

Preference of C2v Symmetry in Low-Spin Hexacarbonyls of Rare-Earth and f Elements

Abstract
:1. Introduction
2. Computational and Experimental Details
2.1. Computational Details
2.2. Matrix Isolation Spectroscopy
3. Results and Discussion
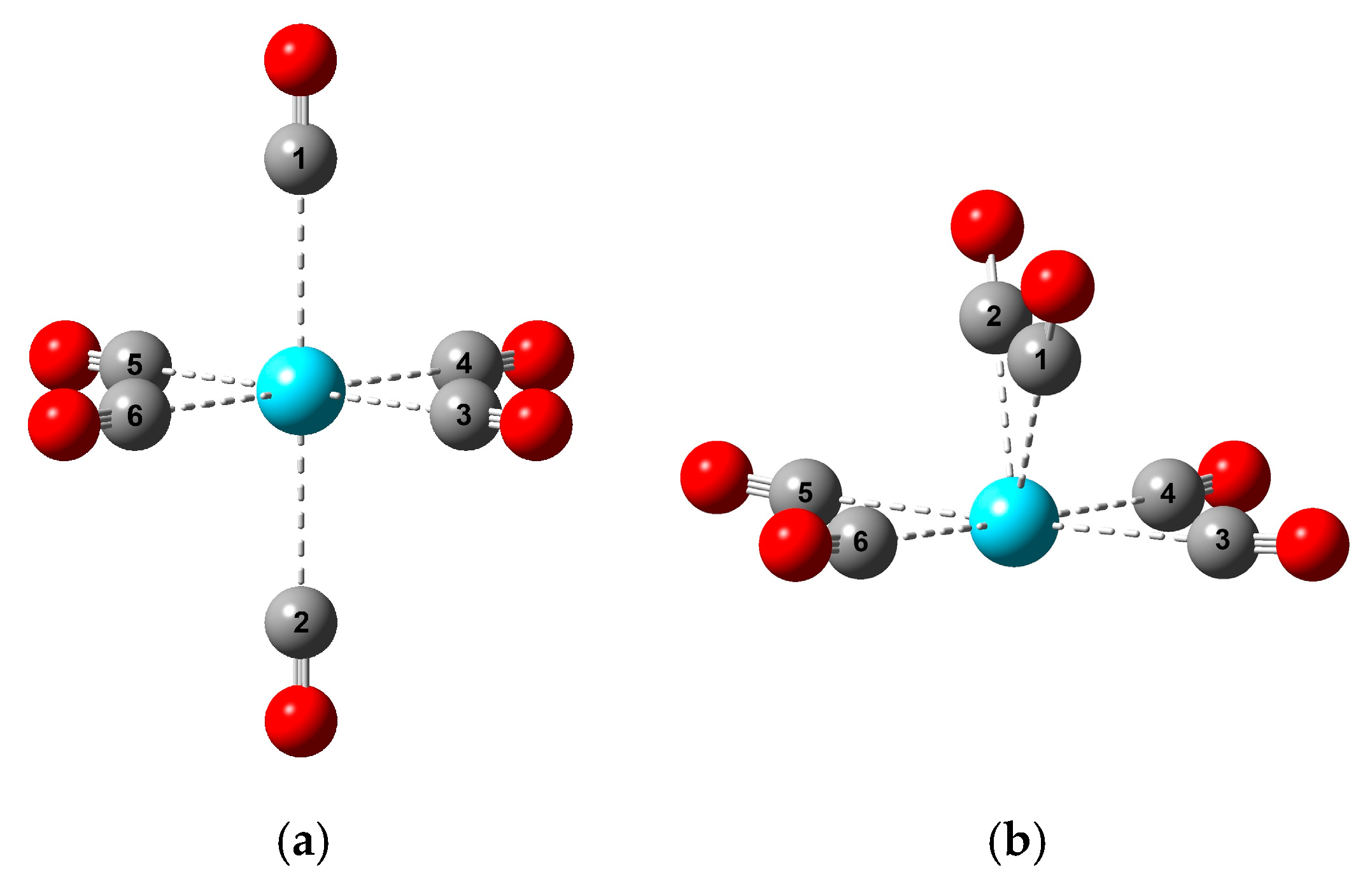
