
Non-Selective Reduction of P-Stereogenic Phosphinoylacetic Acid Esters and 3-Phosphorylated Coumarins to Phosphino-Boranes: Discovery of Unexpected 2,3-Dihydrobenzofuran Derivative


Abstract
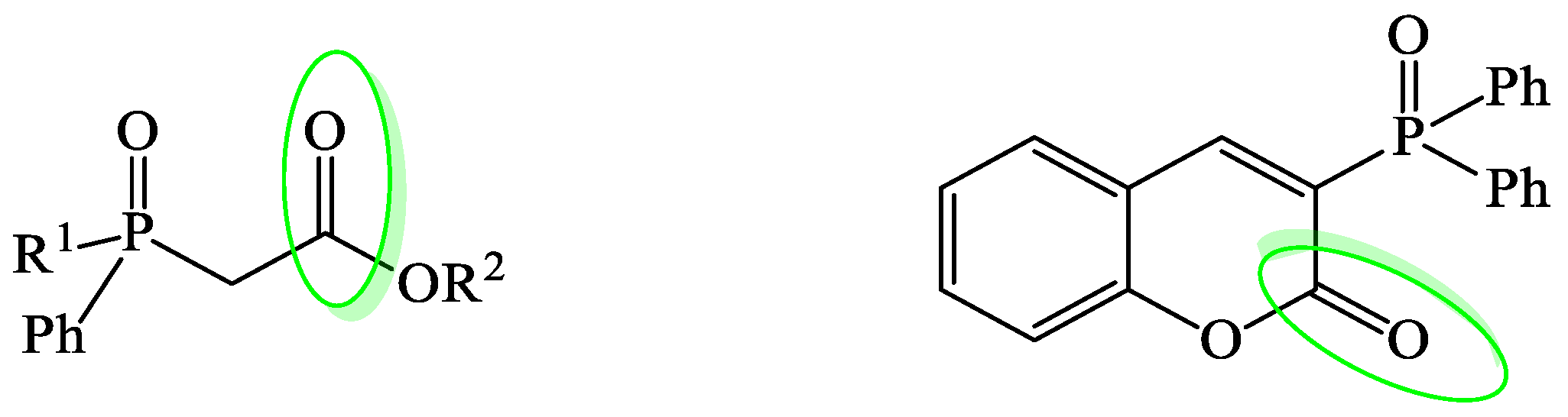
:1. Introduction
2. Materials and Methods
2.1. Instrumentation
2.1.1. General
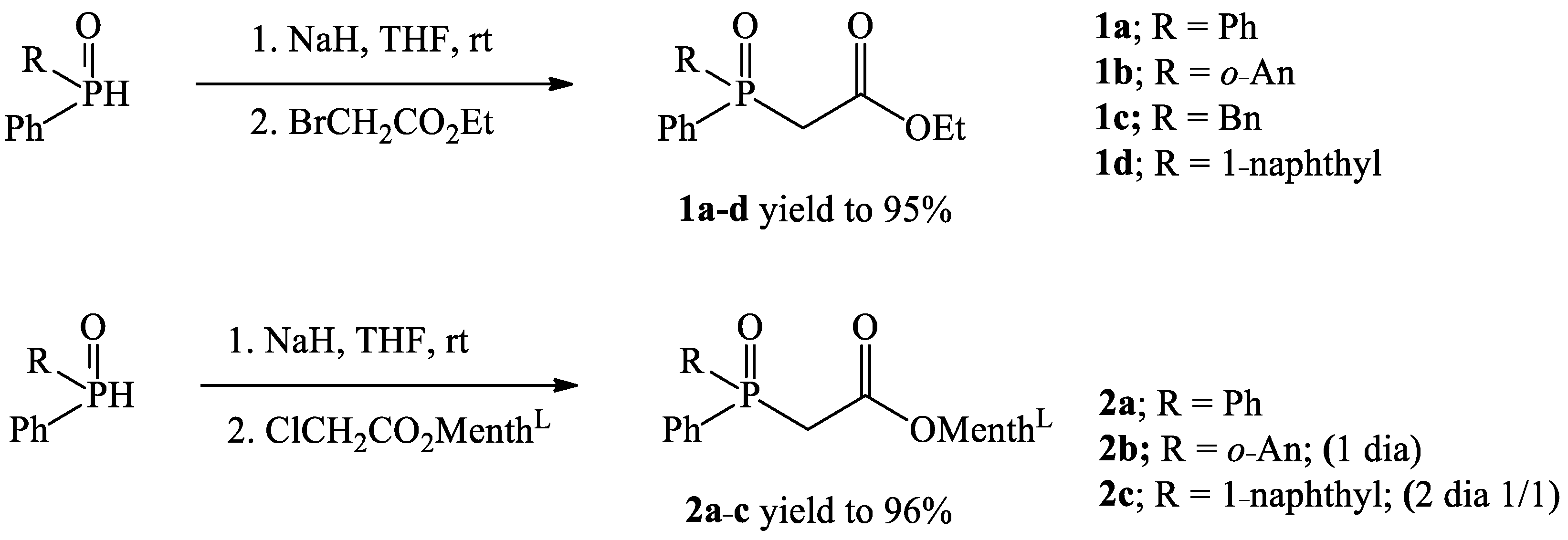
2.1.2. General Procedure for the Synthesis of Phosphinoylacetic Acid Ethyl Esters 1a–d
2.2. General Procedure for the Synthesis of Phosphinoylacetic Acid L-menthyl Esters 2a–c
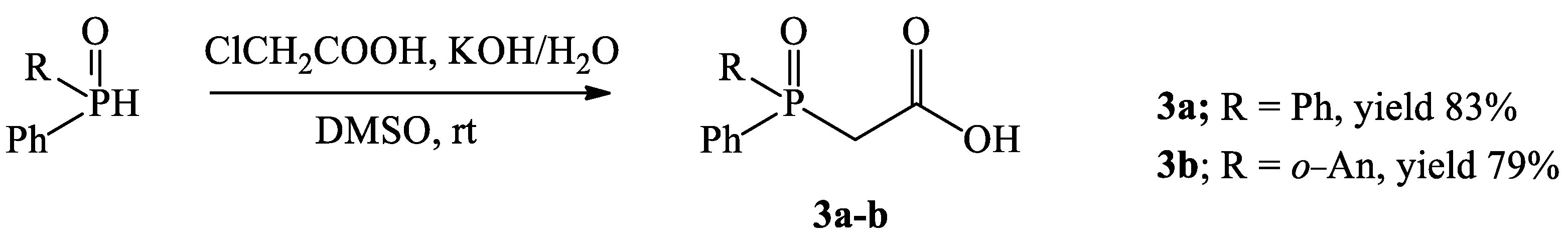
2.3. General Procedure for Synthesis of Phosphinoylacetic Acids 3a–b
2.4. General Procedure for Synthesis of Phosphino-Borane Complexes 4a–d from Phosphinoylacetic Acid Ethyl Esters 1a–d
(2-Hydroxyethyl)(2-metoxyphenyl)phenylphosphino-Borane (4b)
2.5. General Procedure for Synthesis of Phosphino-Borane Complexes 4a–b from Phosphinoylacetic Acid L-menthyl Esters 2a–b
2.6. Procedure for Synthesis of 3-(Diphenylphosphinyl)-2H-Chromen-2-One (5)
3-(Diphenylphosphinyl)-2H-Chromen-2-One (5)
2.7. Procedure for Synthesis of (2-methyl-tetrahydro-[2]furyl)-diphenylphosphine-borane (6)
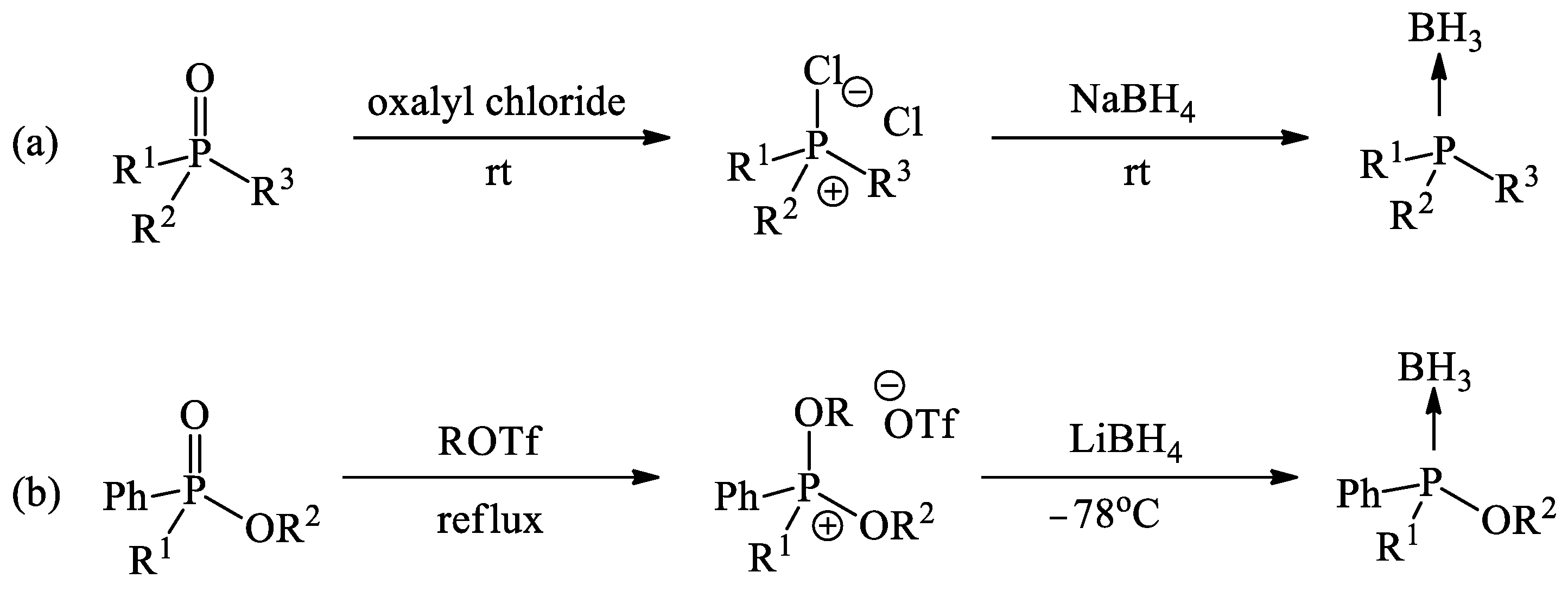
3. Results and Discussion
3.1. Synthesis and Borane Reduction of Phosphinoylacetic Esters and Acids
3.2. Study on the Reduction of 3-Phosphorylated Coumarin
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Methot, J.L.; Roush, W.R. Nucleophilic Phosphine Organocatalysis. Adv. Synth. Catal. 2004, 346, 1035–1050. [Google Scholar] [CrossRef]
