

New High-Pressure Structures of Transition Metal Carbonates with O3C–CO3 Orthooxalate Groups
 , , and
, , and
Abstract
:1. Introduction
2. Computational Methods
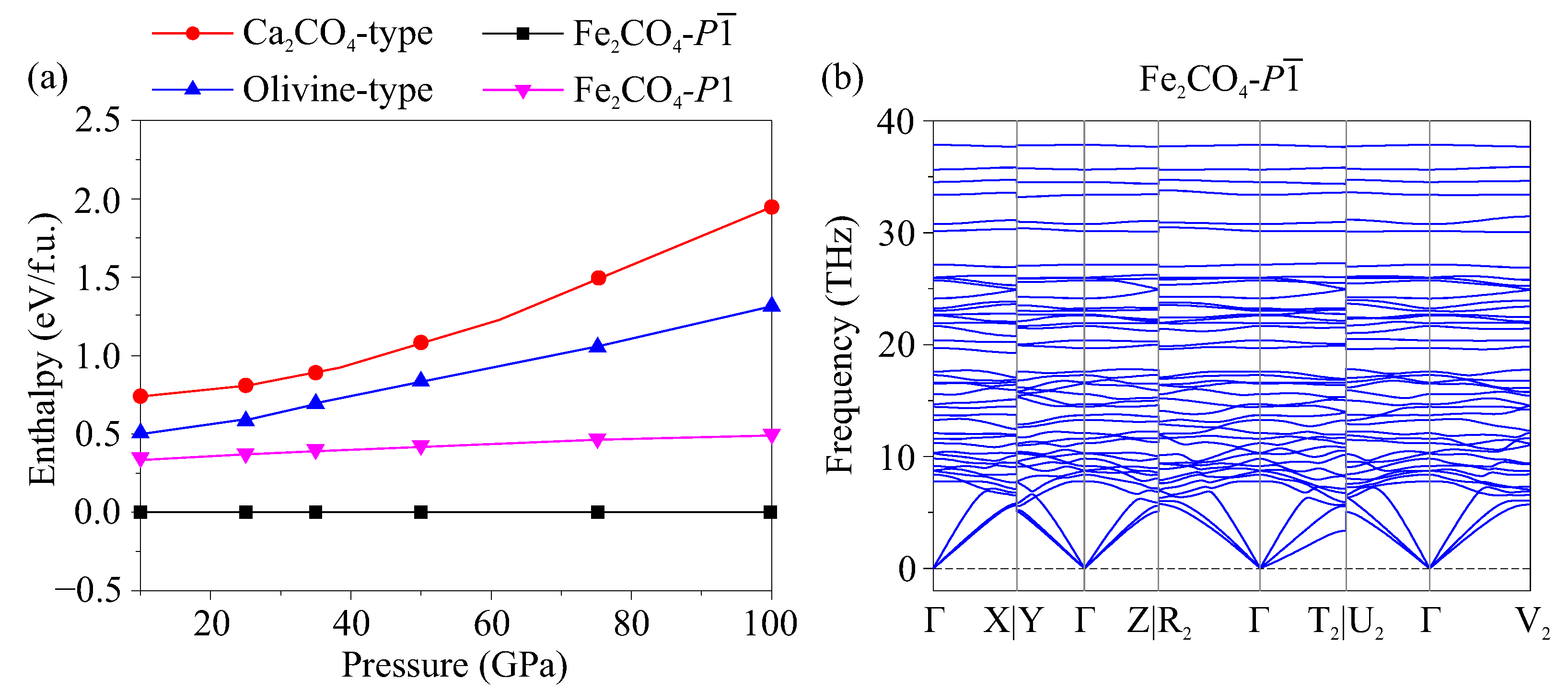
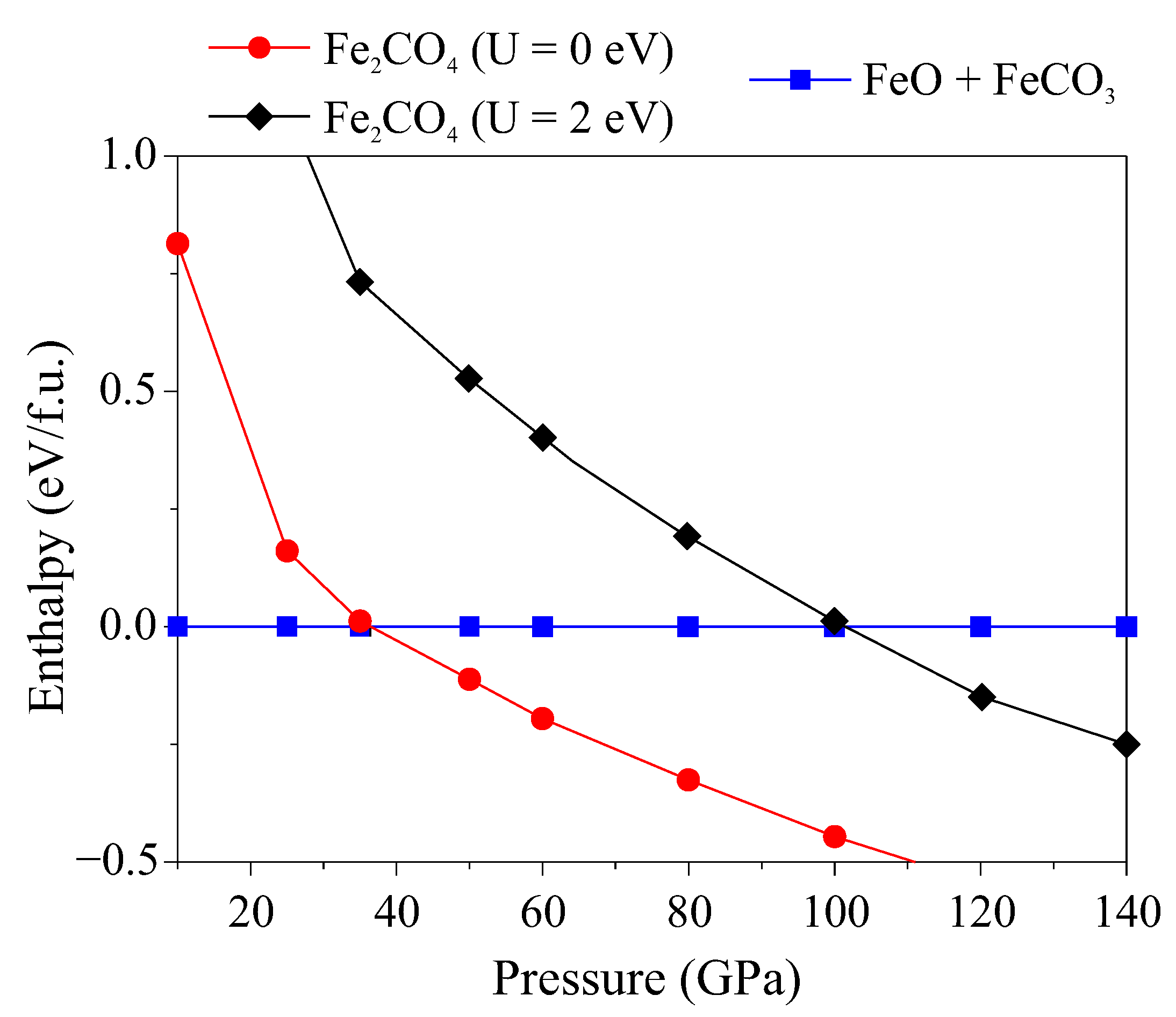
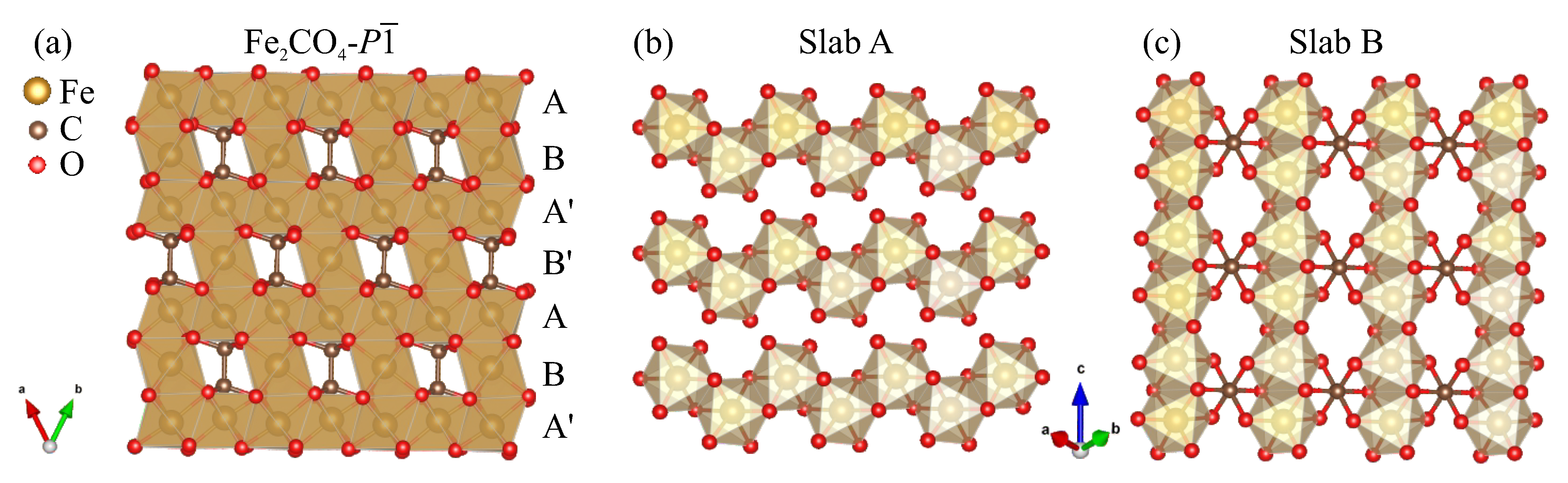
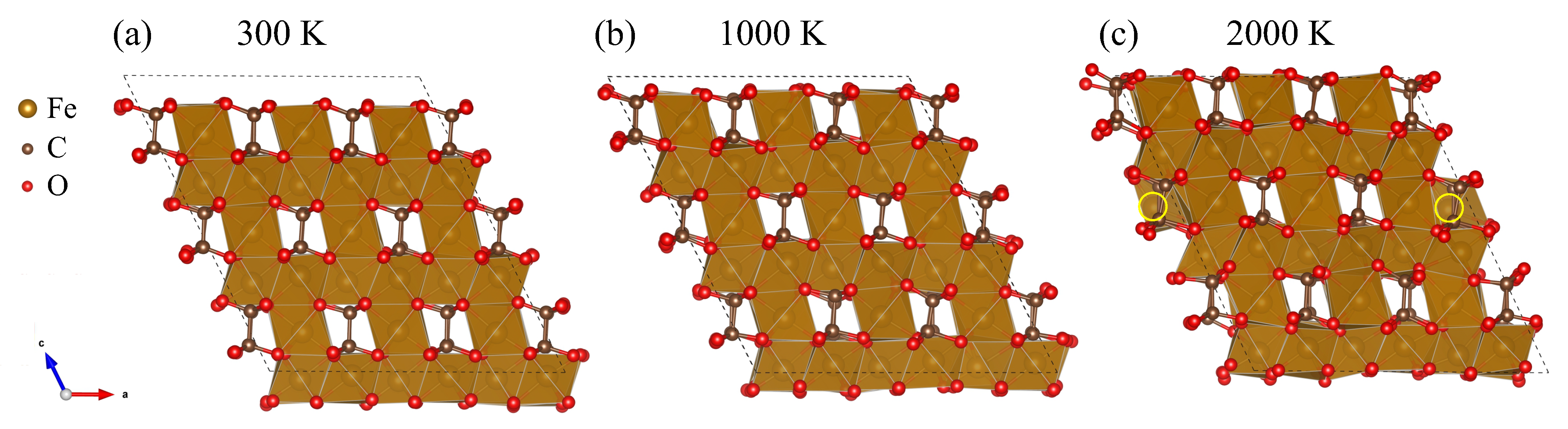
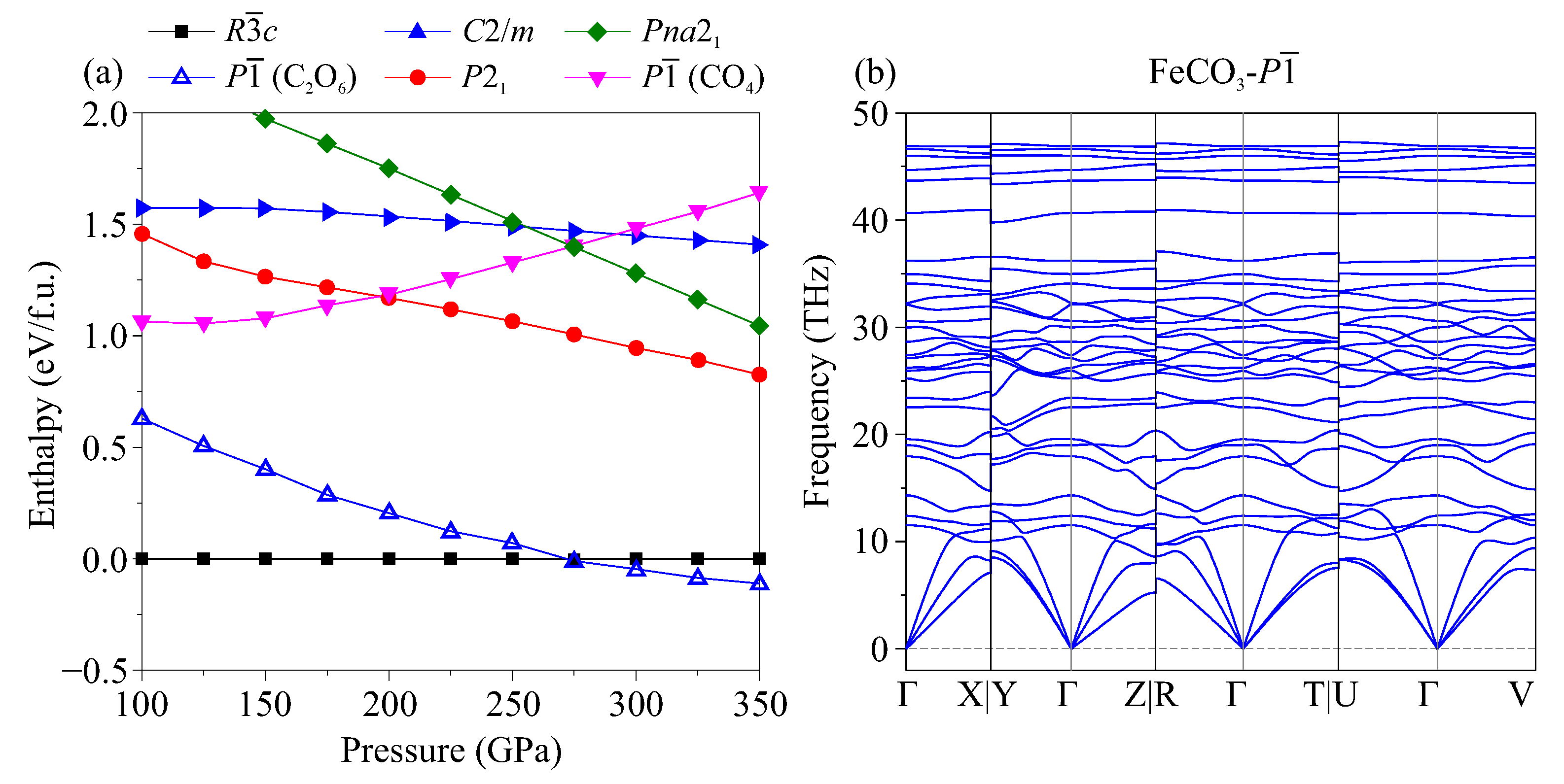
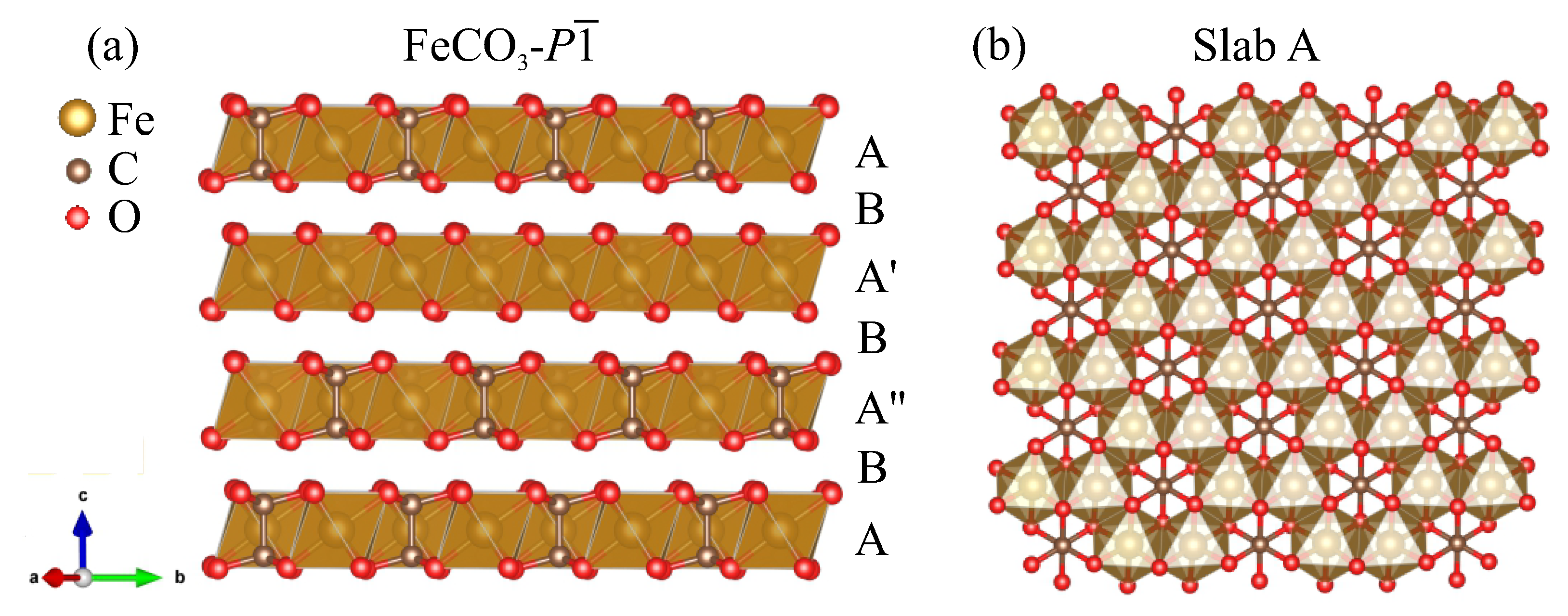
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yao, X.; Xie, C.; Dong, X.; Oganov, A.R.; Zeng, Q. Novel high-pressure calcium carbonates. Phys. Rev. B 2018, 98, 014108. [Google Scholar] [CrossRef]
- Sagatova, D.; Shatskiy, A.; Sagatov, N.; Gavryushkin, P.N.; Litasov, K.D. Calcium orthocarbonate, Ca2CO4-Pnma: A potential host for subducting carbon in the transition zone and lower mantle. Lithos 2020, 370, 105637. [Google Scholar] [CrossRef]
- Gavryushkin, P.N.; Sagatova, D.N.; Sagatov, N.; Litasov, K.D. Formation of Mg-Orthocarbonate through the Reaction MgCO3+ MgO= Mg2CO4 at Earth’s Lower Mantle P–T Conditions. Cryst. Growth Des. 2021, 21, 2986–2992. [Google Scholar] [CrossRef]
- Gavryushkin, P.N.; Sagatova, D.N.; Sagatov, N.; Litasov, K.D. Orthocarbonates of Ca, Sr, and Ba—The Appearance of sp3-Hybridized Carbon at a Low Pressure of 5 GPa and Dynamic Stability at Ambient Pressure. ACS Earth Space Chem. 2021, 5, 1948–1957. [Google Scholar] [CrossRef]