3.1. Characteristic Structures
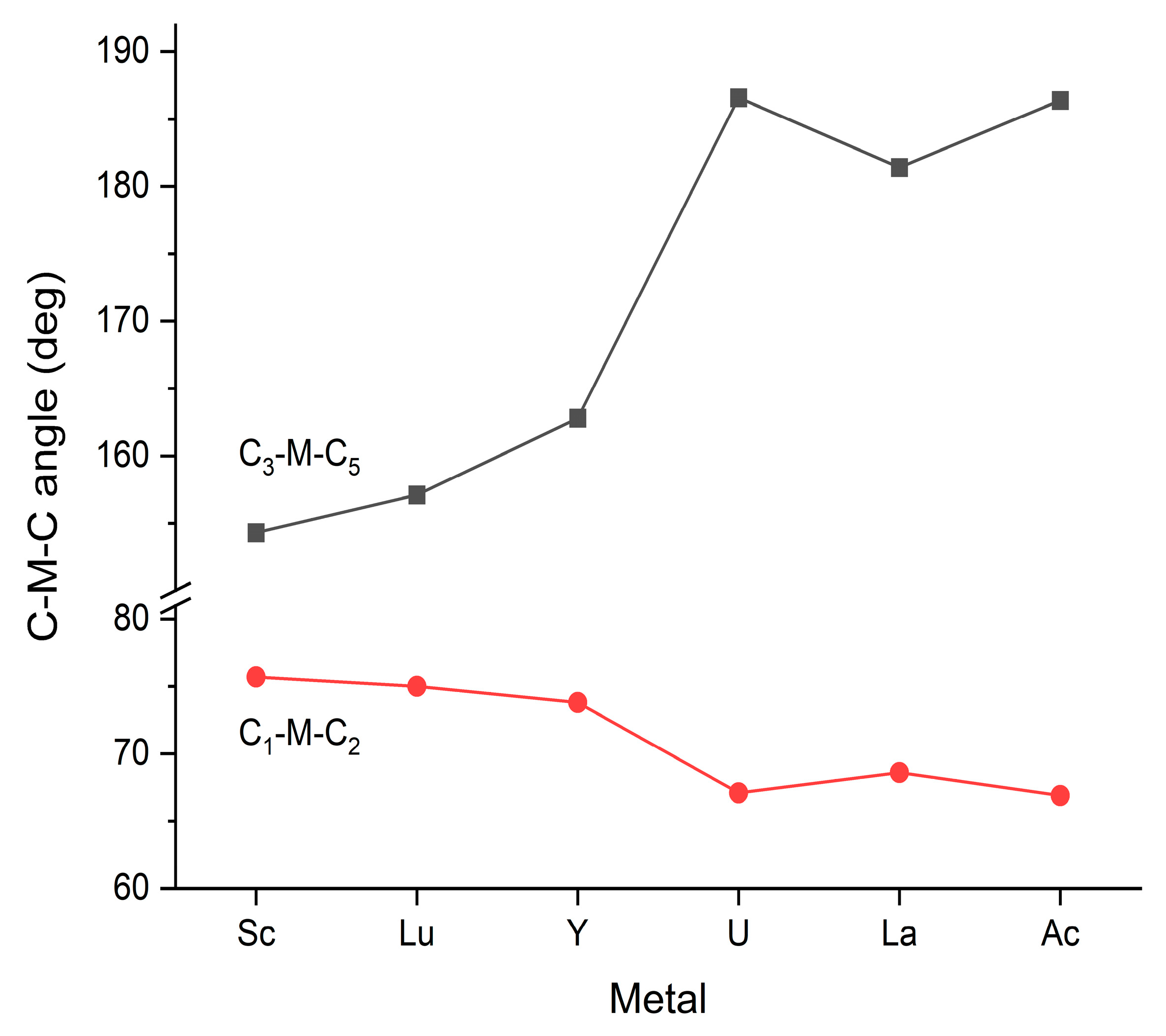
3.2. Steric Conditions
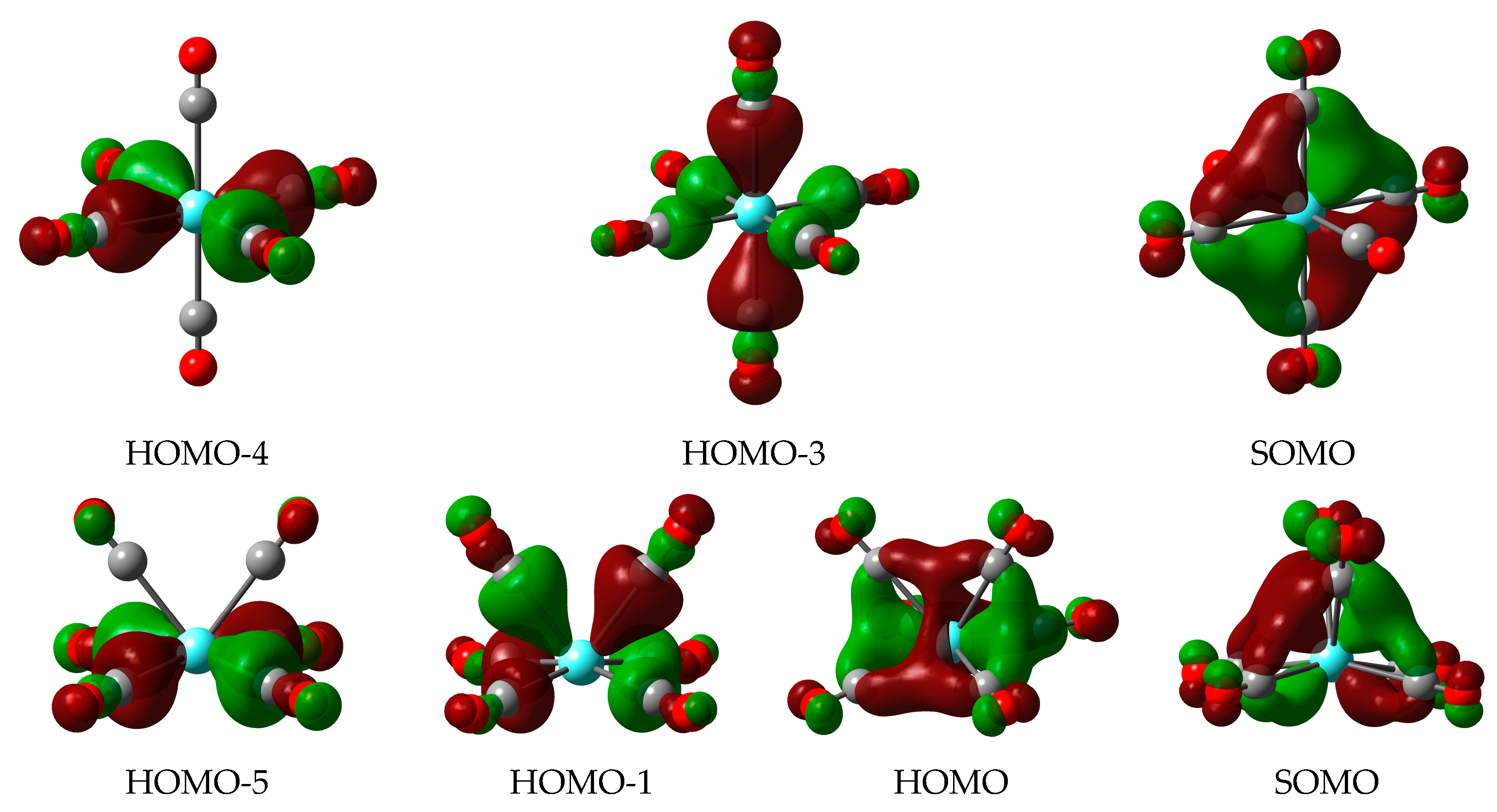
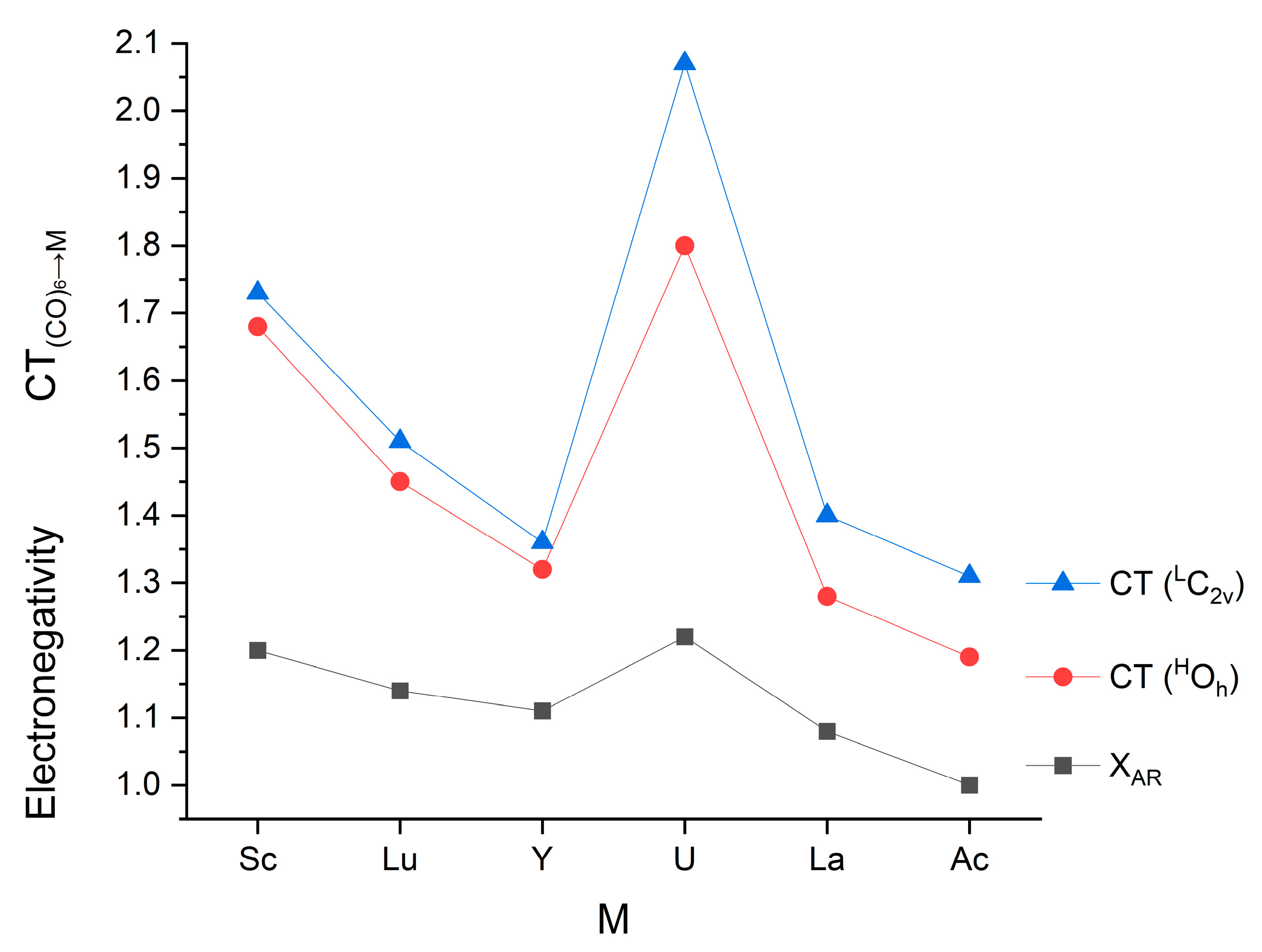
3.3. Donor–Acceptor Interactions
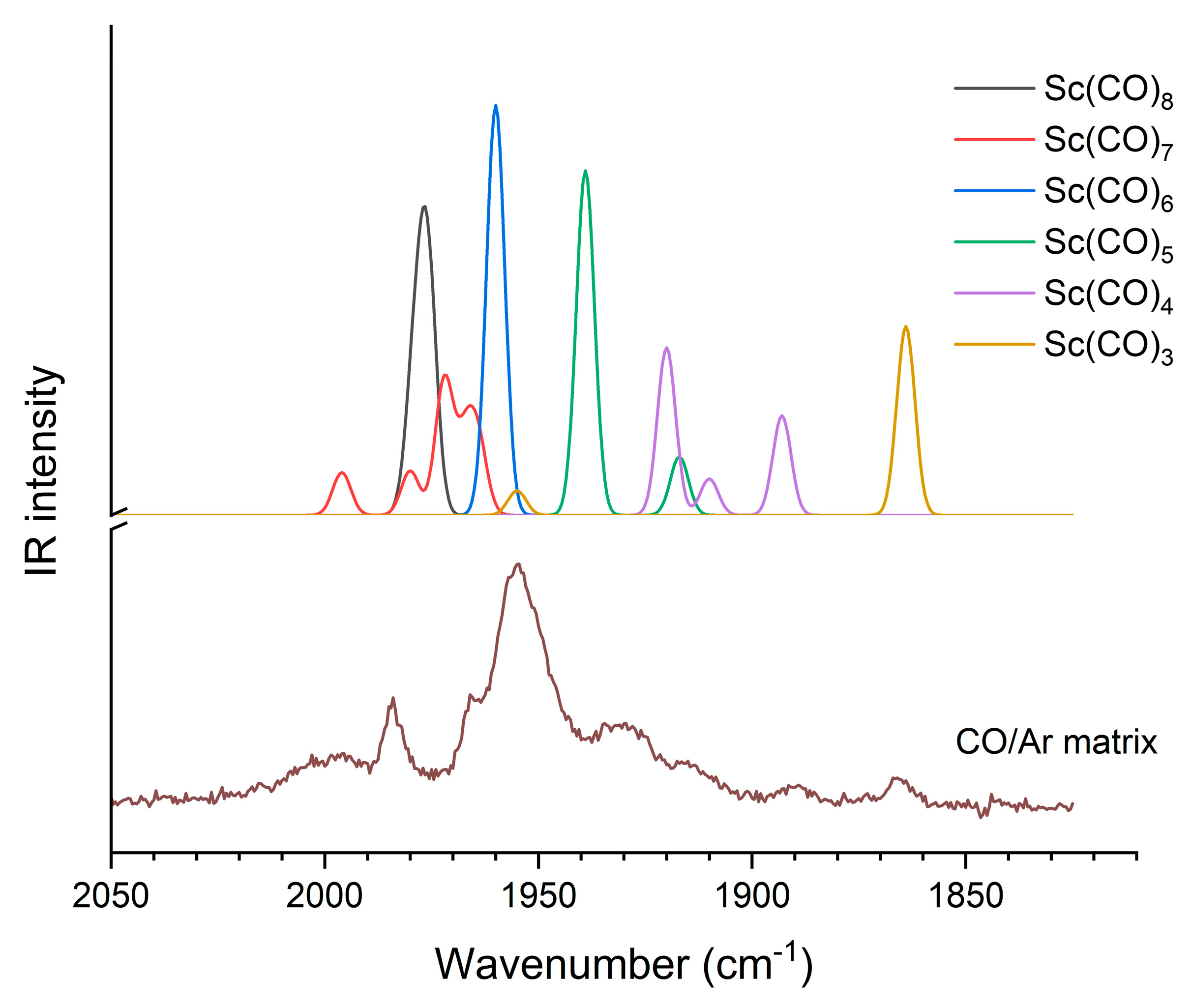
3.4. Matrix Isolation IR
4. Conclusions
- (i)
- Closer M-C distances, implying stronger bonding interactions;
- (ii)
- Weaker steric effects, particularly in the cases of larger M-s;
- (iii)
- Generally stronger CT interactions in terms of transferred electrons,
- (iv)
- Less Pauli repulsion because of the low-spin character.