- Xie, C.; Smaligo, A.J.; Song, X.R.; Kwon, O. Phosphorus-Based Catalysis. ACS Cent. Sci. 2021, 7, 536–558. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Cai, P.; Zhou, H.C.; Madrahimov, S.T. Bridging Homogeneous and Heterogeneous Catalysis: Phosphine-Functionalized Metal-Organic Frameworks. Angew. Chem. Int. Ed. 2024, 63, e202315075. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Fan, Y.C.; Sun, Z.; Wu, Y.; Kwon, O. Phosphine Organocatalysis. Chem. Rev. 2018, 118, 10049–10293. [Google Scholar] [CrossRef] [PubMed]
- Gallen, A.; Riera, A.; Verdaguer, X.; Grabulosa, A. Coordination chemistry and catalysis with secondary phosphine oxides. Catal. Sci. Technol. 2019, 9, 5504–5561. [Google Scholar] [CrossRef]
- Swor, C.D.; Tyler, D.R. Synthesis and coordination chemistry of macrocyclic phosphine ligands. Coord. Chem. Rev. 2011, 255, 2860–2881. [Google Scholar] [CrossRef]
- Fan, Y.C.; Kwon, O. Advances in nucleophilic phosphine catalysis of alkenes, allenes, alkynes, and MBHADs. Chem. Commun. 2013, 49, 11588–11619. [Google Scholar] [CrossRef] [PubMed]
- Byrne, P.A.; Gilheany, D.G. The modern interpretation of the Wittig reaction mechanism. Chem. Soc. Rev. 2013, 42, 6670–6696. [Google Scholar] [CrossRef] [PubMed]
- Appel, R. Tertiary Phosphane/Tetrachloromethane, a Versatile Reagent for Chlorination, Dehydration, and P-N Linkage. Angew. Chem. Int. Ed. 1975, 14, 801–811. [Google Scholar] [CrossRef]
- Del Rio Fuenzalida, N.M.; Alme, E.; Lundevall, F.J.; Bjørsvik, H.R. An environmentally benign and high-rate Appel type reaction. React. Chem. Eng. 2022, 7, 1650–1659. [Google Scholar] [CrossRef]
- Kumara Swamy, K.C.; Bhuvan Kumar, N.N.B.; Balaraman, E.; Pavan Kumar, K.V.P. Mitsunobu and Related Reactions: Advances and Applications. Chem. Rev. 2009, 109, 2551–2651. [Google Scholar] [CrossRef] [PubMed]
- Mitsunobu, O.; Yamada, M. Preparation of Esters of Carboxylic and Phosphoric Acid via Quaternary Phosphonium Salts. BCSJ 1967, 40, 2380–2382. [Google Scholar] [CrossRef]
