Diagnosis of the σ-, π- and (σ+π)-Aromaticity by the Shape of the NICSzz-Scan Curves and Symmetry-Based Selection Rules

1
Laboratory of Inorganic and General Chemistry, Department of Chemistry, University of Ioannina, 451 10 Ioannina, Greece
2
Laboratory of Applied Quantum Chemistry, Faculty of Chemistry, Aristotle University of Thessaloniki, 541 24 Thessaloniki, Greece
*
Author to whom correspondence should be addressed.
Symmetry 2010, 2(1), 284-319; https://doi.org/10.3390/sym2010284
Submission received: 16 December 2009
/
Revised: 21 January 2010
/
Accepted: 24 February 2010
/
Published: 15 March 2010
(This article belongs to the Special Issue Aromaticity and Molecular Symmetry)
Abstract
:The NICSzz-scan curves of aromatic organic, inorganic and “all-metal” molecules in conjunction with symmetry-based selection rules provide efficient diagnostic tools of the σ-, π- and/or double (σ + π)-aromaticity. The NICSzz-scan curves of σ-aromatic molecules are symmetric around the z-axis, having half-band widths approximately less than 3 Å with the induced diatropic ring current arising from Tx,y-allowed transitions involving exclusively σ-type molecular orbitals. Broad NICSzz-scan curves (half-band width approximately higher than 3 Å) characterize double (σ + π)-aromaticity, the chief contribution to the induced diatropic ring current arising from Tx,y-allowed transitions involving both σ- and π-type molecular orbitals. NICSzz-scan curves exhibiting two maxima at a certain distance above and below the molecular plane are typical for (σ + π)-aromatics where the π-diatropic ring current overwhelms the σ-type one. In the absence of any contribution from the σ-diatropic ring current, the NICSzz(0) value is close to zero and the molecule exhibits pure π-aromaticity.
1. Introduction
The collection of recent reviews in the topic of aromaticity/antiaromaticity [1,2,3,4,5,6,7,8,9] shows a rather large consensus appear in the computation or experimental tests for the diagnosis of the aromaticity/antiaromaticity (structural, electronic, magnetic, thermodynamic and kinetic diagnostic tools) whereas the mechanisms themselves still remain open to the debate. Various criteria have been used to judge aromaticity/antiaromaticity, but no one is satisfactory, particularly in predicting the orbital type (s-, p-, d-, f-orbital) of aromaticity [10]. Among the usual diagnostic probes of aromaticity/antiaromaticity developed so far those based on the dynamic magnetic measurements, in particular the Nucleus Independent Chemical Shifts (NICS) was the most widely used as a quantitative measure for aromaticity [11,12,13]. The NICS index is the negative value of the isotropic magnetic shielding computed at chosen points in the vicinity of molecules and, hence, directly related to the shielding function. It is typically computed at ring centers, at points above, and even on grids [14] in and around the molecule. The NICS criterion continues to be enhanced and refined, with the out-of-plane zz-components of the shielding tensor elements NICSzz(0) and NICSzz(1) proposed to be a superior aromaticity descriptor [15,16]. Furthermore, the CMO-NICS criterion [17,18,19] of aromaticity dissects the total shieldings calculated at ring or cage centers, into individual contributions from each canonical molecular orbital (CMO).
It was also established that the shapes of the NICS-scan profiles provide a clear picture of the type of the ring current in aromatic and antiaromatic systems [20,21] and can be used to characterize unequivocally whether an inorganic system is aromatic, non-aromatic and antiaromatic [22,23]. The reliability of the NICS-scan scheme in assessing aromatic behavior of some planar six-membered heteroaromatic systems has been explored recently by Seal and Chakrabarti [24]. These authors using homodesmotic reactions, aromatic stabilization energy (ASE) and NICS-scan to assess aromaticity of several systems concluded that homodesmotic reactions and NICS-scan agree for all systems but ASE does not for some of them. Morao and Cossio [25] introduced a simple ring current model to characterize the in-plane σ- and out-of-plane π-aromaticity in pericyclic reactions. In-plane aromaticity exhibits a maximum diamagnetic shielding σzzd at the ring center, while π-aromaticity exhibits two ring currents moving at a certain distance above and below the molecular plane. The σ-π separation in all metal aromatic and antiaromatic systems has critically been examined and computational strategies to unambiguously determine the aromaticity/antiaromaticity characteristics of such clusters have been presented by Pati et al. [26,27,28,29].
On the other hand, simple symmetry-based selection rules derived from group theory helped rationalize and predict ring currents to classify π systems [30,31,32,33]. Plotting of current-density maps was also suggested as a standard tool for the resolution of debates about aromaticity and for the interpretation of calculated chemical shift values [30,31,32,34,35,36]. Sundholm and coworkers [37,38,39] introduced the aromatic ring current shielding (ARCS) method to determine the strength of the induced ring current and consequently the relative degree of aromaticity in aromatic systems. In addition, they applied the gauge-including magnetically induced current (GIMIC) method to [c-Al4]2- and [c-Al4]4- clusters to elucidate the type of aromaticity in these aromatics [37,38,39]. Heine et al. [40,41] represented graphically with three-dimensional isosurfaces the induced magnetic field of cyclic molecules and showed that the appearance of the magnetic response characterizes aromatic, antiaromatic and non-aromatic molecules. They also showed that the z-component of the induced magnetic field is directly connected to NICSzz. Finally, the recently developed Anisotropy of the Current Induced Density (ACID) mapping was suggested as a better probe of aromaticity [42,43].
Recently, we showed that the NICSzz-scan curves in conjunction with symmetry-based selection rules for the lowest translationally- and rotationally-allowed transitions help rationalize and predict the orbital-type of aromaticity/antiaromaticity in magnetoresponsive three-membered rings of d- and f-block elements [44], and unequivocally probe the antiaromaticity in a wide range of antiaromatic organic and inorganic rings/cages [45]. Therefore, we thought it would be advisable to test the efficiency of the NICSzz-scan profiles in conjunction with symmetry-based selection rules to provide a clear picture of the orbital type (s-, p-, d-, f-orbital) of aromaticity in a wide range of aromatic organic and inorganic molecules, proving its general use as a powerful diagnostic tool of the orbital type of aromaticity/antiaromaticity. To achieve this goal, we calculated the NICSzz-scan profiles of a wide range of aromatic organic and inorganic molecules, whose magnetic-response properties have been exhaustively investigated and well-characterized in the framework of the aromaticity concept. The results obtained are thoroughly analyzed and presented herein.
2. Computational Details
In view of the good performance of density functional theory (DFT), we were inclined to use DFT calculations on the compounds we studied using the GAUSSIAN03 program suite [46]. The geometries of all species were fully optimized at the B3LYP level, using 6-311+G(d,p) basis set for all elements, except those of the fifth and sixth row elements for which the SDD basis set was used [47,48]. Full geometry optimization was performed for each structure using Schlegel’s analytical gradient method [49], and the attainment of the energy minimum was verified by calculating the vibrational frequencies that result in the absence of imaginary eigenvalues. All the stationary points were identified for minimum (number of imaginary frequencies NIMAG=0) or transition states (NIMAG=1). Magnetic shielding tensors were computed with the GIAO (gauge independent atomic orbitals) DFT method [50,51] as implemented in the GAUSSIAN03 series of programs [46] employing the B3LYP level of theory. Nucleus-Independent Chemical Shifts (NICS) were computed at the B3LYP level according to the procedure described by Schleyer et al. [11]. The computational protocol used is that applied in the previous studies of the aromaticity/antiaromaticity of most of the aromatic molecules discussed herein. The contribution to the induced ring current from the σ- and π-type diatropic and or paratropic translationally (Tx,y) and rotationally (Rz) allowed transitions was inferred from the estimated excitation energies based on symmetry and energy criteria. For a planar conjugated molecule in which the ring currents are induced by a magnetic field perpendicular to the molecular plane, three factors will determine the existence and strength of the contribution of an occupied-to-unoccupied orbital transition, ψn → ψp transition, namely symmetry, spatial distribution and energy. Symmetry determines whether a ψn → ψp transition contributes at all to the current density, spatial distribution of the orbitals determines whether an allowed transition moment will be large or small and the difference in orbital energies (excitation energies) affects the relative weight of a given transition moment in the total induced ring current. Thus, a ψn → ψp transition makes a conventionally diamagnetic (diatropic) contribution to current density if the direct product of representations Γ(ψn)× Γ(ψp)× Γ(Tx, Ty) contains a totally symmetric component, and a conventionally paramagnetic (paratropic) contribution to current density if the direct product Γ(ψn)× Γ(ψp)× Γ(Rz) contains a totally symmetric component. A mixed contribution results if both direct products have a totally symmetric component, while no contribution results if neither direct product has a totally symmetric component [30,31,32].
3. Results and Discussion
3.1. The NICSzz-scan profiles of 2e- and 6e-σ-aromatics
Let us first examine the shape and most salient features of the NICSzz-scan profiles of 2e- and 6e-σ-aromatics, the [c-H3]+ and c-H6 species being the prototypes [52,53,54,55]. It should be noted that the optimized structures of the [c-H4]2-, [c-H5]- and [c-H6] clusters were obtained under symmetry constrains and correspond to third, second and first order saddle points, respectively. The optimized Dnh symmetric [c-Hx]q aromatic rings are similar to the optimized structures reported previously [52,53,54,55]. The NICSzz-scan profiles of selected 2e- and 6e-σ-aromatics are visualized in Figure 1, while the main features of the NICSzz-scan curves along with the Rav values, e.g. the average distance of the ring current to the center of the ring, are compiled in Table 1.
The half-band width, e.g., the width of the symmetric curve at half the NICSzz value, of the NICSzz-scan curves depends on the ring radius, Rav, evaluated as , where n is the number of the nuclei of the ring, and Ri is the distance from the ring center to the nucleus i. For the rings where the 2p orbitals (radial 2p orbitals) participate also in the bonding, forming molecular orbitals contributing to the diatropic ring current of the σ-aromatics, Rav was evaluated as , where Zi is the atomic number of the i-atom, and a0 is the Bohr radius. The term 4a0/Zi corresponds to the maximum of the probability of the radial function 4πr2Rn,l2(r) for a 2p atomic orbital.
It can be seen that the NICSzz-scan curves are symmetric around the z-axis with the NICSzz values decaying rapidly and monotically with respect to R. The half-band widths of the NICSzz-scan curves for the σ-aromatics exhibiting s-orbital aromaticity range from 1.2 to 1.9 Å (Rav ranging from 0.509 to 0.992 Å) is characteristic of the in-plane pure σ-aromaticity due to cyclic delocalization of two 1s electrons over the three-membered ring in [c-H3]+ and six 1s electrons over the four-, five- and six-membered rings in [c-H4]2-, [c-H5]- and [c-H6]. Analogous symmetric sharp NICSzz-scan curves with half-band widths in the range of 0.9 to 2.1 Å are obtained for the [c-B3]-, [c-B3]+, [c-B2C], [c-C3]2+, [c-B4], [c-B4]2+, [c-B5]+ (Figure 1b,c), as well as for the transition states TS1 and TS2 (Figure 1d) of the concerted transfer of two hydrogen atoms from ethane to ethylene (double group transfer reactions) and the trimerization of acetylene to form benzene respectively. shown in Scheme I. The latter two NICSzz-scan curves have a slightly larger half-band width of 2.3 and 2.8 Å, respectively.
3.2. Magnetotropicity of the 2e- and 6e-σ-aromatics
In the [c-H3]+ species with D3h symmetry, the chief contribution to induced diatropic ring current arises from the Tx,y-allowed HOMO (a1′) → LUMO (e′) transition with excitation energy 18.74 eV. In the [c-H4]2- (D4h) cluster the chief contribution to induced diatropic ring current arises from the Tx,y-allowed HOMO,-1 (eu) → LUMO (b1g) and HOMO,-1 (eu) → LUMO+1 (a1g) transitions having excitation energies 6.33 and 6.77 eV respectively (notice that HOMO,-1 denotes HOMO and HOMO-1, LUMO+1,+2 denotes LUMO+1 and LUMO+2, etc.). In the [c-H5]- (D5h) cluster the chief contribution to induced diatropic ring current arises from the Tx,y-allowed HOMO,-1 (e1′) → LUMO+2 (a1′) with excitation energy 11.02 eV. Finally, in the [c-H6] (D6h) cluster the chief contribution to induced diatropic ring current arises from the Tx,y-allowed HOMO,-1 (e1u) → LUMO+2 (a1g) with excitation energy 14.14 eV. The higher excitation energy of the Tx,y-allowed transition in the [c-H3]+ cluster accounts well for the weaker aromatic character. It should be noted that all orbitals involved in the Tx,y-allowed transitions responsible for the induced diatropic ring current are σ-type MOs.