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hallam, H.E. Vibrational Spectroscopy of Trapped Species Infrared and Raman Studies of Matrix-Isolated Molecules, Radicals and Ions; John Wiley & Sons Ltd.: New York, NY, USA, 1973. [Google Scholar]
- Moskovits, M.; Ozin, G.A. Cryochemistry; Wiley: Hoboken, NJ, USA, 1976. [Google Scholar]
- Turner, J.J.; Burdett, J.K.; Perutz, R.N.; Poliakoff, M. Matrix photochemistry of metal carbonyls. Pure Appl. Chem. 1977, 49, 271–285. [Google Scholar] [CrossRef]
- Burdett, J.K. Matrix isolation studies on transition metal carbonyls and related species. Coord. Chem. Rev. 1978, 27, 1–58. [Google Scholar] [CrossRef]
- Perutz, R.N. Matrix Photochemistry of Transition Metal Complexes: Principles, Applications and Links to Other Methods. In Low Temperature Molecular Spectroscopy; NATO ASI Series, 483; Fausto, R., Ed.; Springer: Dordrecht, The Netherlands, 1996; pp. 95–124. [Google Scholar] [CrossRef]
- Zhou, M.; Andrews, L. Matrix Infrared Spectra and Density Functional Calculations of ScCO, ScCO−, and ScCO+. J. Phys. Chem. A 1999, 103, 2964–2971. [Google Scholar] [CrossRef]
- Turner, J.J.; George, M.W.; Poliakoff, M.; Perutz, R.N. Photochemistry of transition metal carbonyls. Chem. Soc. Rev. 2022, 51, 5300–5329. [Google Scholar] [CrossRef]
- Jin, X.; Jiang, L.; Xu, Q.; Zhou, M. Reactions of Gadolinium Atoms and Dimers with CO: Formation of Gadolinium Carbonyls and Photoconversion to CO Activated Molecules. J. Phys. Chem. A 2006, 110, 12585–12591. [Google Scholar] [CrossRef]
- Jiang, L.; Xu, Q. Reactions of Laser-Ablated La and Y Atoms with CO: Matrix Infrared Spectra and DFT Calculations of the M(CO)x and MCO+ (M = La, Y; x = 1−4) Molecules. J. Phys. Chem. A 2007, 111, 3271–3277. [Google Scholar] [CrossRef]
- Ricks, A.M.; Gagliardi, L.; Duncan, M.A. Infrared Spectroscopy of Extreme Coordination: The Carbonyls of U+ and UO2+. J. Am. Chem. Soc. 2010, 132, 15905–15907. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Wang, J.; Qin, Z.; Shi, L.; Tang, Z.; Xing, X. Octacoordinate Metal Carbonyls of Lanthanum and Cerium: Experimental Observation and Theoretical Calculation. J. Phys. Chem. A 2014, 118, 9380–9385. [Google Scholar] [CrossRef] [PubMed]
- Brathwaite, A.D.; Maner, J.A.; Duncan, M.A. Testing the Limits of the 18-Electron Rule: The Gas-Phase Carbonyls of Sc+ and Y+. Inorg. Chem. 2014, 53, 1166–1169. [Google Scholar] [CrossRef]
- Jin, J.; Yang, T.; Xin, K.; Wang, G.; Jin, X.; Zhou, M.; Frenking, G. Octacarbonyl Anion Complexes of Group Three Transition Metals [TM(CO)8]− (TM=Sc, Y, La) and the 18-Electron Rule. Angew. Chem. Int. Ed. 2018, 57, 6236–6241. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Pan, S.; Jin, X.; Lei, S.; Zhao, L.; Frenking, G.; Zhou, M. Octacarbonyl Anion Complexes of the Late Lanthanides Ln(CO)8− (Ln = Tm, Yb, Lu) and the 32-Electron Rule. Chem. Eur. J. 2019, 25, 3229–3234. [Google Scholar] [CrossRef]
- Chi, C.; Pan, S.; Jin, J.; Meng, L.; Luo, M.; Zhao, L.; Zhou, M.; Frenking, G. Octacarbonyl Ion Complexes of Actinides [An(CO)8]+/− (An = Th, U) and the Role of f Orbitals in Metal–Ligand Bonding. Chem. Eur. J. 2019, 25, 11772–11784. [Google Scholar] [CrossRef]
- Wu, X.; Zhao, L.; Jin, J.; Pan, S.; Li, W.; Jin, X.; Wang, G.; Zhou, M.; Frenking, G. Observation of alkaline earth complexes M(CO)8 (M = Ca, Sr, or Ba) that mimic transition metals. Science 2018, 361, 912–916. [Google Scholar] [CrossRef]
- Bettens, T.; Pan, S.; De Proft, F.; Frenking, G.; Geerlings, P. Alkaline Earth Metals Activate N2 and CO in Cubic Complexes Just Like Transition Metals: A Conceptual Density Functional Theory and Energy Decomposition Analysis Study. Chem. Eur. J. 2020, 26, 12785–12793. [Google Scholar] [CrossRef]
- Yang, X. The 18-electron rule for main-group alkaline earth octacarbonyl complexes. Nat. Sci. Rev. 2019, 6, 8–9. [Google Scholar] [CrossRef]
- Xing, X.; Wang, J.; Xie, H.; Liu, Z.; Qin, Z.; Zhao, L.; Tang, Z. Octacoordinate metal carbonyls of scandium and yttrium: Theoretical calculations and experimental observation. Rapid Commun. Mass Spectrom. 2013, 27, 1403–1409. [Google Scholar] [CrossRef]
- Deng, G.; Lei, S.; Pan, S.; Jin, J.; Wang, G.; Zhao, L.; Zhou, M.; Frenking, G. Filling a Gap: The Coordinatively Saturated Group 4 Carbonyl Complexes TM(CO)8 (TM = Zr, Hf) and Ti(CO)7. Chem. Eur. J. 2020, 26, 10487–10500. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Tian, C.-Y.; Zhao, Y.; Jiang, S.; Wang, T.; Zheng, H.; Yan, W.; Li, G.; Xie, H.; Li, J.; et al. Observation of Confinement-Free Neutral Group Three Transition Metal Carbonyls Sc(CO)7 and TM(CO)8 (TM=Y, La). Angew. Chem. Int. Ed. 2023, 62, e202305490. [Google Scholar] [CrossRef] [PubMed]
- Hillier, I.H.; Saunders, V.R. Ab initio molecular orbital calculations of transition metal complexes. Mol. Phys. 1971, 22, 1025–1034. [Google Scholar] [CrossRef]