- Maryanoff, B.E.; Reitz, A.B. The Wittig olefination reaction and modifications involving phosphoryl-stabilized carbanions. Stereochemistry, mechanism, and selected synthetic aspects. Chem. Rev. 1989, 89, 863–927. [Google Scholar] [CrossRef]
- Hérault, D.; Nguyen, D.H.; Nuel, D.; Buono, G. Reduction of secondary and tertiary phosphine oxides to phosphines. Chem. Soc. Rev. 2015, 44, 2508–2528. [Google Scholar] [CrossRef] [PubMed]
- Pei, M.; Tian, A.; Yang, Q.; Huang, N.; Wang, L.; Li, D. Organophosphorus catalytic reaction based on reduction of phosphine oxide. Green Synth. Catal. 2023, 4, 135–149. [Google Scholar] [CrossRef]
- Podyacheva, E.; Kuchuk, E.; Chusov, D. Reduction of phosphine oxides to phosphines. Tetrahedron Lett. 2019, 60, 575–582. [Google Scholar] [CrossRef]
- Kovács, T.; Keglevich, G. 9. Deoxygenation of phosphine oxides. In Organophosphorus Chemistry; Walter de Gruyter GmbH: Berlin/Munich, Germany, 2018; pp. 179–198. [Google Scholar] [CrossRef]
- Henson, P.D.; Naumann, K.; Mislow, K. Stereomutation of phosphine oxides by lithium aluminum hydride. J. Am. Chem. Soc. 1969, 91, 5645–5646. [Google Scholar] [CrossRef]
- Kovacs, T.; Keglevich, G. The Reduction of Tertiary Phosphine Oxides by Silanes. Curr. Org. Chem. 2017, 21, 569–585. [Google Scholar] [CrossRef]
- Fritzsche, H.; Hasserodt, U.; Korte, F. Reduktion organischer Verbindungen des fünfwertigen Phosphors zu Phosphinen, II. Reduktion tertiärer Phosphinoxyde zu tertiären Phosphinen mit Trichlorsilan. Chem. Ber. 1965, 98, 171–174. [Google Scholar] [CrossRef]
- Wu, H.C.; Yu, J.Q.; Spencer, J.B. Stereospecific Deoxygenation of Phosphine Oxides with Retention of Configuration Using Triphenylphosphine or Triethyl Phosphite as an Oxygen Acceptor. Org. Lett. 2004, 6, 4675–4678. [Google Scholar] [CrossRef]