In the [c-B3]- and [c-C3]2+ species with D3h symmetry, the Tx,y-allowed HOMO (a1′) → LUMO+1,+2 (e′) excitations with excitation energies 3.42 and 4.25 eV respectively are responsible for the induced diatropic ring current. On the other hand, for the [c-B3]+ species the Tx,y-allowed HOMO,-1 (e′) → LUMO (a1′) excitation with excitation energy 4.16 eV contributes primarily to the induced diatropic ring current. The lower magnetic aromaticity of the [c-B3]+ species compared to that of the [c-B3]- 2e-σ-aromatic could be explained by the higher energy difference between the orbitals involved in the respective excitations (4.16 versus 3.42 eV). Both the e′ and a1′ MOs participating in the Tx,y-allowed transitions are σ-type MOs and are primarily responsible for the σ-aromaticity of the [c-B3]-.and [c-B3]+ species. The 1a2″ bonding HOMO-1 in [c-B3]- and [c-C3]2+ and HOMO in [c-B3]+ constructed from the overlap of the 2pz AOs of the ring atoms, being a pure π-bonding MO similar to the π-MO in the cyclopropenyl cation ([c-C3H3]+), is magnetically inactive and therefore cannot contribute to the induced diatropic ring current. The magnetic inactivity of the 1a2″ π-MO is further confirmed from the NICSzz-scan profile of the putative [c-B3]3+ species where, due to the removal of two electrons, the 1a2″ π-MO is vacant. The computed NICSzz(0) and NICSzz(1) values for the [c-B3]3+ species are -50.7 and -6.3 ppm, respectively, while the half-band width is 1.0 Å. However, the higher NICSzz(0) value of the [c-B3]+ species by 15.0 ppm compared to that of the [c-B3]3+ one is probably indicative for a small contribution of the 1a2″ π-MO to the diatropic ring current as well, supported by the Tx,y-allowed transitions HOMO-1 (a2″) → LUMO+5,+6 (e″) in [c-B3]-, HOMO (a2″) → LUMO+3,+4 (e″) in [c-B3]+ and HOMO-1 (a2″) → LUMO+2,+3 (e″) in [c-C3]2+ having relatively high excitation energies of 5.97, 8.77 and 11.20 eV, respectively. As the contribution of the 1a2″ π-MO to the diatropic ring current giving rise to π-aromaticity is small, the NICSzz-scan curve is still narrow with band width less than 3.0 Å. Boldyrev and co-workers [56,57,58] have identified [c-B3]- as 2+2 doubly aromatic system containing a π-type (1a2″) HOMO-1 and radial σ-type (2a1′) HOMO both molecular orbitals contributing to the induced diatropic ring current. The NICSzz-scan curve of the [c-B3]- species in conjunction with symmetry-based selection rules are in support of the double aromaticity, but with the induced π-diatropic ring current much weaker than the dominant induced σ-diatropic ring current. In effect the canonical orbital contributions to the out-of-plane NICS tensor component (CMO-NICSzz) for the [c-B3]- species estimated to be -16.1 ppm from the HOMO-4 (1a1′), -36.0 ppm from the degenerate HOMO-2,-3 (1e′), -11.5 ppm from the HOMO-1 (1a2″), the π-type MO and -11.7 ppm from the HOMO (2a1′) clearly illustrate the much weaker induced π-diatropic ring current (-11.5 ppm) than the dominant σ-diatropic ring current (-63.8 ppm). Therefore, the [c-B3]- species could be considered practically as a σ-aromatic species. Similarly, the estimated CMO-NICSzz values for the [c-B3]+ species found to be -16.6 ppm from the HOMO-3 (1a1′), -37.2 ppm from the degenerate HOMO-1,-2 (1e′) and only -12.3 ppm from the π-type HOMO (1a2″). Compare again the much weaker induced π-diatropic ring current (-12.3 ppm) than the dominant σ-diatropic ring current (-53.8 ppm). Finally, the estimated CMO-NICSzz values for the putative [c-B3]3+ species found to be -18.1 ppm from the HOMO-1 (1a1′) and -31.2 ppm from the degenerate HOMO,-1 (1e′) contribute exclusively to the induced σ-diatropic ring current. Notice that the 1a1′ and 1e′ bonding and nonbonding MOs respectively are formed primarily from the overlap of the filled 2s AOs of the boron atoms. It can be concluded that the CMO-NICSzz analysis data reinforce the efficiency of the NICSzz-scan curves and symmetry-based rules model to diagnose the orbital type of aromaticity of the aforementioned 2e- and 6e-σ-aromatics.
In the [c-B4] (D2h) molecule the Tx,y-allowed transitions contributing to the induced diatropic ring current are the HOMO-2 (b3u) → LUMO (ag), HOMO-3 (b2u) → LUMO (ag) and HOMO (ag) → LUMO+4 (b2u) excitations with excitation energies 4.58, 6.93 and 6.96 eV respectively. In addition the Rz-allowed HOMO (ag) → LUMO+3 (b1g) transition with excitation energy 6.27 eV contributes to the long range induced paratropic ring current (Figure 1c). The same holds true for the [c-B4]2+ (D4h) dicationic species where the Tx,y-allowed transitions contributing to the induced diatropic ring current are the HOMO-2,-3 (eu) → LUMO (a1g) and HOMO (a2u) → LUMO+2,+3 (eu) excitations with excitation energies 6.26 and 6.36 eV, respectively, while the Rz-allowed HOMO-1 (b2g) → LUMO+1 (b1g) transition with excitation energy 5.25 eV contributes to the long range induced paratropic ring current. Finally, in the [c-B5]+ (D5h) molecule the Tx,y-allowed transitions contributing to the induced diatropic ring current are the HOMO-1 (a2″) → LUMO,+1 (e1″) and HOMO (a1′) → LUMO+2,+3 (e1′) excitations with excitation energies 4.86 and 4.96 eV, respectively.
It is important to note that the dominant contribution to aromaticity of the aforementioned σ-aromatics due to the in-plane σ-MOs constructed from the overlap of the 2s and/or 2p AOs of the ring atoms is nicely reflected on the excellent linear correlation of the NICSzz(0) with the Rav of the rings (Figure 2).
In-plane aromaticity is a general feature of transition states associated with pericyclic reactions [25,56,57,58]. Cossio et al. [25] using a simple ring current model based on the NICS-scan profiles of the transition states was able to characterize the in-plane and π-aromaticity. For the transition states exhibiting in-plane aromaticity the NICS-scan curves showed a maximum at the molecular plane and decaying rapidly above and below of such plane. In contrast, in π-aromatic systems the NICS-scan curves showed a maximum at a certain distance above and below the molecular plane, which corresponds to the covalent radii of the involved atoms. The shapes of the NICSzz-scan curves of the transition states TS1 and TS2 for the concerted transfer of two hydrogen atoms from ethane to ethylene (double group transfer reactions) [59] and the trimerization of acetylene to form benzene [60,61] respectively (Figure 1d), provide a clear evidence for their in-plane σ-aromatic character. Indeed the NICSzz-scan curves are symmetric sharp curves with half-band widths less than 3 Å.
The predictive power of the NICSzz-scan curves on the orbital type of aromaticity was further corroborated by exploring the orbital type of aromaticity of the [c-B3H7]2- (Cs) [62] and [c-B3(μ2-P)3]2- (D3h) σ-aromatics, both involving a three-membered B3 ring. Indeed for both species the NICSzz-scan curves are symmetric sharp curves with half-band widths of 1.6 and 1.8 Å, respectively. The computed NICSzz(0), NICSzz(1) values of the [c-B3H7]2- and [c-B3(μ2-P)3]2- species are -46.1, -18.2 ppm and -38.9, -18.2, respectively. We also computed the NICSzz-scan curve of diborane, B2H6, which showed all the characteristics of the NICSzz-scan curves of pure σ-aromatics. It is a sharp symmetric curve with half-band width of 1.3 Å, with the NICSzz(0) and NICSzz(1) values being -20.8 and -6.1 ppm, respectively. On the basis of the linear relationship given in Figure 2 and using the Rav value of 0.717 Å, one obtains a NICSzz(0) value of -20.4 ppm, which is in excellent agreement with the computed NICSzz(0) value of -20.8 ppm.
In summary, all 2e- and 6e-σ-aromatics studied herein are characterized by sharp NICSzz-scan curves symmetric around the z-axis (perpendicular to the ring plane) with the NICSzz values decaying rapidly and monotically with respect to R, and having a half-band with approximately less than 3 Å.
3.3. The case of the [c-Li3]+ and cyclopropane 2e-σ-aromatics
For the [c-Li3]+ (D3h) cation, another 2e-σ-aromatic prototype, the NICSzz-scan curve, has also the features of the NICSzz-scan curves of the pure σ-aromatic, but its half-band width is greater than 3 Å (3.7 Å). The broadening of the NICSzz-scan curve of [c-Li3]+ σ-aromatic is probably due to the bigger size of the three-membered Li3 ring (Rav = 1.704 Å) that renders weaker the overlap of the 2s orbitals forming the 3c-2e bond. This is also mirrored on the estimated interaction energy between the Li2 and Li+ fragments (-45.66 kcal/mol), which is much smaller than the interaction energy between the H2 and H+ fragments to form the [c-H3]+ species (-154.03 kcal/mol). Another plausible reason for the broadening of the NICSzz-scan curve of [c-Li3]+ σ-aromatic might be the contribution of the intrinsic local circulation of electrons around the nuclei to the diatropic NICS values. The pure σ-aromaticity in the [c-Li3]+ cation was previously justified by Alexandrova and Boldyrev [63].
Considering the ring current model one would expect the versus z and the versus z curves to be similar:
According to quantum theory for the occurring in the molecular plane (z = 0) for the will have:
For two σ-aromatics exhibiting s-orbital aromaticity it can be easily written:
If we use as a reference σ-aromatic the prototype [c-H3]+ cation we obtain the following relationship:
On the basis of the above relationship we predict for the [c-Li3]+ cation a NICSmax value of -10.0 ppm very close to the estimated NICSzz(0) value of -11.2 ppm.
Cyclopropane being a saturated system was also found to be a strongly σ-aromatic molecule. Cyclopropane exhibits a ring current density induced by a perpendicular external magnetic field arising in the σ framework that is intense, diatropic and annular. The hallmarks of the σ-ring current in cyclopropane is reflected on the shape of the NICSzz-scan curve, which corresponds to a symmetric curve with a maximum at z = 0 and a half-band width around 3.2 Å. Moreover, the main feature of the strong diatropic circulation in [c-C3H6], which reaches maximal intensity (jmax = 0.121 a.u.) outside the line of carbon centers [64,65], accounts well for the slight broadening of the NICSzz-scan curve (half-band width > 3 Å). The calculation of the CMO-NICSzz values for cyclopropane in D3h symmetry showed that the Walsh orbitals HOMO,-1 (2e′), being σ-type MOs, have positive NICSzz value of 22.2 ppm, the HOMO-2,-3 (1e″), being π-type MOs, have also positive NICSzz value of 8.1 ppm and therefore induce σ- and π-type paratropic ring currents, respectively. On the other hand, the HOMO-5 (1a2″), a π-type MO, has negative NICSzz value of -8.7 ppm, which cancels the induced π-paratropic ring current from the 1e″ MOs. The remaining HOMO-4 (2a1′), HOMO-6,-7 (1e′) and HOMO-8 (1a1′) all being σ-type MOs, have negative NICSzz values of -5.4, -18.8 and -21.3 ppm respectively. The CMO-NICSzz results are nicely mirrored on the shape of the NICSzz-scan curve of cyclopropane. It is interesting to notice that Pelloni et al. [65] demonstrated that both the large negative NICS value and anisotropies of cyclopropane are mainly determined by the in-plane components rather than the more relevant out-of-plane contribution. The existence of σ-aromaticity in cyclopropane has recently been challenged by ab initio valence bond (VBSCF/cc-PVTZ) computations which revealed directly that the σ-aromatic stabilization energy of cyclopropane is, at most, 3.5 kcal/mol relative to propane [66]. However, such small energy difference raised the question whether the cyclopropane is really the σ-aromatic paradigm.