- Banaev, M.V.; Sagatov, N.E.; Sagatova, D.N.; Gavryushkin, P.N. High-Pressure Crystal Structures of Pb2CO4 and PbC2O5 with Tetrahedral [CO4] and Pyrocarbonate [C2O5] atomic groups. ChemistrySelect 2022, 7, e202201940. [Google Scholar] [CrossRef]
- Gavryushkin, P.; Martirosyan, N.; Rashchenko, S.; Sagatova, D.; Sagatov, N.; Semerikova, A.; Fedotenko, T.; Litasov, K. First Experimental Synthesis of Mg Orthocarbonate by the MgCO3 + MgO= Mg2CO4 Reaction at Pressures of the Earth’s Lower Mantle. JETP Lett. 2022, 116, 477–484. [Google Scholar] [CrossRef]
- Binck, J.; Laniel, D.; Bayarjargal, L.; Khandarkhaeva, S.; Fedotenko, T.; Aslandukov, A.; Glazyrin, K.; Milman, V.; Chariton, S.; Prakapenka, V.B.; et al. Synthesis of calcium orthocarbonate, Ca2CO4-Pnma at P-T conditions of Earth’s transition zone and lower mantle. Am. Mineral. 2022, 107, 336–342. [Google Scholar] [CrossRef]
- König, J.; Spahr, D.; Bayarjargal, L.; Gavryushkin, P.N.; Sagatova, D.; Sagatov, N.; Milman, V.; Liermann, H.P.; Winkler, B. Novel Calcium sp3 Carbonate CaC2O5-I4d May Be a Carbon Host in Earth’s Lower Mantle. ACS Earth Space Chem. 2022, 6, 73–80. [Google Scholar] [CrossRef]