- Baerends, E.J.; Ros, P. The electronic structure of transition metal carbonyl complexes. Mol. Phys. 1975, 30, 1735–1747. [Google Scholar] [CrossRef]
- Rees, B.; Mitschler, A. Electronic structure of chromium hexacarbonyl at liquid nitrogen temperature. 2. Experimental study (x-ray and neutron diffraction) of σ and π bonding. J. Am. Chem. Soc. 1976, 98, 7918–7924. [Google Scholar] [CrossRef]
- Arratia-Perez, R.; Yang, C.Y. Bonding in metal hexacarbonyls. J. Chem. Phys. 1985, 83, 4005–4014. [Google Scholar] [CrossRef]
- Barnes, L.A.; Bauschlicher, C.W., Jr. Theoretical studies of the transition metal–carbonyl systems MCO and M(CO)2, M=Ti, Sc, and V. J. Chem. Phys. 1989, 91, 314–330. [Google Scholar] [CrossRef]
- Davidson, E.R.; Kunze, K.L.; Machado, F.B.C.; Chakravorty, S.J. The transition metal-carbonyl bond. Acc. Chem. Res. 1993, 26, 628–635. [Google Scholar] [CrossRef]
- Fournier, R. Theoretical study of the monocarbonyls of first-row transition metal atoms. J. Chem. Phys. 1993, 99, 1801–1815. [Google Scholar] [CrossRef]
- Ehlers, A.W.; Frenking, G. Structures and Bond Energies of the Transition Metal Hexacarbonyls M(CO)6 (M = Cr, Mo, W). A Theoretical Study. J. Am. Chem. Soc. 1994, 116, 1514–1520. [Google Scholar] [CrossRef]
- Adamo, C.; Lelj, F. A hybrid density functional study of the first-row transition-metal monocarbonyls. J. Chem. Phys. 1995, 103, 10605–10613. [Google Scholar] [CrossRef]
- Jonas, V.; Thiel, W. Theoretical study of the vibrational spectra of the transition metal carbonyls M(CO)6 [M=Cr, Mo, W], M(CO)5 [M=Fe, Ru, Os], and M(CO)4 [M=Ni, Pd, Pt]. J. Chem. Phys. 1995, 102, 8474–8484. [Google Scholar] [CrossRef]
- Koukounas, C.; Kardahakis, S.; Mavridis, A. Electronic and geometric structure of the 3d-transition metal monocarbonyls MCO, M=Sc, Ti, V, and Cr. J. Chem. Phys. 2005, 123, 074327. [Google Scholar] [CrossRef]
- Gao, S.-M.; Guo, W.-P.; Jin, L.; Ding, Y.-H. Maximum carbonyl-coordination number of scandium. Computational study of Sc(CO)n (n = 1–7), Sc(CO)7− and Sc(CO)63−. Int. J. Quantum Chem. 2013, 113, 1192–1199. [Google Scholar] [CrossRef]
- van der Lubbe, S.C.C.; Vermeeren, P.; Fonseca Guerra, C.; Bickelhaupt, F.M. The Nature of Nonclassical Carbonyl Ligands Explained by Kohn–Sham Molecular Orbital Theory. Chem. Eur. J. 2020, 26, 15690–15699. [Google Scholar] [CrossRef]
- Frenking, G.; Fröhlich, N. The Nature of the Bonding in Transition-Metal Compounds. Chem. Rev. 2000, 100, 717–774. [Google Scholar] [CrossRef]
- Frenking, G. Understanding the nature of the bonding in transition metal complexes: From Dewar’s molecular orbital model to an energy partitioning analysis of the metal–ligand bond. J. Organomet. Chem. 2001, 635, 9–23. [Google Scholar] [CrossRef]
- Frenking, G.; Fernández, I.; Holzmann, N.; Pan, S.; Krossing, I.; Zhou, M. Metal–CO Bonding in Mononuclear Transition Metal Carbonyl Complexes. JACS Au 2021, 1, 623–645. [Google Scholar] [CrossRef]
- Brockway, L.O.; Ewens, R.V.C.; Lister, M. An electron diffraction investigation of the hexacarbonyls of chromium, molybdenum and tungsten. Trans. Faraday Soc. 1938, 34, 1350–1357. [Google Scholar] [CrossRef]
- Jones, L.H. Vibrational spectra and force constants of the hexacarbonyls of chromium, molybdenum and tungsten. Spectrochim. Acta 1963, 19, 329–338. [Google Scholar] [CrossRef]
- Arnesen, S.P.; Seip, H.M. Studies on the Failure of the First Born Approximation in Electron Diffraction. V. Molybdenum- and Tungsten Hexacarbonyl. Acta Chem. Scand. 1966, 20, 2711–2727. [Google Scholar] [CrossRef]
- Jost, A.; Rees, B.; Yelon, W.B. Electronic structure of chromium hexacarbonyl at 78 K. I. Neutron diffraction study. Acta Cryst. Sect. B 1975, 31, 2649–2658. [Google Scholar] [CrossRef]
- Tevault, D.; Nakamoto, K. Matrix isolation and computer simulation spectra of chromium hexacarbonyl Cr(CO)6 and molybdenum hexacarbonyl Mo(CO)6. Inorg. Chem. 1975, 14, 2371–2373. [Google Scholar] [CrossRef]
- Perutz, R.N.; Turner, J.J. Photochemistry of the Group VI hexacarbonyls in low-temperature matrices. II. Infrared spectra and structures of carbon-13 monoxide-enriched hexacarbonyls and pentacarbonyls of chromium, molybdenum, and tungsten. Inorg. Chem. 1975, 14, 262–270. [Google Scholar] [CrossRef]
- Hawkins, N.J.; Mattraw, H.C.; Sabol, W.W.; Carpenter, D.R. Spectroscopy of Gaseous Carbonyls. I. Infrared Spectra and Thermodynamic Properties of Chromium and Molybdenum Hexacarbonyls. J. Chem. Phys. 2004, 23, 2422–2427. [Google Scholar] [CrossRef]
- Langmuir, I. Types of Valence. Science 1921, 54, 59–67. [Google Scholar] [CrossRef]
- Tolman, C.A. The 16 and 18 electron rule in organometallic chemistry and homogeneous catalysis. Chem. Soc. Rev. 1972, 1, 337–353. [Google Scholar] [CrossRef]
- Jensen, W.B. The Origin of the 18-Electron Rule. J. Chem. Educ. 2005, 82, 28. [Google Scholar] [CrossRef]