3.4. The NICSzz-scan curves of the M3(μ2-CO)3(CO)3 (M = Ni, Pd, Pt) molecules predict their σ-aromaticity
Let us now explore the efficiency of the NICSzz-scan profiles to predict the pure σ-aromaticity characterizing the M3(μ2-CO)3(CO)3 (M = Ni, Pd, Pt) molecules. Recently, R. B. King [67] applied the σ-aromaticity model used for cyclopropane to Group 8 trinuclear metal-carbonyl complexes, [M3(CO)12] (M = Fe, Ru, Os). The σ-aromaticity of these molecules was further verified by a structural and NICS analysis reported recently [68]. According to this model the bonding in the M3 triangles is composed by a core 3c-2e bond of Hückel topology formed by overlap of inward pointing radial hybrid orbitals of M and a surface (in-plane) 3c-4e bond of Möbius topology formed by tangential p-orbitals of M. Notice that for the M3(μ2-CO)3(CO)3 (M = Ni, Pd, Pt) molecules the presence of the μ2-CO ligands precludes the 3c-4e Möbius surface bonding orbitals. The NICSzz-scan curves of the Pt3(μ2-CO)3(CO)3 molecule along with the molecular orbital energy level diagram, symmetries, 3D molecular orbital pictures and lowest Tx,y-allowed transitions contributing to the diamagnetic ring current are visualized in Figure 3, while the most salient features of the NICSzz-scan curves along with the half-band widths and Rav values of the rings of the M3(μ2-CO)3(CO)3 (M = Ni, Pd, Pt) molecules are compiled in Table 2.
It can be seen that the NICSzz-scan curves of the M3 (M = Ni, Pd, Pt) clusters are sharp symmetric curves with half-band width less than 2 Å, illustrating their pure σ-aromatic character. The broadening of the NICSzz-scan curves of the M3(μ2-CO)3(CO)3 (M = Ni, Pd, Pt) molecules (half-band width of 2.7-4.6 Å), could be attributed to the contribution of the diatropic NICS values of the strongly σ-aromatic c-M2C rings to the diatropic NICS values of the M3 core rings.
3.5. The NICSzz-scan curves of triangular main group clusters exhibiting double (σ + π)-aromaticity
Let us further examine the NICSzz-scan curves of three-membered rings of main group atoms, which have been extensively studied and characterized as double (σ + π)-aromatics. All these molecules showed NICSzz-scan curves symmetric around the z-axis with the NICSzz values decaying monotonically with respect to R, but with half-band width greater than 3 Å. The NICSzz-scan profiles in the form of NICSzz/NICSzzmax versus R curves for selected triangular main group clusters exhibiting double (σ + π)-aromaticity are given in Figure 4, while the most salient features of the NICSzz-scan curves along with the Rav values of the rings are compiled in Table 3.
3.5.1. Magnetotropicity of [c-Be3]2- and [c-Mg3]2- clusters
The 1A1′(σ) ground-state occupation in D3h symmetry of 8 valence electrons for the [c-Be3]2- and [c-Mg3]2- clusters was found to be HOMO-3 (a1′), HOMO-1,2 (e′), HOMO (a1′). The lowest Tx,y-allowed transitions are the HOMO (a1′) → LUMO+1,+2 (e′) and HOMO (a1′) → LUMO+3,+4 (e′) with excitation energies of 1.22 and 1.76 eV for [c-Be3]2- cluster and 0.67 and 0.94 eV for [c-Mg3]2- cluster respectively. In these transitions a σ-type (a1′) MO is involved in the excitation inducing a σ-diatropic ring current and therefore [c-Be3]2- and [c-Mg3]2- clusters in their 1A1′(σ) ground-states exhibit σ-aromaticity in line with the results concerning the tuning of aromaticity in trigonal alkaline earth metal clusters reported recently [69]. The NICSzz-scan curves of the [c-Be3]2- and [c-Mg3]2- clusters in their 1A1′(σ) ground-states have the features of the NICSzz-scan curves of pure σ-aromatics but their half-band width is greater than 3 Å (3.7 Å). The broadening of the NICSzz-scan curves could be explained as in the case of the [c-Li3]+ σ-aromatic.
We have also investigated the aromaticity/antiaromaticity of the 1A1′(π) electronic states of the [c-Be3]2- and [c-Mg3]2- clusters found as local minima at 4.6 and 5.3 kcal/mol higher in energy than the 1A1′(σ) ground states at the B3LYP/6-311+G(d) level in line with the results reported by Matito et al. [68]. The 1A1′(π) electronic state occupation in D3h symmetry of 8 valence electrons for the [c-Be3]2- and [c-Mg3]2- clusters was found to be HOMO-3 (a1′), HOMO-1,2 (e′), HOMO (a2″). It can be seen that the lowest Tx,y-allowed transitions are the HOMO-1,2 (e′) → LUMO (a1′) and HOMO (a2″) → LUMO+5,6 (e″). In the first transition a σ-type (a1′) MO is involved in the excitation inducing a σ- diatropic ring current, while in the second transition a π-type (a2″) MO is involved inducing a π-diatropic ring current.
In the [c-Be3]2- cluster the σ-diatropic ring current overwhelms the π-diatropic ring current because of the lower excitation energy (1.59 eV) of the first transition relative to the second one (1.67 eV). This observation accounts well for the broadening of the NICSzz-scan curve of the [c-Be3]2- cluster having its maximum at z = 0. On the other hand, in the [c-Mg3]2- cluster the π-diatropic ring current is much stronger than the σ-diatropic ring current because of the much lower excitation energy (0.94 eV) of the π-type transition relative to the σ-type one (2.19 eV). It is obvious then why the NICSzz-scan curve of the 1A1′(π) [c- Mg3]2- exhibits two maxima at a certain distance above and below the molecular plane. Kuznetsov and Boldyrev showed that the unusual [c-Mg3]2- trigonal planar cluster exhibits an extraordinary type of aromaticity, where π-aromaticity occurs without initial formation of the σ-framework [70].
3.5.2. Magnetotropicity of the [c-E3]- (E = Al, Ga, In, Tl), [c-E3]2- (E = C, Si, Ge), [c-E3]+ (E = N, P, As, Sb, Bi) and [c-E3] (E = S, Se, Te, Po) rings
The NICSzz-scan curves of the [c-Al3]-, [c-Ga3]-, [c-In3]- and [c-Tl3]- clusters looks like the mirror image of a typical scan of double (σ + π)-aromatic molecules (Figure 4b). The multiple aromaticity of the [c-X3]- (X = Al, and Ga) clusters was recently verified by Boldyrev et al. [56,62] through a molecular orbital analysis, and by Dixon et al. [71] by estimating the resonance energy using high-level ab initio quantum chemical calculations. The estimated excitation energies of the lowest Tx,y- and Rz-allowed transitions of the three-membered rings of main group atoms are compiled in Table 4.
The lowest Tx,y-allowed transitions of the three-membered rings of main group atoms are the a1′ → e′ (σ-type) and a2″ → e″ (π-type) excitations. The coexistence of both the σ- and π-type transitions accounts well for the double (σ + π)-aromatic character of the aforementioned rings. The extent of contribution of the two types of transitions to the induced diatropic ring current is determined by the excitation energies of the σ- and (π-type) transitions (Table 4). Furthermore, removal of two electrons from the [c-Al3]- and [c-Ga3]- clusters affords [c-Al3]+ and [c-Ga3]+ respectively, where the lowest σ-type transition disappears and therefore the π-diatropic ring current exceeds the σ-type one, a fact imprinted on the NICSzz-scan curves of [c-Al3]+ and [c-Ga3]+ clusters which exhibit two maxima at a certain distance above and below the molecular plane (Table 3). The estimated excitation energies also illustrate that the induced π-diatropic ring current slightly overwhelms the σ-diatropic ring current in the [c-N3]+ and [c-P3]+ clusters, which unequivocally exhibit double (σ + π)-aromaticity. In effect, the NICSzz-scan curves (Fig. 4d) for the [c-N3]+ and [c-P3]+ clusters are typical for double (σ + π)-aromatic molecules with slightly stronger π- than σ-aromatic character. According to the estimated excitation energies the [c-As3]+, [c-Sb3]+ and [c-Bi3]+ clusters are doubly (σ + π)-aromatic molecules with slightly higher π- than σ-aromatic character. Finally, the NICSzz-scan curves of the 18 valence electron [c-S3], [c-Se3], [c-Te3] and [c-Po3] clusters are also typical for double (σ + π)-aromatics with varying magnitude of π- and σ-aromatic character (Fig. 4e). For [c-S3] and [c-Se3] the π-component should be slightly stronger than the σ-one, while the opposite is true for the [c-Te3] and [c-Po3] clusters. This is compatible with the excitation energies of the lowest Tx,y-allowed transitions for the neutral triangular chalcogen clusters (Table 4). Computed GIAO-SCF NICS and localized (LMO) NICS analysis of the An, (A = O, S, Se; n = 3-6) clusters carried out by Schleyer et al. [72] indicated that the σ-ring electrons are chiefly responsible for the zigzac NICS(0) behavior of the An rings with the A3 clusters being aromatic. It is important to notice that the double (σ + π)-aromaticity of all three-membered rings of main group atoms studied is reflected on the excellent linear correlation of the NICSzz(0) values with the Rav values of the rings shown in Figure 5.
In summary all triangular main group clusters exhibiting double (σ + π)-aromaticity studied herein are characterized by broad NICSzz-scan curves with half-band width >3 Å indicating double (σ + π)-aromaticity with comparable contribution to the induced total diatropic ring current from the σ- and π-diatropic ring currents.
3.6. The NICSzz-scan curves of four- and five-membered rings of main group atoms exhibiting double (σ + π)-aromaticity
Next we examined the NICSzz-scan curves of four- and five -membered rings of main group atoms, which have been extensively studied and characterized as double (σ + π)-aromatics. The NICSzz-scan profiles in the form of NICSzz/NICSzzmax versus R curves of the [c-B4]2-, [c-Al4]2-, [c-Ga4]2-, [c-In4]2-, [c-Tl4]2-, [c-Si4]2+ and [c-Mg4]2+ clusters given in Figure 6 are symmetric around the z-axis with maximum at z = 0 and half-band width (except [c-B4]2-) greater than 3 Å.
The NICSzz-scan curves of the [c-Al4]4- species as well as of the five-membered pnictogen [c-Pn5]- (Pn = N, P, As, Sb, Bi) clusters exhibit two maxima at a certain distance above and below the molecular plane (Figure 6), which are typical for double (σ + π)-aromatics where the π-diatropic ring current overwhelms the σ-type one. The most salient features of the NICSzz-scan curves of all these clusters along with the Rav values of the rings are compiled in Table 5.
3.6.1. Magnetotropicity of the [c-E4]2- (E = B, Al, Ga, In, Tl), [c-Si4]2+, [c-Mg4]2+, [c-Li2Mg2], [c-C5H5]-, and [c-E5]- (E = N, P, As, Sb, Bi) rings
Let us now analyze the shapes of the NICSzz-scan curves of the four- and five-membered rings of main group atoms in relation with the relative ratio of the induced σ- and π-diatropic ring currents. The ground-state (1A1g) occupation in D4h symmetry of 14 valence electrons for the [c-Al4]2-, [c-Ga4]2-, [c-In4]2- and [c-Tl4]2- clusters was found to be 1a1g21eu41b1g21b2g22a1g21a2u2. In [c-In4]2- cluster there is an inversion of the 1a2u and 2a1g. For the [c-B4]2- cluster the electron occupation follows a different order, e.g. 1a1g21eu41a2u21b2g22a1g21b1g2. The computed excitation energies of the main translational transitions of [c-Al4]2-, [c-Ga4]2-, [c-In4]2-, [c-Tl4]2- and [c-Si4]2+ clusters are given in Table 6.