- Laniel, D.; Binck, J.; Winkler, B.; Vogel, S.; Fedotenko, T.; Chariton, S.; Prakapenka, V.; Milman, V.; Schnick, W.; Dubrovinsky, L.; et al. Synthesis, crystal structure and structure–property relations of strontium orthocarbonate, Sr2CO4. Acta Crystallogr. Sect. B 2021, 77, 131–137. [Google Scholar] [CrossRef]
- Spahr, D.; König, J.; Bayarjargal, L.; Gavryushkin, P.N.; Milman, V.; Liermann, H.P.; Winkler, B. Sr3[CO4]O Antiperovskite with Tetrahedrally Coordinated sp3-Hybridized Carbon and OSr6 Octahedra. Inorg. Chem. 2021, 60, 14504–14508. [Google Scholar] [CrossRef]
- Spahr, D.; König, J.; Bayarjargal, L.; Milman, V.; Perlov, A.; Liermann, H.P.; Winkler, B. Sr[C2O5] is an Inorganic Pyrocarbonate Salt with [C2O5]2– Complex Anions. J. Am. Chem. Soc. 2022, 144, 2899–2904. [Google Scholar] [CrossRef] [PubMed]
- Spahr, D.; König, J.; Bayarjargal, L.; Luchitskaia, R.; Milman, V.; Perlov, A.; Liermann, H.P.; Winkler, B. Synthesis and Structure of Pb[C2O5]: An Inorganic Pyrocarbonate Salt. Inorg. Chem. 2022, 61, 9855–9859. [Google Scholar] [CrossRef] [PubMed]
- Sagatova, D.N.; Gavryushkin, P.N.; Sagatov, N.E.; Banaev, M.V. High-pressure transformations of CaC2O5—A full structural trend from double [CO3] triangles through the isolated group of [CO4] tetrahedra to framework and layered structures. Phys. Chem. Chem. Phys. 2022, 24, 23578–23586. [Google Scholar] [CrossRef] [PubMed]