- Kapuśniak, Ł.; Plessow, P.N.; Trzybiński, D.; Woźniak, K.; Hofmann, P.; Jolly, P.I. A Mild One-Pot Reduction of Phosphine(V) Oxides Affording Phosphines(III) and Their Metal Catalysts. Organometallics 2021, 40, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Naumann, K.; Zon, G.; Mislow, K. Use of hexachlorodisilane as a reducing agent. Stereospecific deoxygenation of acyclic phosphine oxides. J. Am. Chem. Soc. 1969, 91, 7012–7023. [Google Scholar] [CrossRef]
- Gevorgyan, A.; Mkrtchyan, S.; Grigoryan, T.; Iaroshenko, V.O. Disilanes as oxygen scavengers and surrogates of hydrosilanes suitable for selective reduction of nitroarenes, phosphine oxides and other valuable substrates. Org. Chem. Front. 2017, 4, 2437–2444. [Google Scholar] [CrossRef]
- Coumbe, T.; Lawrence, N.J.; Muhammad, F. Titanium (IV) catalysis in the reduction of phosphine oxides. Tetrahedron Lett. 1994, 35, 625–628. [Google Scholar] [CrossRef]
- Marsi, K.L. Phenylsilane reduction of phosphine oxides with complete stereospecificity. J. Org. Chem. 1974, 39, 265–267. [Google Scholar] [CrossRef]
- Nicolas, E.; Guerriero, A.; Lyaskovskyy, V.; Peruzzini, M.; Lammertsma, K.; Gonsalvi, L.; Slootweg, J.C. Metal-Free Reduction of Phosphine Oxides Using Polymethylhydrosiloxane. Inorganics 2016, 4, 34. [Google Scholar] [CrossRef]
- Keglevich, G.; Kovács, T.; Csatlós, F. The Deoxygenation of Phosphine Oxides under Green Chemical Conditions. Heteroatom Chem. 2015, 26, 199–205. [Google Scholar] [CrossRef]
- Bouhadir, G.; Amgoune, A.; Bourissou, D. Phosphine-Boranes and Related Ambiphilic Compounds: Synthesis, Structure, and Coordination to Transition Metals. Adv. Organomet. Chem. 2010, 58, 1–107. [Google Scholar] [CrossRef]