Based on the estimated excitation energies of the lowest Tx,y- allowed σ- and π-type 3ag → 3eu (1.42 eV) and 1a2u → 1eg (4.74 eV) transitions compiled in Table 6 we predict that the [c-B4]2- posses rather pure σ-aromatic character, which is also mirrored on the shape of the symmetric sharp (half-band width = 2.3 Å) NICSzz-scan curve (Figure 6a). The same holds true for the [c-Mg4]2+ and [c-Li2Mg2] clusters where the occupation of 6 valence electrons was found to be 1eu41a1g2 for [c-Mg4]2+ (D4h) and 1ag21b1u21b2u2 for [c-Li2Mg2] (D2h). The estimated excitation energies of the lowest Tx,y-allowed transitions found to be 1.12 eV for the σ-type eu → a1g in [c-Mg4]2+ (D4h) and 2.06 eV for the σ-type b2u → a1g and 4.92 eV for the π-type ag → b3u in [c-Li2Mg2] (D2h) accounts well for the shape of the NICSzz-scan curves (Fig. 6a). As in the case of the [c-Li3]+ (D3h) cation the broadening of the NICSzz-scan curves of [c-Mg4]2+ and [c-Li2Mg2] clusters is probably due to the bigger size of the four-membered rings (Rav = 2.213 and 2.066 Å in c-Mg4]2+ and [c-Li2Mg2] rings respectively) and/or to the contribution of the intrinsic local circulation of electrons around the nuclei to the diatropic NICS values. The pure σ-aromaticity in the [c-Mg4]2+ and [c-Li2Mg2] clusters was previously justified by Alexandrova and Boldyrev [57]. In all these double (σ + π)-aromatic clusters the σ-aromatic character overwhelms the π-aromatic one enforcing the broadening of the NICSzz-scan curves, which have half-band widths greater than 4 Å (Fig. 6). Of particular importance is the excellent linear correlation of the NICSzz(0) values of the four-membered rings of 13 group atoms with the Rav values of the rings (Figure 7).
The double (σ + π)-aromaticity of the [c-Al4]2- cluster was first introduced in a seminal work published in Science by Boldyrev et al. [73] and subsequently justified by the use of different criteria of aromaticity, such as the induced magnetic field analysis [74], the aromatic ring current shielding (ARCS) [39,75], the ring current maps [76,77,78,79], NICS [80], resonance energy (RE) estimations [39,81], valence bond (VB) calculations [82], bifurcation analysis of electron localization function (ELF) [83], conceptual DFT descriptors [84], and quantum theory of atoms in molecules (QTAIM) calculations [85]. It should also be noticed that Sundholm et al. [75] based on ARCS calculations on the [c-Al4]2-, [c-Ga4]2-, [c-In4]2- and [c-Tl4]2- indicated that all these species are aromatic, the calculated ring-current susceptibilities and the (ring radius) found to be 16.2 nA/T (194 pm), 17.2 nA/T (190 pm), 19.3 nA/T (210 pm) and 18.4 nA/T (241 pm) respectively, at the 3-VE+sp2df HF level.
The NICSzz-scan curve of the [c-Al4]4- cluster exhibits two maxima at a distance of 0.9 Å above and below the molecular plane (Fig. 6a), which is typical for double (σ + π)-aromatics where the π-type diatropic ring current exceeds slightly the σ-type one. In effect, this is compatible with the estimated excitation energies of the Tx,y-allowed σ-type ag → b2u (1.27 eV) and π-type b3u → ag (1.58 eV) and ag → b3u (2.05 eV) transitions. Notice the contribution of two Tx,y-allowed π-transitions to the π-diatropic ring current in [c-Al4]4- cluster.
The NICSzz-scan curve of the [c-Si4]2+ cluster, which is isoelectronic to [c-Al4]2- one, is typical for double (σ + π)-aromatics where the σ-type diatropic ring current overwhelms remarkably the π-type one. The curve is symmetric around the z-axis with a half-band width of 3.1 Å (Figure 6a). The stronger σ- than the π-aromatic character in [c-Si4]2+ cluster is reflected on the estimated excitation energies (Table 6) for the a1g → eu, b2g → eu, b1g → eu σ-type transitions, compared to the a2u → eg π-type transition. The [c-Si4]2+ has previously been characterized as triply aromatic, the triple aromaticity coming from the πp-z-, σp-r- and σp-t-MOs (p-z, p-r and p-t are the vertical, radial and tangential 3p AOs of the Si atoms in the ring).
Finally, the NICSzz-scan curves of the 26 valence electron five-membered pnictogen [c-Pn5]- (Pn = N, P, As, Sb, Bi) clusters in D5h symmetry are typical for double (σ + π)-aromatics with the π-aromatic character exceeding at a different extent the σ-aromatic character (Figure 6b). There is an increase of the π-aromatic character as one goes down in the 15 group pnictogen atom five-membered rings. The NICSzz-scan curve of the five-membered cyclopentadienyl [c-C5H5]- ligand, a prototype of π-aromatic organic molecule, is also shown in Figure 6b.
The estimated excitation energies of the lowest Tx,y-allowed σ- and π-type transitions of the five-membered pnictogen [c-Pn5]- (Pn = N, P, As, Sb, Bi) rings in D5h symmetry tabulated in Table 7 account well for the shape of the NICSzz-scan curves, which are characteristic for the double (σ + π)-aromaticity of the rings.
It can be seen that all five-membered pnictogen [c-Pn5]- (Pn = N, P, As, Sb, Bi) rings are double (σ + π)-aromatic with stronger π- than σ-aromatic character. The relative strength of the π- and σ-aromatic character of the rings can be expressed qualitatively by the ratio of the excitation energies of the π- and σ-type Tx,y-allowed transitions which were found to be 1.13, 1.37, 1.42, 1.73 and 1.88 for the [c-N5]-, [c-P5]-, [c-As5]-, [c-Sb5]- and [c-Bi5]- respectively. It is interesting to notice that for the series of the five-membered pnictogen [c-Pn5]- clusters the NICSzz(0) values are linearly correlated with the Rav values of the rings (Figure 8) following a similar pattern with that established for other series of aromatic clusters discussed herein.
The double (σ + π)-aromaticity of the pentagonal structures (D5h symmetry) of the [c-Pn5]- clusters was justified by Boldyrev et al. [86] using molecular orbital and photoelectron spectra analysis and compared with the aromaticity of the isovalent cyclopentadiene, [c-C5H5]- ligand. More recently, De Proft et al. [87] calculated the maps for the current density of the [c-P5]- and [c-As5]- clusters, which showed an apparently classic example of a diatropic ring current, but not solely π current. The current density at z = 1.0a0 has approximately equal contribution from the four electrons in the 2e1″ HOMO (π-MO) and the four electrons in the 6e2′ HOMO-2 (σ-MO). The pattern of the current maps and orbital contributions of the [c-P5]- and [c-As5]- clusters are compatible with the NICSzz-scan profiles.
In summary the four- and five-membered rings of main group clusters exhibiting double (σ + π)-aromaticity studied herein are characterized, as in the case of the three-membered rings, by broad NICSzz-scan curves with half-band width approximately > 3 Å indicating double (σ + π)-aromaticity with comparable contribution to the induced total diatropic ring current from the σ- and π-diatropic ring currents.
3.7. The NICSzz-scan curves versus the orbital-type of aromaticity in organic molecules
We next tested the predictive power of the NICSzz-scan curves of the orbital-type of aromaticity for selected organic double (σ + π)- and pure π-aromatic molecules computed at the B3LYP/6-311+G(d,p) level. The NICSzz-scan profiles in the form of NICSzz/NICSzzmax versus R curves of the selected organic molecules are depicted schematically in Figure 9, while the main features of the NICSzz-scan curves of all molecules along with the Rav values of the rings are compiled in Table 8. The estimated excitation energies of the lowest Tx,y-allowed σ- and π-type transitions of the selected aromatic organic molecules are compiled in Table 9, Table 10 and Table 11.
3.7.1. The NICSzz-scan curves and magnetotropicity of the [C3H3]+, C6H6, [C6H6]4-, [C6H6]6-, o-C6H4, m-C6H4, and p-C6H4 molecules
For the [C3H3]+, C6H6, [C6H6]4-, [C6H6]6-, o-C6H4 (o-benzyne), m-C6H4 (m-benzyne), and p-C6H4 (p-benzyne) molecules the estimated excitation energies of the lowest Tx,y- and Rz-allowed σ- and π-type transitions are indicative of their double (σ + π)-aromatic character with varying σ-/π- ratio.
For the [C3H3]+, C6H6, [C6H6]4- and [C6H6]6- molecules the contribution to the ring current from π-delocalized electrons is significantly higher than from σ-delocalized electrons (compare the excitation energies of the σ- and π-type transitions given in Table 9), a fact that is clearly reflected on the shape of the NICSzz-scan curves (Figure 9a). It is important to notice that the occupation of the antibonding MOs of benzene with four and six electrons affording the anionic [C6H6]4- and [C6H6]6- species, which correspond to local minima in the PES, increase the aromaticity with respect to benzene molecule, despite the bonding from the bonding MOs will be offset by the effect of the occupied antibonding counterparts. This observation illustrates that the dynamic magnetic response of the aromatic species expressed by the NICS values could not be outweighed from the occupation of the antibonding MOs, since the induced ring current from the occupied bonding MOs could not be equal and of opposite sign with that from the occupied antibonding counterparts. However, the anionic [C6H6]4- and [C6H6]6- species should be unstable with respect to electron autodetachment in the gas phase and the results for these metastable species should be approached with caution as it is the case of the “all-metal aromatic” [c-Al4]2- dianions [88].
For the singlet o- and m-benzyne isomers and the open-shell singlet p-benzyne the contribution to the ring current from π-delocalized electrons is also higher than from σ-delocalized electrons (compare the excitation energies of the σ- and π-type transitions given in Table 9). However, in the three isomers the induced paratropic current from π-delocalized electrons via the Rz-allowed transitions diminishes the induced π-diatropic ring current, thus rendering the contribution to the induced ring current from σ-delocalized electrons higher than from π-delocalized electrons, which is clearly reflected on the shape of the NICSzz-scan curves (Figure 9a). It should be noticed that the relative aromaticities of the singlet o- and m-benzyne isomers and the open-shell singlet p-benzyne have previously been probed with a diverse aromaticity indicators [89]. Unlike benzene, the most negative NICS value occurs in the ring planes of the benzynes, which might indicate that the largest diamagnetic ring current is in the molecular plane. In effect, this is mirrored on the shapes of the NICSzz-scan curves reported herein, which once again correctly predict the orbital-type of aromaticity in the isomeric benzyne molecules.
3.7.2. The NICSzz-scan curves and magnetotropicity of the c-C6, [c-C6]4+, [c-C6H3]+, c-C5BH3, c-C7 and c-C10 molecules
The double (σ + π)-aromaticity is also present in the c-C6, [c-C6]4+, [c-C6H3]+, c-C5BH3, c-C7 and c-C10 molecules. The double (σ + π)-aromaticity of the c-C6 cluster was first discussed by Schleyer et al. [16]. It was claimed that the c-C6 cluster is also σ-antiaromatic due to cyclic delocalization of its four lone-pair electrons. In effect, the estimated excitation energies of the lowest Tx,y- and Rz-allowed transitions in the c-C6 cluster (Table 10) indicate a strong induced σ-diatropic ring current, which is partly outweighed from the induced σ-paratropic ring current due to the σ-Rz transition but still the induced σ-diatropic ring current seems to be stronger than the π-diatropic ring current, which is compatible with the shape of the NICSzz-scan curve (Figure 9b). The estimated excitation energies of the lowest Tx,y-allowed transitions in the cationic [c-C6]4+ species clearly indicate that it is double (σ + π)-aromatic with σ-aromatic character much stronger than the π-aromatic one in line with the shape of the NICSzz-scan curve (Figure 9b) having a half-band width of 2.1 Å, which is typical for completely σ-aromatic molecules.
The NICSzz-scan curves of the [c-C6H3]+ and c-C5BH3 have the shape of the typical curves for double (σ + π)-aromatic molecules with σ-aromatic character much stronger than the π-aromatic one. Actually, the estimated excitation energies of the [c-C6H3]+ and c-C5BH3 molecules (Table 10) are indicative of the much stronger σ- than π-aromatic character. For the c-C5BH3 molecule the contribution to the diatropic ring current from π-delocalized electrons is further decreased from the induced paratropic ring current due to the π-Rz transition. Hofmann and Berndt [90] showed that the the NICS(1) values of the double aromatic isoelectronic [c-C6H3]+ and c-C5BH3 molecules are significantly larger than that of the benzene molecule due to the contribution of an additional aromatic system, namely the σ-aromatic component.