- Oganov, A.R.; Glass, C.W.; Ono, S. High-pressure phases of CaCO3: Crystal structure prediction and experiment. Earth Planet. Sci. Lett. 2006, 241, 95–103. [Google Scholar] [CrossRef]
- Pickard, C.J.; Needs, R.J. Structures and stability of calcium and magnesium carbonates at mantle pressures. Phys. Rev. B 2015, 91, 104101. [Google Scholar] [CrossRef]
- Smith, D.; Lawler, K.V.; Martinez-Canales, M.; Daykin, A.W.; Fussell, Z.; Smith, G.A.; Childs, C.; Smith, J.S.; Pickard, C.J.; Salamat, A. Postaragonite phases of CaCO3 at lower mantle pressures. Phys. Rev. Mater. 2018, 2, 013605. [Google Scholar] [CrossRef]
- Binck, J.; Bayarjargal, L.; Lobanov, S.S.; Morgenroth, W.; Luchitskaia, R.; Pickard, C.J.; Milman, V.; Refson, K.; Jochym, D.B.; Byrne, P.; et al. Phase stabilities of MgCO3 and MgCO3-II studied by Raman spectroscopy, x-ray diffraction, and density functional theory calculations. Phys. Rev. Mater. 2020, 4, 055001. [Google Scholar] [CrossRef]
- Cerantola, V.; Bykova, E.; Kupenko, I.; Merlini, M.; Ismailova, L.; McCammon, C.; Bykov, M.; Chumakov, A.I.; Petitgirard, S.; Kantor, I.; et al. Stability of iron-bearing carbonates in the deep Earth’s interior. Nat. Commun. 2017, 8, 1–9. [Google Scholar] [CrossRef]
- Nagai, T.; Ishido, T.; Seto, Y.; Nishio-Hamane, D.; Sata, N.; Fujino, K. Pressure-induced spin transition in FeCO3-siderite studied by X-ray diffraction measurements. J. Phys. Conf. Ser. 2010, 215, 012002. [Google Scholar] [CrossRef]
