznado, W.; Pérez-Donoso, J.M.; Bravo, D.; Yáñez, O. Steady State Kinetics for Enzymes with Multiple Binding Sites Upstream of the Catalytic Site. Symmetry 2023, 15, 2176. https://doi.org/10.3390/sym15122176
Osorio MI, Petrache M, Salinas DG, Valenzuela-Ibaceta F, González-Nilo F, Tiznado W, Pérez-Donoso JM, Bravo D, Yáñez O. Steady State Kinetics for Enzymes with Multiple Binding Sites Upstream of the Catalytic Site. Symmetry. 2023; 15(12):2176. https://doi.org/10.3390/sym15122176
Chicago/Turabian StyleOsorio, Manuel I., Mircea Petrache, Dino G. Salinas, Felipe Valenzuela-Ibaceta, Fernando González-Nilo, William Tiznado, José M. Pérez-Donoso, Denisse Bravo, and Osvaldo Yáñez. 2023. "Steady State Kinetics for Enzymes with Multiple Binding Sites Upstream of the Catalytic Site" Symmetry 15, no. 12: 2176. https://doi.org/10.3390/sym15122176
APA StyleOsorio, M. I., Petrache, M., Salinas, D. G., Valenzuela-Ibaceta, F., González-Nilo, F., Tiznado, W., Pérez-Donoso, J. M., Bravo, D., & Yáñez, O. (2023). Steady State Kinetics for Enzymes with Multiple Binding Sites Upstream of the Catalytic Site. Symmetry, 15(12), 2176. https://doi.org/10.3390/sym15122176