Akin to c-C6 (D3h) the c-C10 (D5h) cluster was also proved to be a double (σ + π)-aromatic molecule with enhanced aromaticity due to an additional diatropic subsystem (the diatropic in-plane radial system) [16]. This is consistent with the estimated excitation energies of the σ- and π-type transitions (Table 10), which indicate that the σ-aromatic component should be larger than the π-aromatic one and is mirrored on the shape of the NICSzz-scan curve shown in Figure 9b. Schleyer et al. [16] showed that the c-C7 (C2v) cluster has mixed aromatic (π) and antiaromatic (radial) systems. According to our calculations for the c-C7 (C2v) cluster the lowest σ- and π-Tx,y-allowed transitions have much lower excitation energies than the σ-Rz-allowed ones (Table 10) and therefore it is expected to be a double(σ + π)-aromatic molecule involving stronger π- than σ-aromatic component, in line with the shape of the NICSzz-scan curve (Figure 9b). The calculation of the CMO-NICSzz values for the c-C7 (C2v) cluster showed that six σ-type MOs have positive NICSzz values of totally 122.6 ppm and the remaining eighteen MOs have negative NICSzz values of totally -154.9 ppm, thus rendering the c-C7 (C2v) cluster aromatic. The σ-type MOs (totally fifteen) contribute to diatropic CMO-NICSzz values by -118.3 ppm and the π-type MOs (HOMO-3,-4,-6) contribute to diatropic CMO-NICSzz values by -36.6 ppm. It is evident that the net effect would be a stronger π- than σ-aromatic component as it is reflected on the shape of the NICSzz-scan curve of c-C7 (C2v) cluster. It should be noticed that the NICSzz-scan curve of c-C7 (C2v) cluster is similar to that of the benzene molecule. This could be understood on the basis of the CMO-NICSzz analysis data for benzene, which showed that five σ-type MOs have positive NICSzz values of totally 85.9 ppm and the remaining sixteen MOs have negative NICSzz values of totally -100.4 ppm. Among these MOs, nine σ-type MOs contribute to diatropic CMO-NICSzz values by -39.0 ppm and seven π-type MOs contribute to diatropic CMO-NICSzz values by -61.4 ppm. It is again evident that the net effect would be the dominant π-aromatic component for benzene.
3.7.3. The NICSzz-scan curves and magnetotropicity of Fulvalene2-, Phenalenyl+, Oxazole, and [c-C8H24]2+molecules
Very recently Mills and Benish [91] evaluated the aromaticity of the tetrabenzo[5.5]fulvalene dianion through magnetic criteria and found that all rings exhibit appreciable aromaticity. The NICSzz-scan curve (Figure 9c) justifies the aromaticity of the tetrabenzo[5.5]fulvalene dianion, indicating that it is totally due to the π-aromatic system.
The lowest Tx,y-allowed transitions of the tetrabenzo[5.5]fulvalene dianions are only of the π-type (Table 11). The same holds also true for the 12π electron phenalenyl cation which supports diatropic perimeter ring current. The NICSzz-scan curve (Figure 9c) of the phenalenyl cation is typical for aromatic molecules exhibiting pure π-aromatic character. Very recently, Fowler et al. [92] using the ipsocentric approach predicted the diatropicity of the phenalenyl cation which arises mainly from the contribution of the doubly degenerate 2e HOMO-2 pair.
Morao and Cosio [25] applied a simple ring current model for describing the in-plane aromaticity in pericyclic reactions to oxazole, a five-membered π-aromatic heterocycle containing two heteroatoms. They found that this kind of aromatic compound can be described using two ring currents. The NICSzz-scan curve (Figure 9c) of oxazole in conjunction with the estimated excitation energies for the lowest Tx,y-allowed σ- and π-type transitions (Table 11) clearly indicate the double (σ + π)-aromatic character of oxazole involving stronger π- than σ-aromatic component.
Finally, both the shape of the NICSzz-scan curve (Figure 9c) and the estimated excitation energies for the Tx,y-allowed transitions (Table 11) for the hypothetical [c-C8H24]2+ species with delocalized electrons in a cyclic array of p orbitals arranged tangentially in σ-bonding fashion are in support of the pure σ-aromatic character in line with the ring-current maps calculated by Fowler et al. [93] It should be noted that the [c-C8H24]2+ species correspond to a sixth order saddle point in the PES. The broadening of the NICSzz-scan curve is what it is expected for an eigth-membered ring with Rav = 3.072 Å.
3.8. Predicting the peculiar aromaticity of borazine
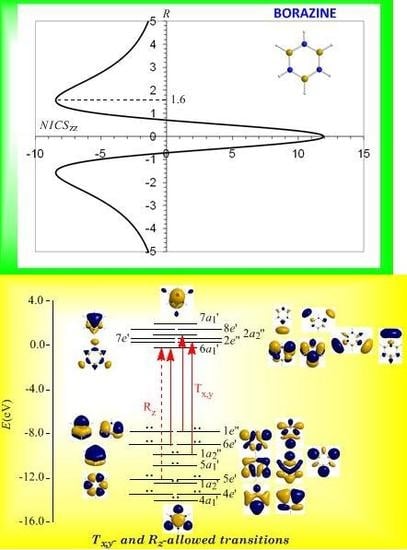
Borazine, c-B3N3H6 - the boron-nitrogen six-membered ring compound often referred to as “inorganic benzene - has been subject of controversial discussions with respect to the aromaticity of this molecule. Very recently Islas et al. [94] in their publication entitled Borazine: to be or not to be aromatic based on a detailed analysis of two molecular fields, the induced magnetic field (Bind) and the electron localization function (ELF) indicated that borazine can be described as a π-aromatic compound but it is not a globally aromatic species, since the delocalized system is not as homogeneous as that of benzene. The induced magnetic field in borazine is weaker than that of benzene but nevertheless pronounced and long-ranged. The NICSzz-scan curve of borazine shown in Figure 10 clearly demonstrates the long-range π-aromaticity and the in-plane σ-antiaromaticity of borazine. It shows a maximum negative (diatropic) NICSzz value of -8.4 ppm at z = 1.6 Å above and below the ring and a maximum positive (paratropic) NICSzz value of 12.0 ppm at z = 0.
The estimated excitation energies of the lowest Tx,y-allowed transitions are for the π-type e″ → a2″ and a2″ → e″ excitations 8.57 eV and 10.19 eV respectively and for the σ-type e′ → a1′ excitation 9.05 eV. The latter is outweighed by the Tx,y-allowed a2′ → a1′ transition with excitation energy 12.04 eV. The relatively high excitation energies lead to weak induced π-diatropic ring currents, which nicely explain the substantial decrease of the π-electron delocalization relative to benzene (compare the NICSzzmax values of -8.4 and -29.2 ppm, for the borazine and benzene molecules, respectively). It is interesting to notice that the local contributions of the σ electrons in borazine according to the calculations carried out by Islas et al. [94] generate a short-range response and a paratropic (antiaromatic) region in the center of the ring, while the π-orbital contribution to Bind shows the typical response of an aromatic system. This is exactly the topology of the total ring currents represented by the NICSzz-scan curve of borazine (Figure 10).
3.9. Applying the magnetic NICSzz-scan criterion to diagnose aromaticity/antiaromaticity in the non-planar corannulene, corannulene dianion and sumarene molecules
The bowl-shaped corannulene, C20H10, corannulene dianion, [C20H10]2- and sumarene, C21H12, polycyclic hydrocarbons, which share a fullerene substructure, have been studied extensively in recent years [95,96]. Such studies have emphasized the description of their aromaticity and electron interactions on curved surfaces from both the experimental and theoretical perspectives.
The counter-rotating ring currents in coronene and corannulene have been investigated by Jenneskens et al. [97] Very recently Monaco and Zanasi [98] calculated the π current of aromatic polycyclic hydrocarbons placed in a uniform magnetic field and found substantial localization on subunits in some cases. Moreover, Zanasi et al. [99] in their recent publication entitled Magnetic Euripi in Corannulene computed ab initio current densities for corannulene dianion, dication and tetraanion and found large π-ring currents with respect to benzene which undergo remarkable changes in response to variation in the oxidation state. The three corannulene ions along with the neutral species constitute a full set that spans all of the possible patterns of rim and hub circulations. Thus, for the neutral corannulene the magnetotropicity corresponds to paratropic/hub-diatropic/rim pattern, while for the corannulene dianion to diatropic/hub-paratropic/rim pattern.
The NICSzz-scan curves of corannulene, corannulene dianion and sumanene computed for both the inner (hub) and outer (rim) rings are shown in Figure 11.
The NICSzz-scan curve of the inner (hub) five-membered ring of corannulene clearly demonstrates the antiaromatic character of the ring (maximum NICSzz(0.2) value of 54.5 ppm) in line with the computed current-density maps for the σ, π, and total (σ + π) electrons at 1 a0 above the molecular planes showing a paramagnetic counter-circulation on the inner hub [97,98,99]. The NICSzz-scan curve of the inner (hub) five-membered ring of corannulene is similar to the NICSzz-scan curve of the highly antiaromatic cross-conjugated annelated five-membered ring system, acepentalene, [45] corresponding to relatively narrow curve with half-band width of 1.3 Å. On the other hand, the NICSzz-scan curve of the outer (rim) six-membered rings of corannulene exhibiting two maxima at a certain distance above and below the molecular plane and having NICSzz(0) value close to zero indicates their pure π-type aromatic character (minimum NICSzz(1.1) and NICSzz(-1.3) values of -24.1 and -17.1 ppm respectively), which is also consistent with the computed current-density maps showing a clear diamagnetic circulation around the outer rim [97,98,99]. It is important to be noticed that both the central paramagnetic (paratropic) and the outer diamagnetic (diatropic) induced ring currents are enhanced inside the bowl but depleted on the outside as a result of ring current superpositions.
The NICSzz-scan curve of the inner (hub) five-membered ring exhibiting two maxima at a certain distance above and below the molecular plane and having NICSzz(0) value close to zero clearly demonstrate the high π-aromaticity (diatropicity) of the hub (minimum NICSzz(0.4) value of -293.8 ppm). The NICSzz-scan curve of the outer (rim) six-membered rings of corannulene dianion is typical of highly in-plane antiaromatic systems [45] (maximum NICSzz(0.2) value of 216.6 ppm). Notice again that the inner (hub) diamagnetic (diatropic) induced ring current is strongly enhanced inside the bowl but depleted on the outside. It is important to be noted that the NICSzz-scan curves of corannulene and corannulene dianion correctly predict the paratropic/hub-diatropic/rim and diatropic/hub-paratropic/rim patterns respectively obtained by the ab initio computed probability current densities [99].
The NICSzz-scan curve of the inner six-membered ring of sumanene indicates paratropicity at the ring center with a maximum NICSzz(-0.2) value of 15.3 ppm and a long range diatropicity with minimum NICSzz(1.7) and NICSzz(-1.9) values of -14.5 and -8.2 ppm respectively. Therefore the inner six-membered ring of sumanene can be considered practically as non aromatic. Notice again the enhancement of the induced diatropic ring currents inside the bowl. The NICSzz-scan curve of the outer five-membered ring of sumanene (a typical curve for in-plane antiaromatic systems) [45] clearly illustrates the antiaromatic character of the ring, while the NICSzz-scan curve of the outer six-membered ring of sumanene exhibiting two maxima at a certain distance above and below the molecular plane and having NICSzz(0) value close to 0 is characteristic of the pure π-aromaticity enhanced inside the bowl (NICSzz(1.1) = -29.8 ppm), but depleted on the outside (NICSzz(-1.4) = -16.2 ppm).
The 3D molecular orbital pictures of the MOs participating in the lowest Tx,y- and Rz-allowed transitions in corannulene, corannulene dianion and sumanene are shown in Figure 12.
In the corannulene molecule with C5v symmetry, the chief contribution to induced diatropic ring current arises from the Tx,y-allowed HOMO,-1 (e1) → LUMO+2 (a1) and HOMO,-1 (e1) → LUMO+3 (a2) transitions with excitation energies 5.75 and 6.23 eV respectively. On the other hand, the chief contribution to induced paratropic ring current arises from the Rz-allowed HOMO,-1 (e1) → LUMO,+1 (e1) transition with excitation energy 4.35 eV. For the corannulene dianion with Cs symmetry, the chief mixed contribution to induced paratropic and diatropic ring currents arises from the lowest Rz- and Tx,y-allowed HOMO (a″) → LUMO (a′), HOMO (a″) → LUMO+1 (a″) and HOMO (a″) → LUMO+2 (a′) transitions with excitation energies 0.83, 0.96 and 1.07 eV respectively.
In the sumanene molecule with C3v symmetry, the chief contribution to induced diatropic ring current arises from the lowest Tx,y-allowed HOMO,-1 (e) → LUMO+2 (a1) transition with excitation energy 4.66 eV. Furthermore, the chief contribution to induced paratropic ring current arises from the lowest Rz-allowed HOMO,-1 (e) → LUMO,+1 (e) and HOMO-2 (a2) → LUMO+2 (a1) transitions with excitation energies 4.65 and 5.36 eV, respectively.