- Bellard, S.; Rubinson, K.A.; Sheldrick, G.M. Crystal and molecular structure of vanadium hexacarbonyl. Acta Cryst. Sect. B 1979, 35, 271–274. [Google Scholar] [CrossRef]
- Elschenbroich, C.; Salzer, A. Organometallics: A Concise Introduction, 2nd ed.; Wiley-VCH: Weinheim, Germany, 1992. [Google Scholar]
- Busby, R.; Klotzbuecher, W.; Ozin, G.A. Titanium hexacarbonyl, Ti(CO)6, and titanium hexadinitrogen, Ti(N2)6. 1. Synthesis using titanium atoms and characterization by matrix infrared and ultraviolet-visible spectroscopy. Inorg. Chem. 1977, 16, 822–828. [Google Scholar] [CrossRef]
- Calderazzo, F.; Englert, U.; Pampaloni, G.; Pelizzi, G.; Zamboni, R. Studies on carbonyl derivatives of early transition elements. A convenient method for the preparation of the [Nb(CO)6]- anion at atmospheric pressure and room temperature. Crystal and molecular structure of [M(CO)6]- (M = Nb, Ta) as their bis(triphenylphosphine) nitrogen(1+) derivatives. Inorg. Chem. 1983, 22, 1865–1870. [Google Scholar] [CrossRef]
- Holloway, J.H.; Senior, J.B.; Szary, A.C. Rhenium carbonyl fluorides: Preparation of [Re(CO)6][ReF6] and some reactions of [Re(CO)5F·ReF5]. J. Chem. Soc. Dalton Trans. 1987, 741–745. [Google Scholar] [CrossRef]
- Ellis, J.E.; Chi, K.M. Highly reduced organometallics. 28. Synthesis, isolation, and characterization of [K(cryptand 2.2.2)]2[Hf(CO)6], the first substance to contain hafnium in a negative oxidation state. Structural characterization of [K(cryptand 2.2.2)]2[M(CO)6]•pyridine (M = Ti, Zr, and Hf). J. Am. Chem. Soc. 1990, 112, 6022–6025. [Google Scholar] [CrossRef]
- Bernhardt, E.; Bach, C.; Bley, B.; Wartchow, R.; Westphal, U.; Sham, I.H.T.; von Ahsen, B.; Wang, C.; Willner, H.; Thompson, R.C.; et al. Homoleptic, σ-Bonded Octahedral [M(CO)6]2+ Cations of Iron(II), Ruthenium(II), and Osmium(II): Part 1: Syntheses, Thermochemical and Vibrational Characterizations, and Molecular Structures as [Sb2F11]− and [SbF6]− Salts. A Comprehensive, Comparative Study. Inorg. Chem. 2005, 44, 4189–4205. [Google Scholar] [CrossRef] [PubMed]
- Geier, J.; Willner, H.; Lehmann, C.W.; Aubke, F. Formation of Hexacarbonylmanganese(I) Salts, [Mn(CO)6]+X-, in Anhydrous HF. Inorg. Chem. 2007, 46, 7210–7214. [Google Scholar] [CrossRef] [PubMed]
- Kovács, A.; Klotzbücher, W. A DFT and Matrix-Isolation IR/UV-Visible Study of High-Coordinated Lanthanide-CO Complexes. Molecules 2023, 28, 5043. [Google Scholar] [CrossRef] [PubMed]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.R. Valency and Bonding: A Natural Bond Orbital Donor-Acceptor Perspective; Cambridge University Press: Cambridge, UK, 2005. [Google Scholar] [CrossRef]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. Natural bond orbital methods. WIREs Comput. Mol. Sci. 2012, 2, 1–42. [Google Scholar] [CrossRef]
- Weinhold, F. Natural bond orbital analysis: A critical overview of relationships to alternative bonding perspectives. J. Comput. Chem. 2012, 33, 2363–2379. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.R.; Glendening, E.D. What is NBO analysis and how is it useful? Int. Rev. Phys. Chem. 2016, 35, 399–440. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Balabanov, N.B.; Peterson, K.A. Systematically convergent basis sets for transition metals. I. All-electron correlation consistent basis sets for the 3d elements Sc–Zn. J. Chem. Phys. 2005, 123, 064107. [Google Scholar] [CrossRef] [PubMed]
- Andrae, D.; Haeussermann, U.; Dolg, M.; Stoll, H.; Preuss, H. Energy-adjusted ab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Dolg, M.; Stoll, H.; Preuss, H. Energy-adjusted ab initio pseudopotentials for the rare earth elements. J. Chem. Phys. 1989, 90, 1730–1734. [Google Scholar] [CrossRef]
- Martin, J.M.L.; Sundermann, A. Correlation consistent valence basis sets for use with the Stuttgart–Dresden–Bonn relativistic effective core potentials: The atoms Ga–Kr and In–Xe. J. Chem. Phys. 2001, 114, 3408–3420. [Google Scholar] [CrossRef]
- Cao, X.; Dolg, M. Segmented contraction scheme for small-core lanthanide pseudopotential basis sets. J. Mol. Struct. Theochem 2002, 581, 139–147. [Google Scholar] [CrossRef]
- Küchle, W.; Dolg, M.; Stoll, H.; Preuss, H. Energy-Adjusted Pseudopotentials for the Actinides. Parameter Sets and Test Calculations for Thorium and Thorium Monoxide. J. Chem. Phys. 1994, 100, 7535–7542. [Google Scholar] [CrossRef]
- Cao, X.; Dolg, M.; Stoll, H. Valence basis sets for relativistic energy-consistent small-core actinide pseudopotentials. J. Chem. Phys. 2003, 118, 487–496. [Google Scholar] [CrossRef]
- Cao, X.; Dolg, M. Segmented contraction scheme for small-core actinide pseudopotential basis sets. J. Mol. Struct. Theochem 2004, 673, 203–209. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parameterization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Bursch, M.; Mewes, J.-M.; Hansen, A.; Grimme, S. Best-Practice DFT Protocols for Basic Molecular Computational Chemistry. Angew. Chem. Int. Ed. 2022, 61, e202205735. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6169. [Google Scholar] [CrossRef]