- Pellon, P. Phosphine-boranes in synthesis. Borane as an efficient protecting group in the preparation of functionalized phosphines. Tetrahedron Lett. 1992, 33, 4451–4452. [Google Scholar] [CrossRef]
- Staubitz, A.; Robertson, A.P.; Sloan, M.E.; Manners, I. Amine− and Phosphine–Borane Adducts: New Interest in Old Molecules. Chem. Rev. 2010, 110, 4023–4078. [Google Scholar] [CrossRef]
- Ohff, M. Borane Complexes of Trivalent Organophosphorus Compounds. Versatile Precursors for the Synthesis of Chiral Phosphine Ligands for Asymmetric Catalysis. Synthesis 1998, 1998, 1391–1415. [Google Scholar] [CrossRef]
- Rajendran, K.V.; Gilheany, D.G. Simple unprecedented conversion of phosphine oxides and sulfides to phosphine boranes using sodium borohydride. Chem. Commun. 2012, 48, 779–781. [Google Scholar] [CrossRef] [PubMed]
- Kenny, N.P.; Rajendran, K.V.; Gilheany, D.G. Chemoselective reduction of the phosphoryl bond of O-alkyl phosphinates and related compounds: An apparently impossible transformation. Chem. Commun. 2015, 51, 16561–16564. [Google Scholar] [CrossRef] [PubMed]
- Keglevich, G.; Fekete, M.; Chuluunbaatar, T.; Dobó, A.; Böcskei, Z.; Töke, L. Convenient Method for the Reduction of the Double-Bond of Cyclic Vinylphosphine Oxides Using Borane. Synth. Commun. 2000, 30, 4221–4231. [Google Scholar] [CrossRef]
- Keglevich, G.; Chuluunbaatar, T.; Ludányi, K.; Tőke, L. Phosphine-Boranes Based on the 7-Phosphanorbornene Framework: A Regioselective Approach to the Monoboranes of the Dimers of Phospholes. Tetrahedron 2000, 56, 1–6. [Google Scholar] [CrossRef]
- Keglevich, G.; Fekete, M.; Chuluunbaatar, T.; Dobó, A.; Harmat, V.; Tőke, L. One-pot transformation of cyclic phosphine oxides to phosphine–boranes by dimethyl sulfide–borane. J. Chem. Soc. Perkin Trans. 1 2000, 4451–4455. [Google Scholar] [CrossRef]