The estimated excitation energies of the lowest Tx,y- and Rz-allowed transitions of the corannulene, corannulene dianion and sumanene molecules in conjunction with the 3D molecular orbital pictures of the relevant MOs account well for the magnetotropicity patterns derived from the shapes of the NICSzz-scan curves.
4. Conclusions
To summarize, in the present computational study we showed that the NICSzz-scan curves computed at the GIAO/B3LYP level in conjunction with symmetry-based selection rules provide efficient diagnostic tools of the orbital-type, σ-, π- and double (σ + π)- of aromaticity in a wide range of aromatic organic and main group inorganic rings.
Thus, the NICSzz-scan curves of any aromatic molecule exhibiting pure σ-aromatic character are symmetric around the z-axis (perpendicular to the ring plane) with the NICSzz values decaying rapidly and monotically with respect to R and having a half-band width approximately less than 3 Å. This is the case of σ-aromatics exhibiting s-orbital aromaticity due to cyclic delocalization of two 1s electrons over the three-membered ring in [c-H3]+ and six 1s electrons over the four-, five- and six-membered rings in [c-H4]2-, [c-H5]- and [c-H6] species, the [c-B3]-, [c-B3]+, [c-B2C], [c-C3]2+, [c-B4], [c-B4]2+, [c-B5]+ aromatics with in-plane σ-MOs of Hückel topology constructed from the overlap of the 2p AOs of the ring atoms, the M3(μ2-CO)3(CO)3 (M = Ni, Pd, Pt) molecules, as well as for the transition states of the concerted transfer of two hydrogen atoms from ethane to ethylene (double group transfer reactions) and the trimerization of acetylene to form benzene, respectively. In all cases, the chief contribution to induced diatropic ring current arises from the lowest Tx,y-allowed occupied-to-unoccupied molecular orbital transitions involving exclusively σ-type molecular orbitals.; the estimated excitation energies accounting for the relative strength of their aromatic character.
A broad NICSzz-scan curve with half-band width approximately > 3 Å is typical for double (σ + π)-aromatic molecules with comparable contribution to the induced total diatropic ring current from the σ- and π-diatropic ring currents. This is the case of a number of three-membered rings of main group atoms including the [c-Be3]2-, [c-Mg3]2-, [c-E3]- (E = Al, Ga, In, Tl), [c-E3]2- (E = C, Si, Ge), [c-E3]+ (E = N, P, As, Sb, Bi) and [c-E3] (E = S, Se, Te, Po), four-membered rings of main group atoms including the [c-E4]2- (E = B, Al, Ga, In, Tl), [c-Si4]2+, [c-Mg4]2+ and [c-Li2Mg2] and five-membered rings of main group atoms including the [c-E5]- (E = N, P, As, Sb, Bi) and [c-C5H5]-. The magnitude and type (σ-, π-) of the induced diatropic and paratropic ring currents are determined by the excitation energies of the lowest Tx,y- and Rz-allowed occupied → unoccupied molecular orbital transitions respectively with the coexistence of both the σ- and π-type transitions accounting well for the double (σ + π)-aromatic character of all these clusters.
The NICSzz-scan curves exhibiting two maxima at a certain distance above and below the molecular plane are typical for double (σ + π)-aromatics where the π-diatropic ring current overwhelms the σ-type one. In the absence of any contribution from the σ-diatropic ring current the NICSzz(0) value is close to zero and the molecule is a pure π-aromatic molecule. This is the case of a vast number of organic aromatic molecules. Again the estimated excitation energies of the lowest Tx,y- and Rz-allowed occupied → unoccupied molecular orbital transitions are indicative of the double (σ + π)-aromatic character of the organic aromatics accounting well for the observed varying σ-/π- ratio.
Finally, the NICSzz-scan curves correctly predict the peculiar aromaticity of borazine and the magnetotropicity (diatropicity/paratropicity) patterns of the non-planar corannulene, corannulene dianion and sumanene.
References and Notes
- Masui, H. Metalloaromaticity. Coord. Chem. Rev. 2001, 219, 957–992. [Google Scholar] [CrossRef]
- Tsipis, C.A. DFT study of “all-metal” aromatic compounds. Coord. Chem. Rev. 2005, 249, 2740–2762. [Google Scholar] [CrossRef]
- Boldyrev, A.I.; Wang, L.-S. All-metal aromaticity and antiaromaticity. Chem. Rev. 2005, 105, 3716–3757. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Robinson, G.H. Organometallics of the group 13 M-M bond (M = Al, Ga, In) and the concept of metalloaromaticity. Organometallics 2007, 26, 2–11. [Google Scholar] [CrossRef]
- Lee, V.Y.; Sekiguchi, A. Aromaticity of group 14 organometallics: Experimental aspects. Angew. Chem. Int. Ed. 2007, 46, 6596–6620. [Google Scholar] [CrossRef]
- Zubarev, D.Y.; Averkiev, B.B.; Zhai, H.-J.; Wang, L.-S.; Boldyrev, A.I. Aromaticity and antiaromaticity in transition-metal systems. Phys. Chem. Chem. Phys. 2008, 10, 257. [Google Scholar] [CrossRef] [PubMed]
- Schleyer, P.v.R.; Guest Editor. Thematic Issue on Aromaticity. Chem. Rev. 2001, 101. [Google Scholar]
- Schleyer, P.v.R.; Guest Editor. Thematic Issue on Delocalization - Pi and Sigma. Chem. Rev. 2005, 105. [Google Scholar]
- Special edition on New Perspectives on Aromaticity. Phys. Chem. Chem. Phys. 2004, 6.
- Mirkin, V.I.; Glukhotsev, M.N.; Simkin, B.Y. Aromaticity and Antiaromaticity: Electronic and Structural Aspects; John Wiley & Sons: New York, NY, USA, 1994. [Google Scholar]
- Schleyer, P.v.R.; Maerker, C.; Dransfeld, A.; Jiao, H.; Hommes, N. J. R. V. Nucleus-independent chemical shifts: A simple and efficient aromaticity probe. J. Am. Chem. Soc. 1996, 118, 6317–6318. [Google Scholar] [CrossRef]
- Schleyer, P.v.R.; Manohran, M.; Wang, Z.-X.; Kiran, B.; Jiao, H.; Puchta, R.; Hommes, N.J.R.V.R. Dissected nucleus-independent chemical shift analysis of pi-aromaticity and antiaromaticity. Org. Lett. 2001, 3, 2465–2468. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wannere, C.S.; Corminboeuf, C.; Puchta, R.; Schleyer, P.v.R. Nucleus-independent chemical shifts (NICS) as an aromaticity criterion. Chem. Rev. 2005, 105, 3842–3888. [Google Scholar] [CrossRef] [PubMed]
- Klod, S.; Kleinpeter, E. Ab initio calculation of the anisotropy effect of multiple bonds and the ring current effect of arenes - application in conformational and configurational analysis. J. Chem. Soc. Perkin Trans. 2001, 2, 1893–1898. [Google Scholar]
- Fallah-Bagher-Shaidaei, H.; Wannere, C.S.; Corminboeuf, C.; Puchta, R.; Schleyer, P.v.R. Which NICS aromaticity index for planar pi rings is best? Org. Lett. 2006, 8, 863–866. [Google Scholar] [CrossRef] [PubMed]
- Wodrich, M.D.; Corminboeuf, C.; Park, S.S.; Schleyer, P.v.R. Double aromaticity in monocyclic carbon, boron, and borocarbon rings based on magnetic criteria. Chem. Eur. J. 2007, 13, 4582–4593. [Google Scholar] [CrossRef] [PubMed]
- Bohmann, J.A.; Weinhold, F.; Farrar, T.C. Natural chemical shielding analysis of nuclear magnetic resonance shielding tensors from gauge- including atomic orbital calculations. J. Chem. Phys. 1997, 107, 173. [Google Scholar] [CrossRef]
- Corminboeuf, C.; Heine, T.; Weber, J. Evaluation of aromaticity: A new dissected NICS model based on canonical orbitals. Phys. Chem. Chem. Phys. 2003, 5, 246–251. [Google Scholar] [CrossRef]
- Heine, T.; Schleyer, P.v.R.; Corminboeuf, C.; Seifert, G.; Reviakine, R.; Weber, J. Analysis of aromatic delocalization: Individual molecular orbital contributions to nucleus-independent chemical shifts. J. Phys. Chem. A 2003, 107, 6470–6475. [Google Scholar] [CrossRef]
- Stanger, A. Nucleus-independent chemical shifts (NICS): Distance dependence and revised criteria for aromaticity and antiaromaticity. J. Org. Chem. 2006, 71, 883. [Google Scholar] [CrossRef]
- Stanger, A. Can substituted cyclopentadiene become aromatic or antiaromatic? Chem. Eur. J. 2006, 12, 2745–2751. [Google Scholar] [CrossRef]
- Poater, J.; Bofill, J.M.; Alemany, P.; Solà, M. Role of electron density and magnetic couplings on the nucleus-independent chemical shift (NICS) profiles of [2.2]paracyclophane and related species. J. Org. Chem. 2006, 71, 1700–1702. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Halla, J.O.C.; Matito, E.; Robles, J.; Solà, M. Nucleus-independent chemical shift (NICS) profiles in a series of monocyclic planar inorganic compounds. J. Organometal. Chem. 2006, 691, 4359–4366. [Google Scholar] [CrossRef]
- Seal, P; Chakrabarti, S. Is nucleus-independent chemical shift scan a reliable aromaticity index for planar heteroatomic ring systems? J. Phys. Chem. A 2007, 111, 9988–9994. [Google Scholar] [CrossRef] [PubMed]
- Morao, I; Cossio, F.P. A simple ring current model for describing in-plane aromaticity in pericyclic reactions. J. Org. Chem. 1999, 64, 1868–1874. [Google Scholar] [CrossRef]
- Data, A.; Mallajosyula, S.S.; Pati, S.K. Nonlocal electronic distribution in metallic clusters: A critical examination of aromatic stabilization. Acc. Chem. Res. 2007, 40, 213–221. [Google Scholar] [CrossRef]
- Datta, A.; Pati, S.K. Limit to puckering of benzene with sterically crowded molecules: Hexaferrocenylbenzene. Chem. Phys. Lett. 2006, 433, 67–70. [Google Scholar] [CrossRef]
- Datta, A.; Pati, S.K. Rationalization of the pi-sigma (anti)aromaticity in all metal molecular clusters. J. Chem. Theory Comput. 2005, 1, 824–826. [Google Scholar] [CrossRef]
- Datta, A.; Pati, S.K. Stable transition metal complexes of an all-metal antiaromatic molecule (Al4Li4): Role of complexations. J. Am. Chem. Soc. 2005, 127, 3496–3500. [Google Scholar] [CrossRef]
- Steiner, E.; Fowler, P.W. Four- and two-electron rules for diatropic and paratropic ring currents in monocyclic pi systems. Chem. Commun. 2001, 2220–2221. [Google Scholar] [CrossRef]