- Roos, B.O. Advances in Chemical Physics, Ab Initio Methods in Quantum Chemistry—II; Lawley, K.P., Ed.; John Wiley & Sons Ltd.: Chichester, UK, 1987; pp. 399–446. [Google Scholar]
- Karlström, G.; Lindh, R.; Malmqvist, P.-Å.; Roos, B.O.; Ryde, U.; Veryazov, V.; Widmark, P.-O.; Cossi, M.; Schimmelpfennig, B.; Neogrady, P.; et al. MOLCAS: A Program Package for Computational Chemistry. Comput. Mat. Sci. 2003, 28, 222–239. [Google Scholar] [CrossRef]
- Aquilante, F.; Autschbach, J.; Carlson, R.K.; Chibotaru, L.F.; Delcey, M.G.; De Vico, L.; Galván, I.F.; Ferré, N.; Frutos, L.M.; Gagliardi, L.; et al. MOLCAS 8: New Capabilities for Multiconfigurational Quantum Chemical Calculations Across the Periodic Table. J. Comput. Chem. 2016, 37, 506–541. [Google Scholar] [CrossRef] [PubMed]
- Douglas, N.; Kroll, N.M. Quantum Electrodynamical Corrections to the Fine Structure of Helium. Ann. Phys. 1974, 82, 89–155. [Google Scholar] [CrossRef]
- Hess, B.A. Relativistic Electronic-Structure Calculations Employing a Two-Component No-Pair Formalism with External-Field Projection Operators. Phys. Rev. A 1986, 33, 3742–3748. [Google Scholar] [CrossRef] [PubMed]
- Roos, B.O.; Lindh, R.; Malmqvist, P.-Å.; Veryazov, V.; Widmark, P.-O. New Relativistic ANO Basis Sets for Transition Metal Atoms. J. Phys. Chem. A 2005, 109, 6575–6579. [Google Scholar] [CrossRef] [PubMed]
- Roos, B.O.; Lindh, R.; Malmqvist, P.-Å.; Veryazov, V.; Widmark, P.-O. New Relativistic Atomic Natural Orbital Basis Sets for Lanthanide Atoms with Applications to the Ce Diatom and LuF3. J. Phys. Chem. A 2008, 112, 11431–11435. [Google Scholar] [CrossRef]
- Roos, B.O.; Lindh, R.; Malmqvist, P.-Å.; Veryazov, V.; Widmark, P.-O. New Relativistic ANO Basis Sets for Actinide Atoms. Chem. Phys. Lett. 2005, 409, 295–299. [Google Scholar] [CrossRef]
- Roos, B.O.; Lindh, R.; Malmqvist, P.-Å.; Veryazov, V.; Widmark, P.-O. Main Group Atoms and Dimers Studied with a New Relativistic ANO Basis Set. J. Phys. Chem. A 2004, 108, 2851–2858. [Google Scholar] [CrossRef]
- Andersson, K.; Malmqvist, P.-Å.; Roos, B.O.; Sadlej, A.; Wolinski, K. Second-Order Perturbation Theory with a CASSCF Reference Function. J. Phys. Chem. 1990, 94, 5483–5488. [Google Scholar] [CrossRef]
- Andersson, K.; Malmqvist, P.-Å.; Roos, B.O. Second-Order Perturbation Theory with a Complete Active Space Self-Consistent Field Reference Function. J. Chem. Phys. 1992, 96, 1218–1226. [Google Scholar] [CrossRef]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Landis, C.R.; Weinhold, F. NBO 6.0; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2013. [Google Scholar]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. NBO 6.0: Natural Bond Orbital Analysis Program. J. Comput. Chem. 2013, 34, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.; Piquemal, J.-P.; Liu, S.; Cisneros, G.A. DFT-steric-based energy decomposition analysis of intermolecular interactions. Theor. Chem. Acc. 2014, 133, 1484. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Kovács, A.; Klotzbücher, W. Octa-coordination in complexes of lanthanides with N2 confirmed by matrix-isolation IR spectroscopy and DFT calculations. J. Mol. Struct. 2023, 1272, 134222. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
- Scalmani, G.; Frisch, M.J. Continuous surface charge polarizable continuum models of solvation. I. General formalism. J. Chem. Phys. 2010, 132, 114110. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides. Acta Cryst. 1976, A32, 751–767. [Google Scholar] [CrossRef]
- Slater, J.C. Atomic Radii in Crystals. J. Chem. Phys. 2004, 41, 3199–3204. [Google Scholar] [CrossRef]
- Liu, S. Steric effect: A quantitative description from density functional theory. J. Chem. Phys. 2007, 126, 244103. [Google Scholar] [CrossRef]
- Weizsäcker, C.F.v. Zur Theorie der Kernmassen. Z. Phys. 1935, 96, 431–458. [Google Scholar] [CrossRef]
- Liu, S.; Govind, N. Toward Understanding the Nature of Internal Rotation Barriers with a New Energy Partition Scheme: Ethane and n-Butane. J. Phys. Chem. A 2008, 112, 6690–6699. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhong, A.-G.; Yang, Q.; Liu, S. Origin of anomeric effect: A density functional steric analysis. J. Chem. Phys. 2011, 134, 084103. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Hu, H.; Pedersen, L.G. Steric, Quantum, and Electrostatic Effects on SN2 Reaction Barriers in Gas Phase. J. Phys. Chem. A 2010, 114, 5913–5918. [Google Scholar] [CrossRef] [PubMed]