- Stankevič, M.; Pietrusiewicz, K.M. An Expedient Reduction of sec-Phosphine Oxides to sec-Phosphine-boranes by BH3·SMe2. Synlett 2003, 2003, 1012–1016. [Google Scholar] [CrossRef]
- Stankevič, M.; Andrijewski, G.; Pietrusiewicz, K.M. Direct Conversion of sec-Phosphine Oxides into Phosphinous Acid-Boranes. Synlett 2004, 2004, 311–315. [Google Scholar] [CrossRef]
- Sowa, S.; Stankevič, M.; Szmigielska, A.; Małuszyńska, H.; Kozioł, A.E.; Pietrusiewicz, K.M. Reduction of Functionalized Tertiary Phosphine Oxides with BH3. J. Org. Chem. 2015, 80, 1672–1688. [Google Scholar] [CrossRef]
- Sowa, S.; Stankevič, M.; Flis, A.; Pietrusiewicz, K.M. Reduction of Tertiary Phosphine Oxides by BH3, Assisted by Neighboring Activating Groups. Synthesis 2018, 50, 2106–21181. [Google Scholar] [CrossRef]
- Sowa, S.; Pietrusiewicz, K.M. Chemoselective Reduction of the P=O Bond in the presence of P-O and P-N Bonds in Phosphonate and Phosphinate Derivatives. Eur. J.Org. Chem. 2019, 5, 923–938. [Google Scholar] [CrossRef]
- Ou, Y.; Huang, Y.; Liu, Y.; Huo, Y.; Gao, Y.; Li, X.; Chen, Q. Iron-Catalyzed and Air-Mediated C(sp3)–H Phosphorylation of 1,3-Dicarbonyl Compounds Involving C–C Bond Cleavage. Adv. Synth. Catal. 2020, 362, 5783–5787. [Google Scholar] [CrossRef]
- Bredikhin, A.A.; Eliseenkova, R.M.; Tarasova, R.I.; Voskresenskaya, O.V.; Balandina, A.A.; Dobrynin, A.B.; Latypov, S.K.; Litvinov, I.A.; Sharafutdinova, D.R.; Efremov, Y.Y. Nonracemic menthyl phosphorylacetates. Russ. Chem. Bull. Int. Ed. 2007, 56, 290–297. [Google Scholar] [CrossRef]
- Dziuba, K.; Lubańska, M.; Pietrusiewicz, K.M. Enantiodivergent Synthesis of Both PAMPO Enantiomers Using l-Menthyl Chloroacetate and Stereomutation at P in Classical Quaternisation Reactions. Synthesis 2020, 52, 909–916. [Google Scholar] [CrossRef]