- Steiner, E.; Fowler, P.W. Patterns of ring currents in conjugated molecules: A few-electron model based on orbital contributions. J. Phys. Chem. A 2001, 105, 9553–9562. [Google Scholar] [CrossRef]
- Fowler, P.W.; Steiner, E.; Havenith, R.W.A.; Jenneskens, L.W. Current density, chemical shifts and aromaticity. Magn. Reson. Chem. 2004, 42, S68–S78. [Google Scholar] [CrossRef] [PubMed]
- Corminboeuf, C.; King, R.B.; Schleyer, P.v.R. Implications of molecular orbital symmetries and energies for the electron delocalization of inorganic clusters. ChemPhysChem 2007, 8, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Soncini, A.; Teale, A.M.; Helgaker, T.; De Proft, F.; Tozer, D.J. Maps of current density using density-functional methods. J. Chem. Phys. 2008, 129, 074101. [Google Scholar] [CrossRef] [PubMed]
- Viglione, R.G.; Zanasi, R.; Lazzeretti, P. Are ring currents still useful to rationalize the benzene proton magnetic shielding? Org. Lett. 2004, 6, 2265–2267. [Google Scholar] [CrossRef] [PubMed]
- Pelloni, S.; Faglioni, F.; Zanasi, R.; Lazzeretti, P. Topology of magnetic-field-induced current-density field in diatropic monocyclic molecules. Phys. Rev. 2006, 74, 012506. [Google Scholar] [CrossRef]
- Jusélius, J.; Sundholm, D. Ab initio determination of the induced ring current in aromatic molecules. Phys. Chem. Chem. Phys. 1999, 1, 3429–3435. [Google Scholar] [CrossRef]
- Jusélius, J.; Sundholm, D.; Gauss, J. Calculation of current densities using gauge-including atomic orbitals. J. Chem. Phys. 2004, 121, 3952–3963. [Google Scholar] [CrossRef]
- Lin, Y.-C.; Jusélius, J.; Sundholm, D.; Gauss, J. Magnetically induced current densities in Al42- and Al44- species studied at the coupled-cluster level. J. Chem. Phys. 2005, 122, 214308. [Google Scholar] [CrossRef] [PubMed]
- Merino, G.; Heine, T.; Seifert, G. The induced magnetic field in cyclic molecules. Chem. Eur. J. 2004, 10, 4367–4371. [Google Scholar] [CrossRef] [PubMed]
- Heine, T.; Islas, R.; Merino, G. sigma and pi contributions to the induced magnetic field: Indicators for the mobility of electrons in molecules. J. Comput. Chem. 2007, 28, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Periyasamy, G.; Burton, N.A.; Hillier, I.H.; Thomas, J.M.H. Electron delocalization in the metallabenzenes: A computational analysis of ring currents. J. Phys. Chem. A 2008, 112, 5960–5972. [Google Scholar] [CrossRef] [PubMed]
- Geuenich, D.; Hess, K.; Kohler, F.; Herges, R. Anisotropy of the induced current density (ACID), a general method to quantify and visualize electronic delocalization. Chem. Rev. 2005, 105, 3758–3772. [Google Scholar] [CrossRef] [PubMed]
- Tsipis, A.C.; Depastas, I.G.; Karagiannis, E.E.; Tsipis, C.A. Diagnosis of Magnetoresponsive Aromatic and Antiaromatic Zones in Three-Membered Rings of d- and f-Block Elements. J. Comput. Chem. 2009. [Google Scholar] [CrossRef] [PubMed]
- Tsipis, A.C. Efficiency of the NICSzz-scan Curves to Probe the Antiaromaticity of Organic and Inorganic Rings/Cages. Phys. Chem. Chem. Phys. 2009, 11, 8244–8261. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Schlegel, H.B.; Scusseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Zakrzewski, V.G.; Montgomery, J.A.; Vreven, T.; Kudin, K.N.; Burant, J.C.; Millan, J.M.; Iyengar, S.S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G.A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J.E.; Hratchian, H.P.; Cross, J.B.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R.E.; Yazyev, O.; Austin, A J.; Cammi, R.; Pomelli, C.; Ochterski, J.W.; Ayala, P.Y.; Morokuma, K.; Voth, G.A.; Salvador, P.; Dannenberg, J.J.; Zakrzewski, V.G.; Dapprich, S.; Daniels, A.D.; Strain, M.C.; Farkas, O.; Malick, D.K.; Rabuck, A.D.; Raghavachari, K.; Foresman, J.B.; Ortiz, J.V.; Cui, Q.; Baboul, A.G.; Clifford, S.; Cioslowski, J.; Stefanov, B.B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R.L.; Fox, D.J.; Keith, T.; Al-Laham, M.A.; Peng, C.Y.; Nanayakkara, A.; Challacombe, M.; Gill, P.M.W.; Johnson, B.; Chen, W.; Wong, M.W.; Gonzalez, C.; Pople, J.A. Gaussian 03, Revision E. 01; Gaussian, Inc.: Pittsburgh, PA, USA, 2004. [Google Scholar]
- Dolg, M.; Stoll, H.; Preuss, H.; Pitzer, R. M. Relativistic and correlation-effects for element 105 (Hahnium, Ha) - A comparative-study of M and MO (M = Nb, Ta, Ha) using energy-adjusted ab initio pseudopotentials. J. Phys. Chem. 1993, 97, 5852–5859. [Google Scholar] [CrossRef]
- Stuttgart RSC 1997 ECP EMSL Basis Set Exchange Library, https://bse.pnl.gov/bse/portal, accessed on 9 June 2008.
- Schlegel, H.B. Optimization of equilibrium geometries and transition structures. J. Comput. Chem. 1982, 3, 214–218. [Google Scholar] [CrossRef]
- Ditchfield, R.D. Self-consistent perturbation theory of diamagnetism I. A gauge-invariant LCAO method for N.M.R. chemical shifts. Mol. Phys. 1974, 27, 789–807. [Google Scholar] [CrossRef]
- Gauss, J. Effects of electron correlation in the calculation of nuclear-magnetic-resonance chemical-shifts. J. Chem. Phys. 1993, 99, 3629–3643. [Google Scholar] [CrossRef]
- Jiao, H.; Schleyer, P.v.R.; Glukhovtsev, M.N. Are the Dmh symmetric Hxq rings with 4n+2 sigma-electrons and hydrogen clusters aromatic? J. Phys. Chem. 1996, 100, 12299–12304. [Google Scholar] [CrossRef]
- Jursic, S.B. Hybrid density functional theory study of hydrogen cluster aromaticity by computing their relative energies and magnetic properties. Int. J. Quantum Chem. 1999, 73, 451–458. [Google Scholar] [CrossRef]
- Havenith, R.W.A.; De Proft, F.; Fowler, P.W.; Geerlings, P. sigma-Aromaticity in H3+ and Li3+: Insights from ring-current maps. Chem. Phys. Lett. 2005, 407, 391–396. [Google Scholar] [CrossRef]
- Yong, L.; Wu, S.D.; Chi, X.X. Theoretical study of aromaticity in small hydrogen and metal cation clusters X3+ (X = H, Li, Na, K, and Cu). Int. J. Quantum Chem. 2007, 107, 722–728. [Google Scholar] [CrossRef]
- Kuznetsov, A.E.; Boldyrev, A.I. Theoretical evidence of aromaticity in X3- (X = B, Al, Ga) species. Struct. Chem. 2002, 13, 141–148. [Google Scholar] [CrossRef]
- Zhai, H.-J.; Wang, L.-S.; Alexandrova, A.N.; Boldyrev, A.I.; Zakrzewski, V.G. Photoelectron spectroscopy and ab initio study of B3- and B4- anions and their neutrals. J. Phys. Chem. A 2003, 107, 9319–9328. [Google Scholar] [CrossRef]
- Alexandrova, A.N.; Boldyrev, A.I.; Zhai, H.-J.; Wang, L.-S. All-boron aromatic clusters as potential new inorganic ligands and building blocks in chemistry. Coord. Chem. Rev. 2006, 250, 2811–2866, and references therein. [Google Scholar] [CrossRef]
- Fernández, I.; Sierra, M.A.; Cossio, F.P. In-plane aromaticity in double group transfer reactions. J. Org. Chem. 2007, 72, 1488–1491. [Google Scholar] [CrossRef] [PubMed]
- Morao, I.; Lecea, B.; Cossio, F.P. In-plane aromaticity in 1,3-dipolar cycloadditions. J. Org. Chem. 1997, 62, 7033–7036. [Google Scholar] [CrossRef]
- Jiao, H.; Schleyer, P.v.R. Aromaticity of pericyclic reaction transition structures: magnetic evidence. J. Phys. Org. Chem. 1998, 11, 655–662. [Google Scholar] [CrossRef]
- Scheschkewitz, D.; Hofmann, M.; Ghaffari, A.; Amseis, P.; Praesang, C.; Mesbah, W.; Geiseler, G.; Massa, W.; Berndt, A. Very strong anionic homoaromaticity in (deloc-1,3,4)-1-sila-3,4-diboracyclopentane-1-ides, the importance of the energy of the reference system for homoaromatic stabilization energies. J. Organomet. Chem. 2002, 646, 262–270. [Google Scholar] [CrossRef]
- Alexandrova, A.N.; Boldyrev, A.I. sigma-aromaticity and sigma-antiaromaticity in alkali metal and alkaline earth metal small clusters. J. Phys. Chem. A 2003, 107, 554–560. [Google Scholar] [CrossRef]
- Fowler, P.W.; Baker, J.; Lillington, M. The ring current in cyclopropane. Theor. Chem. Acc. 2007, 118, 123–127. [Google Scholar] [CrossRef]
- Pelloni, S.; Lazzeretti, P.; Zanasi, R. Assessment of sigma-diatropicity of the cyclopropane molecule. J. Phys. Chem. A 2007, 111, 8163–8169. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Ma, B.; Wu, J.I-C.; Schleyer, P.v.R.; Mo, Y. Is cyclopropane really the σ-aromatic paradigm? Chem. Eur. J. 2009, 15, 9730–9736. [Google Scholar] [CrossRef] [PubMed]
- King, R.B. Metal cluster topology. 21. Sigma aromaticity in triangular metal carbonyl clusters. Inorg. Chim. Acta 2003, 350, 126–130. [Google Scholar] [CrossRef]
- Corminboeuf, C.; Schleyer, P.v.R.; King, R.B. Aromaticity of tri- and tetranuclear metal-carbonyl clusters based on magnetic criteria. Chem. Eur. J. 2007, 13, 978–984. [Google Scholar] [CrossRef]
- Jimenez-Halla, J.O.C.; Matito, E.; Blancafort, L.; Robles, J.; Sola, M. Tuning Aromaticity in Trigonal Alkaline Earth Metal Clusters and Their Alkali Metal Salts. J. Comput. Chem. 2009. [Google Scholar] [CrossRef]
- Kuznetsov, A.E.; Boldyrev, A.I. A single pi-bond captures 3, 4 and 5 atoms. Chem. Phys. Lett. 2004, 388, 452–456. [Google Scholar] [CrossRef]
- Zhan, C.-G.; Zheng, F.; Dixon, D.A. Electron affinities of Aln clusters and multiple-fold aromaticity of the square Al42- structure. J. Am. Chem. Soc. 2002, 124, 14795–14803. [Google Scholar] [CrossRef]
- Li, Z.-H.; Moran, D.; Fan, K.-N.; Schleyer, P.v.R. sigma-Aromaticity and sigma-antiaromaticity in saturated inorganic rings. J. Phys. Chem. A 2005, 109, 3711–3716. [Google Scholar] [CrossRef]
- Li, X.; Kuznetsov, A.E.; Zhang, H.-F.; Boldyrev, A.I.; Wang, L.-S. Observation of All-Metal Aromatic Systems. Science 2001, 291, 859. [Google Scholar] [CrossRef]
- Islas, R.; Heine, T.; Merino, G. sigma and pi contributions to the induced magnetic field: Indicators for the mobility of electrons in molecules. J. Chem. Theory Comput. 2007, 3, 302–309. [Google Scholar]