- Zhao, C.; Xu, L.; Gui, W.; Liu, J. Phase Stability and Vibrational Properties of Iron-Bearing Carbonates at High Pressure. Minerals 2020, 10, 1142. [Google Scholar] [CrossRef]
- Oganov, A.R.; Glass, C.W. Crystal structure prediction using ab initio evolutionary techniques: Principles and applications. J. Chem. Phys. 2006, 124, 244704. [Google Scholar] [CrossRef] [PubMed]
- Oganov, A.R.; Lyakhov, A.O.; Valle, M. How Evolutionary Crystal Structure Prediction Works–and Why. Accounts Chem. Res. 2011, 44, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Lyakhov, A.O.; Oganov, A.R.; Stokes, H.T.; Zhu, Q. New developments in evolutionary structure prediction algorithm USPEX. Comput. Phys. Commun. 2013, 184, 1172–1182. [Google Scholar] [CrossRef]
- Bushlanov, P.V.; Blatov, V.A.; Oganov, A.R. Topology-based crystal structure generator. Comput. Phys. Commun. 2019, 236, 1–7. [Google Scholar] [CrossRef]
- Pickard, C.J.; Needs, R.J. High-Pressure Phases of Silane. Phys. Rev. Lett. 2006, 97, 045504. [Google Scholar] [CrossRef] [PubMed]
- Pickard, C.J.; Needs, R.J. Ab initio random structure searching. J. Phys. Condens. Matter 2011, 23, 053201. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Dudarev, S.L.; Botton, G.A.; Savrasov, S.Y.; Humphreys, C.J.; Sutton, A.P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 1998, 57, 1505–1509. [Google Scholar] [CrossRef]