- Dziuba, K.; Szwaczko, K.; Frynas, S. Knoevenagel Condensation of Phosphinoylacetic Acids with Aldehydes: An Efficient One-Pot Strategy for the Synthesis of P-Functionalized Alkenyl Compounds. Synthesis 2021, 53, 2142–2154. [Google Scholar] [CrossRef]
- Dziuba, K.F.; Frynas, S.; Kozioł, A.E.; Szwaczko, K. Synthesis and Structural Elucidation of P-stereogenic Coumarins. Symmetry 2024, 16, 73. [Google Scholar] [CrossRef]
- Tsvetkov, E.N.; Bondarenko, A.N.; Malakhova, I.G.; Kabachnik, M.I. A Simple Synthesis and Some Synthetic Applications of Substituted Phosphide and Phosphinite Anions. Synthesis 1986, 1986, 198–208. [Google Scholar] [CrossRef]
- Arnott, G.E. Reduction of Carboxylic Acids and their Derivatives to Alcohols, Ethers, and Amines. In Comprehensive Organic Synthesis, 2nd ed.; Knochel, P., Ed.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 368–409. [Google Scholar] [CrossRef]
- Ohashi, A.; Imamoto, T. Highly Enantioselective Hydrogenation of α-Dehydroamino Acids by Rhodium Complexes with New Unsymmetric P–Chirogenic Bisphosphine Ligands. Org. Lett. 2001, 3, 373–375. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, A.; Kikuchi, S.-I.; Yasutake, M.; Imamoto, T. Unsymmetrical P-Chirogenic Bis(phosphane) Ligands: Their Preparation and Use in Rhodium-Catalyzed Asymmetric Hydrogenation. Eur. J. Org. Chem. 2002, 15, 2535–2546. [Google Scholar] [CrossRef]
- Morisaki, Y.; Imoto, H.; Hirano, K.; Hayashi, T.; Chujo, Y. Synthesis of Enantiomerically Pure P-Stereogenic Diphosphacrowns and Their Palladium Complexes. J. Org. Chem. 2011, 76, 1795–1803. [Google Scholar] [CrossRef]
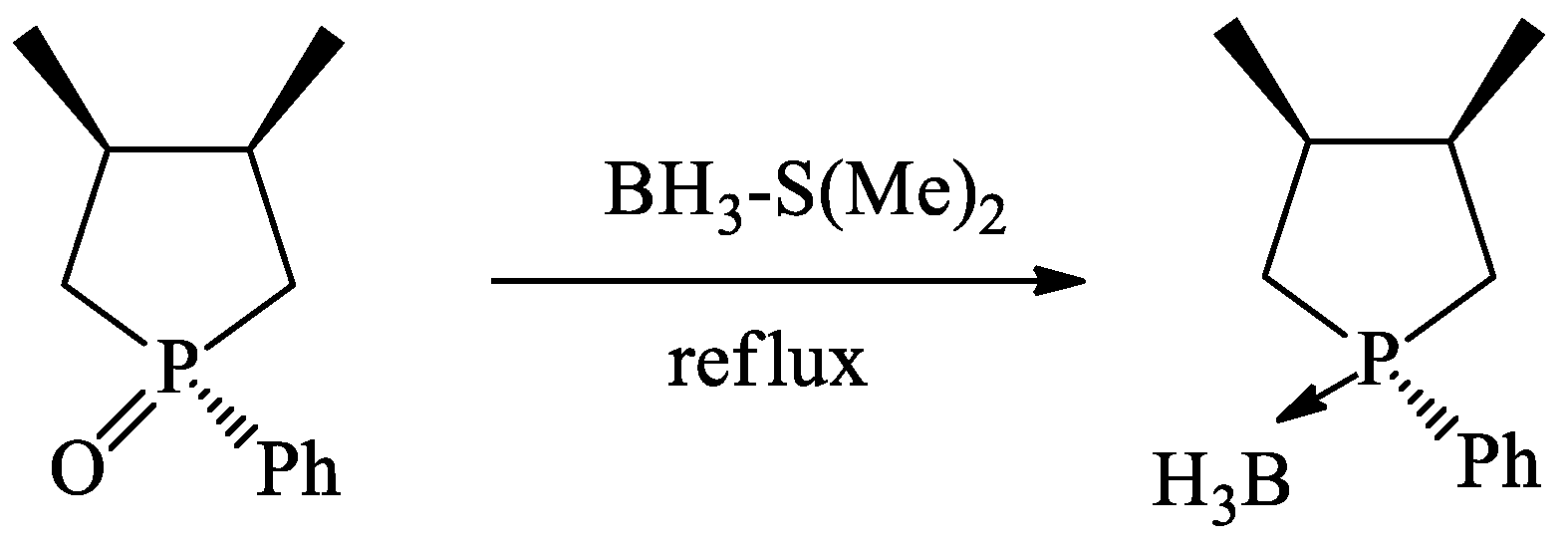



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
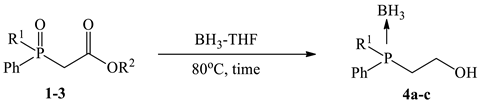 | |||||
| Entry | R1 | R2 | BH₃-THF (equiv.) | Time, h | Conv., % [a] (Yield), % [b] |