- Goldman, A.S.; Krogh-Jespersen, K. Why Do Cationic Carbon Monoxide Complexes Have High C−O Stretching Force Constants and Short C−O Bonds? Electrostatic Effects, Not σ-Bonding. J. Am. Chem. Soc. 1996, 118, 12159–12166. [Google Scholar] [CrossRef]
- Lupinetti, A.J.; Frenking, G.; Strauss, S.H. Nonclassical Metal Carbonyls: Appropriate Definitions with a Theoretical Justification. Angew. Chem. Int. Ed. 1998, 37, 2113–2116. [Google Scholar] [CrossRef]
- Lupinetti, A.J.; Jonas, V.; Thiel, W.; Strauss, S.H.; Frenking, G. Trends in Molecular Geometries and Bond Strengths of the Homoleptic d10 Metal Carbonyl Cations [M(CO)n]x+ (Mx+=Cu+, Ag+, Au+, Zn2+, Cd2+, Hg2+; n=1–6): A Theoretical Study. Chem. Eur. J. 1999, 5, 2573–2583. [Google Scholar] [CrossRef]
- Willner, H.; Aubke, F. σ-Bonded Metal Carbonyl Cations and Their Derivatives: Syntheses and Structural, Spectroscopic, and Bonding Principles. Organometallics 2003, 22, 3612–3633. [Google Scholar] [CrossRef]
- Slater, J.L.; De Vore, T.C.; Calder, V. Detection of neodymium and ytterbium carbonyls using matrix isolation. Inorg. Chem. 1973, 12, 1918–1921. [Google Scholar] [CrossRef]
- Slater, J.L.; DeVore, T.C.; Calder, V. Detection of praseodymium, europium, gadolinium, and holmium carbonyls using matrix isolation. Inorg. Chem. 1974, 13, 1808–1812. [Google Scholar] [CrossRef]
- Klotzbücher, W.E.; Petrukhina, M.A.; Sergeev, G.B. Reactions of Samarium Atoms in Inert and Reactive Matrices. Mendeleev Commun. 1994, 4, 5–7. [Google Scholar] [CrossRef]
- Ermilov, A.Y.; Nemukhin, A.V.; Kovba, V.M. Characterization of structures and spectra of holmium complexes formed in the CO and N2 matrices. Mendeleev Commun. 1999, 9, 88–89. [Google Scholar] [CrossRef]
- Zhou, M.; Andrews, L.; Li, J.; Bursten, B.E. Reactions of Th Atoms with CO: The First Thorium Carbonyl Complex and an Unprecedented Bent Triplet Insertion Product. J. Am. Chem. Soc. 1999, 121, 12188–12189. [Google Scholar] [CrossRef]
- Zhou, M.; Andrews, L.; Li, J.; Bursten, B.E. Reaction of Laser-Ablated Uranium Atoms with CO: Infrared Spectra of the CUO, CUO-, OUCCO, (η2-C2)UO2, and U(CO)x (x = 1−6) Molecules in Solid Neon. J. Am. Chem. Soc. 1999, 121, 9712–9721. [Google Scholar] [CrossRef]
- Allred, A.L.; Rochow, E.G. A scale of electronegativity based on electrostatic force. J. Inorg. Nucl. Chem. 1958, 5, 264–268. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| M | Spin | Symmetry | Character 1 | ΔE 2 | ΔG10K | ΔG298K |
|---|---|---|---|---|---|---|
| Sc | 4 | Oh | min | 0.0 | 0.0 | 0.0 |
| 2 | D4h | min | 11.0 | 10.6 | 7.1 | |
| 2 | C2v | min | 8.5 | 10.9 | 9.0 | |
| Lu | 4 | Oh | min | 2.8 | 0.2 | 0.0 |
| 2 | D4h | min | 11.9 | 8.9 | 2.2 | |
| 2 | C2v | min | 0.0 | 0.0 | 1.7 | |
| Y | 4 | Oh | min | 5.9 | 2.9 | 0.0 |
| 2 | D4h | i(2) | 14.9 | |||
| 2 | C2v | min | 0.0 | 0.0 | 0.8 | |
| U 3 | 7 | Oh | i(3−) | 36.6 | ||
| 7 | C2 | min | 22.3 | 17.0 | 1.4 | |
| 5 | D4h | i(2−) | 72.2 | |||
| 5 | C2v | min | 0.0 | 0.0 | 0.0 | |
| 3 | Cs | min | 12.4 | 15.6 | 19.0 | |
| 1 | C2h | min | 93.6 | 91.2 | 86.9 | |
| La | 4 | Oh | min | 21.8 | 17.6 | 6.6 |
| 2 | D4h | i(2−) | 31.1 | |||
| 2 | C2v | min | 0.0 | 0.0 | 0.0 | |
| Ac | 4 | Oh | min | 25.1 | 20.3 | 4.5 |
| 2 | D4h | i(2−) | 33.2 | |||
| 2 | C2h | min | 25.4 | 22.6 | 15.4 | |
| 2 | C2v | i(1−) | 0.3 | |||
| 2 | Cs 4 | min | 0.0 | 0.0 | 0.0 |
| M | XAR 2 | Spin | Sym | qM | CT | ΣCT | Pop | |||
|---|---|---|---|---|---|---|---|---|---|---|
| (CO)6→M | M→(CO)6 | s | d | f | ||||||
| Sc | 1.20 | 4 | Oh | −0.34 | 1.68 | 1.34 | 3.02 | 0.44 | 2.90 | - |
| 2 | D4h | −0.33 | 1.69 | 1.36 | 3.05 | 0.45 | 2.88 | - | ||
| 2 | C2v | −0.25 | 1.73 | 1.48 | 3.21 | 0.40 | 2.85 | - | ||
| Lu | 1.14 | 4 | Oh | 0.15 | 1.45 | 1.60 | 3.06 | 0.45 | 2.41 | - |
| 2 | C2v | 0.23 | 1.51 | 1.74 | 3.25 | 0.41 | 2.37 | - | ||
| Y | 1.11 | 4 | Oh | 0.21 | 1.32 | 1.53 | 2.85 | 0.42 | 2.36 | - |
| 2 | C2v | 0.33 | 1.36 | 1.68 | 3.04 | 0.37 | 2.29 | - | ||
| U | 1.22 | 7 | C2 | −0.10 | 1.80 | 1.70 | 3.50 | 0.41 | 2.61 | 3.04 |
| 5 | C2v | −0.16 | 2.07 | 1.91 | 3.98 | 0.33 | 2.63 | 3.15 | ||
| La | 1.08 | 4 | Oh | 0.08 | 1.28 | 1.36 | 2.64 | 0.33 | 2.54 | 0.04 |
| 2 | C2v | 0.16 | 1.40 | 1.56 | 2.96 | 0.27 | 2.48 | 0.09 | ||
| Ac | 1.00 | 4 | Oh | 0.31 | 1.19 | 1.50 | 2.69 | 0.35 | 2.26 | 0.07 |
| 2 | Cs 3 | 0.41 | 1.31 | 1.72 | 3.03 | 0.30 | 2.16 | 0.11 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovács, A.; Klotzbücher, W. Preference of C2v Symmetry in Low-Spin Hexacarbonyls of Rare-Earth and f Elements. Symmetry 2024, 16, 178. https://doi.org/10.3390/sym16020178
Kovács A, Klotzbücher W. Preference of C2v Symmetry in Low-Spin Hexacarbonyls of Rare-Earth and f Elements. Symmetry. 2024; 16(2):178. https://doi.org/10.3390/sym16020178
Chicago/Turabian StyleKovács, Attila, and Werner Klotzbücher. 2024. "Preference of C2v Symmetry in Low-Spin Hexacarbonyls of Rare-Earth and f Elements" Symmetry 16, no. 2: 178. https://doi.org/10.3390/sym16020178