- Li, Z.; Stackhouse, S. Iron-rich carbonates stabilized by magnetic entropy at lower mantle conditions. Earth Planet. Sci. Lett. 2020, 531, 115959. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA: A three-dimensional visualization system for electronic and structural analysis. J. Appl. Crystallogr. 2008, 41, 653–658. [Google Scholar] [CrossRef]
- Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO–the Open Visualization Tool. Model. Simul. Mater. Sci. Eng. 2009, 18, 015012. [Google Scholar] [CrossRef]
- Aped, P.; Fuchs, B.; Goldberg, I.; Senderowitz, H.; Tartakovsky, E.; Weinman, S. Structure and conformation of heterocycles. 21. Probing the anomeric effect in orthoesters. Structure, conformation, and dynamic behavior of a unique orthooxalate: 2, 5, 7, 10, 11, 14-hexaoxa [4.4. 4] propellane. J. Am. Chem. Soc. 1992, 114, 5585–5590. [Google Scholar] [CrossRef]
- Junk, P.C. Supramolecular interactions in the X-ray crystal structure of potassium tris (oxalato) ferrate (III) trihydrate. J. Coord. Chem. 2005, 58, 355–361. [Google Scholar] [CrossRef]
- Belonoshko, A.B.; Lukinov, T.; Fu, J.; Zhao, J.; Davis, S.; Simak, S.I. Stabilization of body-centred cubic iron under inner-core conditions. Nat. Geosci. 2017, 10, 312–316. [Google Scholar] [CrossRef]
- Ishizawa, N. Calcite V: A hundred-year-old mystery has been solved. Powder Diffr. 2014, 29, S19–S23. [Google Scholar] [CrossRef]
- Gavryushkin, P.N.; Martirosyan, N.S.; Inerbaev, T.M.; Popov, Z.I.; Rashchenko, S.V.; Likhacheva, A.Y.; Lobanov, S.S.; Goncharov, A.F.; Prakapenka, V.B.; Litasov, K.D. Aragonite-II and CaCO3-VII: New High-Pressure, High-Temperature Polymorphs of CaCO3. Cryst. Growth Des. 2017, 17, 6291–6296. [Google Scholar] [CrossRef]
- Solomatova, N.V.; Caracas, R.; Manning, C.E. Carbon sequestration during core formation implied by complex carbon polymerization. Nat. Commun. 2019, 10, 789. [Google Scholar] [CrossRef] [Green Version]
- Kuang, H.; Tse, J.S. High-Temperature, High-Pressure Reactions of H2 with CaCO3 Melts. Phys. Status Solidi B 2022, 259, 2100644. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phase (#s.g.) | P (GPa) | Lattice Parameters (Å, deg) | Atom | Coordinates | ||||
|---|---|---|---|---|---|---|---|---|
| x | y | z | ||||||
| FeCO- | 50 | Fe | 0.7775 | 0.3775 | 0.0286 | |||
| = 103.63 | = 115.32 | = 90.81 | Fe | 0.4953 | 0.2401 | 0.4757 | ||
| C | −0.0905 | −0.0435 | 0.3287 | |||||
| O | 0.2903 | 0.2940 | 0.7483 | |||||
| O | 0.2825 | 0.8464 | 0.7596 | |||||