| 1 | Ph | Et | 5 | 24 | 4a; 40 (32) |
| 2 | Ph | Et | 10 | 72 | 4a; 98 (91) |
| 3 | o-An | Et | 10 | 24 | 4b; 44 |
| 4 | o-An | Et | 10 | 72 | 4b; 90 (85) |
| 5 | Bn | Et | 10 | 72 | 4c; 76 (68) |
| 6 | 1-Naphthyl | Et | 10 | 72 | 4d; 55 (49) |
| 7 | Ph | Menthyl | 10 | 72 | 4a; 66 (58) |
| 8 | o-An | Menthyl | 10 | 72 | 4b; 79 (73) |
| 9 | 1-Naphthyl | Menthyl | 10 | 72 | ND |
| 10 [c] | 1-Naphthyl | Menthyl | 10 | 72 | ND |
| 11 | Ph | H | 10 | 72 | 4a; 66 (60) |
| 12 | o-An | H | 10 | 72 | 4b; 64 (54) |
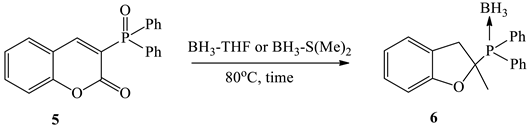 | ||||
| Entry | Borane Complexes (equiv.) | Time, h | Conv., % [a] | Yield, % [b] |
| 1 | BH3-THF (5.0) | 24 | 35 | complex mixture |
| 2 | BH3-THF (10.0) | 24 | 70 | 62 |
| 3 | BH3-THF (10.0) | 72 | 100 | 89 |
| 4 | BH₃-S(Me)₂ (5.0) | 24 | 100 | 90 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dziuba, K.; Walczak, N.; Szwaczko, K. Non-Selective Reduction of P-Stereogenic Phosphinoylacetic Acid Esters and 3-Phosphorylated Coumarins to Phosphino-Boranes: Discovery of Unexpected 2,3-Dihydrobenzofuran Derivative. Symmetry 2024, 16, 976. https://doi.org/10.3390/sym16080976
Dziuba K, Walczak N, Szwaczko K. Non-Selective Reduction of P-Stereogenic Phosphinoylacetic Acid Esters and 3-Phosphorylated Coumarins to Phosphino-Boranes: Discovery of Unexpected 2,3-Dihydrobenzofuran Derivative. Symmetry. 2024; 16(8):976. https://doi.org/10.3390/sym16080976
Chicago/Turabian StyleDziuba, Kamil, Natalia Walczak, and Katarzyna Szwaczko. 2024. "Non-Selective Reduction of P-Stereogenic Phosphinoylacetic Acid Esters and 3-Phosphorylated Coumarins to Phosphino-Boranes: Discovery of Unexpected 2,3-Dihydrobenzofuran Derivative" Symmetry 16, no. 8: 976. https://doi.org/10.3390/sym16080976