| O | 0.2239 | 0.4483 | 0.2571 | |||||
| O | 0.8404 | 0.0695 | 0.7651 | |||||
| FeCO- | 250 | Fe | 0.6729 | 0.8428 | 0.4959 | |||
| = 91.75 | = 116.73 | = 118.79 | C | 0.8665 | 0.4328 | 0.2999 | ||
| O | 0.5147 | 0.6023 | 0.7874 | |||||
| O | 0.8863 | 0.2857 | 0.7919 | |||||
| O | 0.1883 | −0.0950 | 0.8004 | |||||
| FeCO | FeCO | ||
|---|---|---|---|
| Atom | Bader Charge () | Atom | Bader Charge () |
| Fe | +1.195 | Fe1 | +1.282 |
| Fe2 | +1.215 | ||
| C | +2.228 | C | +1.609 |
| O | −1.141 | O1 | −1.045 |
| O2 | −1.078 | ||
| O3 | −0.924 | ||
| O4 | −1.059 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sagatov, N.E.; Sagatova, D.N.; Gavryushkin, P.N.; Litasov, K.D. New High-Pressure Structures of Transition Metal Carbonates with O3C–CO3 Orthooxalate Groups. Symmetry 2023, 15, 421. https://doi.org/10.3390/sym15020421
Sagatov NE, Sagatova DN, Gavryushkin PN, Litasov KD. New High-Pressure Structures of Transition Metal Carbonates with O3C–CO3 Orthooxalate Groups. Symmetry. 2023; 15(2):421. https://doi.org/10.3390/sym15020421
Chicago/Turabian StyleSagatov, Nursultan E., Dinara N. Sagatova, Pavel N. Gavryushkin, and Konstantin D. Litasov. 2023. "New High-Pressure Structures of Transition Metal Carbonates with O3C–CO3 Orthooxalate Groups" Symmetry 15, no. 2: 421. https://doi.org/10.3390/sym15020421