- Juselius, J.; Straka, M.; Sundholm, D. Magnetic-shielding calculations on Al42- and Analogues. A new family of aromatic molecules? J. Phys. Chem. A 2001, 105, 9939–9944. [Google Scholar] [CrossRef]
- Fowler, P.W.; Havenith, R.W.A.; Steiner, E. Unconventional ring currents in an ‘all-metal aromatic’, Al42-. Chem. Phys. Lett. 2001, 342, 85–90. [Google Scholar] [CrossRef]
- Fowler, P.W.; Havenith, R.W.A.; Steiner, E. Ring current and electron delocalisation in an all-metal cluster, Al42-. Chem. Phys. Lett. 2002, 359, 530–536. [Google Scholar] [CrossRef]
- Havenith, R.W.A.; Fowler, P.W.; Steiner, E.; Shetty, S.; Kanhere, D.; Pal, S. Aromaticity and antiaromaticity of LixAl4 clusters: Ring current patterns versus electron counting. Phys. Chem. Chem. Phys. 2004, 6, 285–288. [Google Scholar] [CrossRef]
- Havenith, R.W.A.; Fowler, P.W. The origin of the ring current in the all-metal aromatic, Al42. Phys. Chem. Chem. Phys. 2006, 8, 3383–3386. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Corminboeuf, C.; Heine, T.; Bohmann, J.; Schleyer, P.v.R. Do all-metal antiaromatic clusters exist? J. Am. Chem. Soc. 2003, 125, 13930–13931. [Google Scholar] [CrossRef]
- Boldyrev, A.I.; Kuznetsov, A.E. On the resonance energy in new all-metal aromatic molecules. Inorg. Chem. 2002, 41, 532–537. [Google Scholar] [CrossRef] [PubMed]
- Havenith, R.W.A.; van Lenthe, J.H. A valence bond study of the sigma and pi aromatic species Al42-. Chem. Phys. Lett. 2004, 385, 198–201. [Google Scholar] [CrossRef]
- Santos, J.C.; Tiznado, W.; Conteras, R. Fuentealba, Sigma-pi separation of the electron localization function and aromaticity. J. Chem. Phys. 2004, 120, 1670–1673. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Roy, D.R.; Elango, M.; Subramanian, V. Chemical reactivity descriptor based aromaticity indices applied to Al42- and Al44- systems. J. Mol. Struct.(THEOCHEM) 2006, 759, 109–110. [Google Scholar] [CrossRef]
- Mandado, M.; Krishtal, A.; van Alensoy, C.; Bultinck, P.; Hermida-Ramon, J. M. Bonding study in all-metal clusters containing Al4 units. J. Phys. Chem. A 2007, 111, 11885–11893. [Google Scholar] [CrossRef] [PubMed]
- Zhai, H.-J.; Wang, L.-S.; Kuznetsov, A.E.; Boldyrev, A.I. Probing the electronic structure and aromaticity of pentapnictogen cluster anions Pn5- (Pn = P, As, Sb, and Bi) using photoelectron spectroscopy and ab initio calculations. J. Phys. Chem. A 2002, 106, 5600–5606. [Google Scholar] [CrossRef]
- De Proft, F.; Fowler, P.W.; Havenith, W.A.; Schleyer, P.v.R.; van Lier, G.; Geerlings, P. Ring Currents as Probes of the Aromaticity of Inorganic Monocycles: P5-, As5-, S2N2, S3N3-, S4N3+, S4N42+, S5N5+, S42+ and Se42+. Chem. Eur. J. 2004, 10, 940–950. [Google Scholar] [CrossRef]
- Lambrecht, D.S.; Fleig, T.; Sommerfeld, T. Instability of the Al-4(2-) “All-Metal aromatic” ion and its implications. J. Phys. Chem. A 2008, 112, 2855–2862. [Google Scholar] [CrossRef] [PubMed]
- De Proft, F.; Schleyer, P.v.R.; van Lenthe, J.H.; Stahl, F.; Geerlings, P. Magnetic properties and aromaticity of o-, m-, and p-benzyne. Chem. Eur. J. 2002, 8, 3402–3410. [Google Scholar] [CrossRef]
- Hofmann, M.; Berndt, A. (pi+sigma)-double aromatic and pi,sigma-mixed aromatic boron compounds with two electrons delocalized over three centers. Heteroatom. Chem. 2006, 17, 224–237. [Google Scholar] [CrossRef]
- Mills, N.S.; Benish, M. The aromaticity/antiaromaticity continuum. 1. Comparison of the aromaticity of the dianion and the antiaromaticity of the dication of tetrabenzo[5.5]fulvalene via magnetic measures. J. Org. Chem. 2006, 71, 2207–2213. [Google Scholar] [CrossRef]
- Cryański, M.K.; Havenith, R.W.A.; Dobrowolski, M.A.; Gray, B.R.; Krygowski, T.M.; Fowler, P.W.; Jenneskens, L.W. The phenalenyl motf: A magnetic chameleon. Chem. Eur. J. 2007, 13, 2201–2207. [Google Scholar] [CrossRef]
- Fowler, P.W.; Rogowska, A.; Soncini, A.; Lillington, M.; Olson, L. P. Currents in tangentially p-p bonded sigma-aromatic systems. J. Org. Chem. 2006, 71, 6459–6467. [Google Scholar] [CrossRef]
- Islas, R.; Chamorro, E.; Robles, J.; Heine, T.; Santos, J.C.; Merino, G. Borazine: to be or not to be aromatic. Struct. Chem. 2007, 18, 833–839. [Google Scholar] [CrossRef]
- Wu, Y.-T.; Siegel, J.S. Aromatic Molecular-Bowl Hydrocarbons: Synthetic Derivatives, Their Structures, and Physical Properties. Chem. Rev. 2006, 106, 4833–4867. [Google Scholar] [CrossRef] [PubMed]
- Tsefrikas, V.M.; Scott, L.T. Geodesic Polyarenes by Flash Vacuum Pyrolysis. Chem. Rev. 2006, 106, 4868–4884. [Google Scholar] [CrossRef] [PubMed]
- Steiner, E.; Fowler, P.W.; Jenneskens, L.W. Counter-Rotating Ring Currents in Coronene and Corannulene. Angew. Chem. Int. Ed. 2001, 40, 362–366. [Google Scholar] [CrossRef]
- Monaco, G.; Zanasi, R. On the additivity of current density in polycyclic aromatic hydrocarbons. J. Chem. Phys. 2009, 131, 044126–044135. [Google Scholar] [CrossRef]
- Monaco, G.; Scott, L.T.; Zanasi, R. Magnetic Euripi in Corannulene. J. Phys. Chem. 2008, 112, 8136–8147. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
NICSzz-scan profiles (NICSzz in ppm, R in Å) of selected 2e-σ- and 6e-σ-aromatics computed at the B3LYP/6-311+G(d,p) level.
Figure 1.
NICSzz-scan profiles (NICSzz in ppm, R in Å) of selected 2e-σ- and 6e-σ-aromatics computed at the B3LYP/6-311+G(d,p) level.

Scheme I.
The transition states TS1 and TS2 of the concerted transfer of two hydrogen atoms from ethane to ethylene and the trimerization of acetylene to form benzene, respectively.
Scheme I.
The transition states TS1 and TS2 of the concerted transfer of two hydrogen atoms from ethane to ethylene and the trimerization of acetylene to form benzene, respectively.

Figure 2.
Linear relationship between NICSzz(0) (ppm) and Rav (Å) for σ-aromatic molecules with radial in-plane σ-MOs of Hückel topology constructed from the overlap of the 2p AOs of the ring atoms.
Figure 2.
Linear relationship between NICSzz(0) (ppm) and Rav (Å) for σ-aromatic molecules with radial in-plane σ-MOs of Hückel topology constructed from the overlap of the 2p AOs of the ring atoms.

Figure 3.
NICSzz-scan profiles (NICSzz in ppm, R in Å) of the [c-Pt3(μ2-CO)3(CO)3] complex, along with the molecular orbital energy level diagram, symmetries, 3D molecular orbital pictures and lowest Tx,y- and Rz-allowed transitions computed at the B3LYP/ SDD(Pt)∪6-311+G(d,p) (C,O) level (solid and dash arrows indicate Tx,y- and Rz-allowed transitions respectively).
Figure 3.
NICSzz-scan profiles (NICSzz in ppm, R in Å) of the [c-Pt3(μ2-CO)3(CO)3] complex, along with the molecular orbital energy level diagram, symmetries, 3D molecular orbital pictures and lowest Tx,y- and Rz-allowed transitions computed at the B3LYP/ SDD(Pt)∪6-311+G(d,p) (C,O) level (solid and dash arrows indicate Tx,y- and Rz-allowed transitions respectively).

Figure 4.
The NICSzz-scan profiles in the form of NICSzz/NICSzzmax versus R (in Å) curves for selected triangular main group clusters exhibiting double (σ + π)-aromaticity.
Figure 4.
The NICSzz-scan profiles in the form of NICSzz/NICSzzmax versus R (in Å) curves for selected triangular main group clusters exhibiting double (σ + π)-aromaticity.

Figure 5.
Linear relationship between NICSzz(0) (in ppm) and Rav (in Å) for triangular (D3h) main group double (σ + π)-aromatic clusters.
Figure 5.
Linear relationship between NICSzz(0) (in ppm) and Rav (in Å) for triangular (D3h) main group double (σ + π)-aromatic clusters.

Figure 6.
The NICSzz-scan profiles in the form of NICSzz/NICSzzmax versus R (in Å) curves for selected four- and five-membered rings of main group atoms exhibiting double (σ + π)-aromaticity.
Figure 6.
The NICSzz-scan profiles in the form of NICSzz/NICSzzmax versus R (in Å) curves for selected four- and five-membered rings of main group atoms exhibiting double (σ + π)-aromaticity.

Figure 7.
Linear relationship between NICSzz(0) (in ppm) and Rav (in Å) for the four-membered rings of 13 group atoms.
Figure 7.
Linear relationship between NICSzz(0) (in ppm) and Rav (in Å) for the four-membered rings of 13 group atoms.

Figure 8.
Linear relationship between NICSzz(0) (in ppm) and Rav (in Å) for the five-membered rings of 15 group atoms.
Figure 8.
Linear relationship between NICSzz(0) (in ppm) and Rav (in Å) for the five-membered rings of 15 group atoms.

Figure 9.
The NICSzz-scan profiles in the form of NICSzz/NICSzzmax versus R curves for selected aromatic organic molecules computed at the B3LYP/6-311+G(d,p) level.
Figure 9.
The NICSzz-scan profiles in the form of NICSzz/NICSzzmax versus R curves for selected aromatic organic molecules computed at the B3LYP/6-311+G(d,p) level.

Figure 10.
The NICSzz-scan curve of the borazine molecule along with the molecular orbital energy level diagram, symmetries, 3D molecular orbital pictures and significant Tx,y- and Rz-allowed transitions leading to the diamagnetic and paramagnetic ring currents respectively, computed at the B3LYP/6-311+G(d,p) level.
Figure 10.
The NICSzz-scan curve of the borazine molecule along with the molecular orbital energy level diagram, symmetries, 3D molecular orbital pictures and significant Tx,y- and Rz-allowed transitions leading to the diamagnetic and paramagnetic ring currents respectively, computed at the B3LYP/6-311+G(d,p) level.
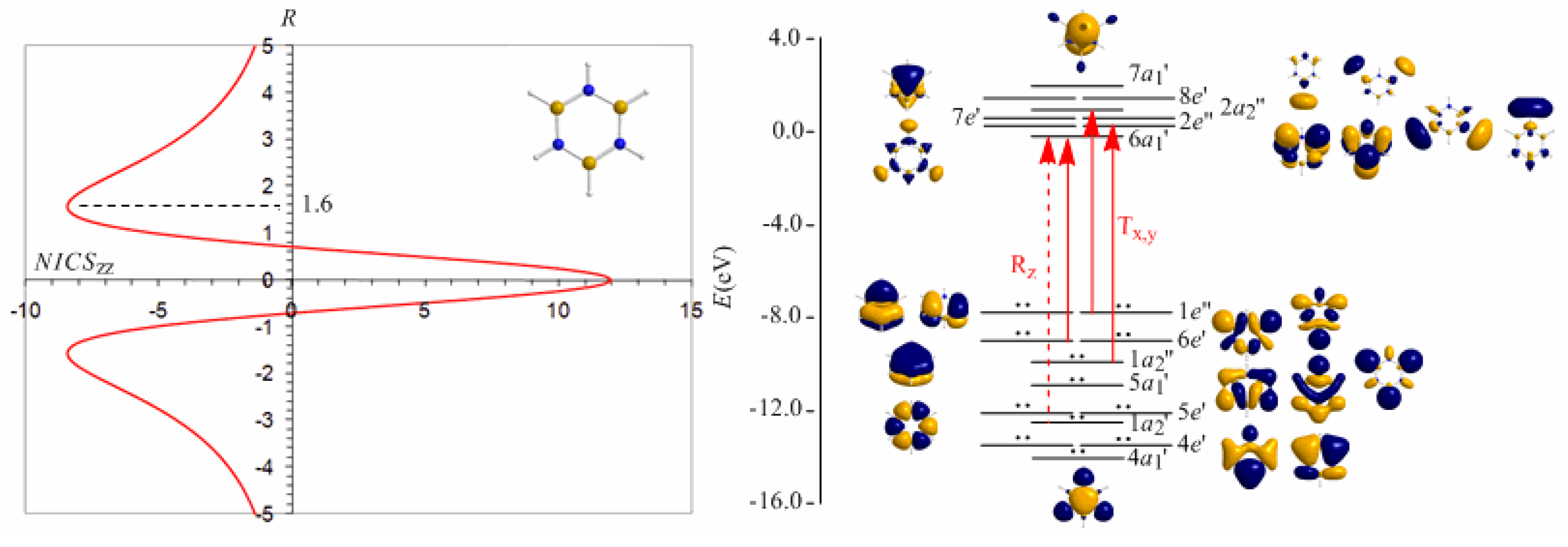
Figure 11.
The NICSzz-scan curves of corannulene, corannulene dianion and sumanene computed for both the inner (hub) and outer (rim) rings at the B3LYP/6-311+G(d,p) level.
Figure 11.
The NICSzz-scan curves of corannulene, corannulene dianion and sumanene computed for both the inner (hub) and outer (rim) rings at the B3LYP/6-311+G(d,p) level.

Figure 12.
3D contour surfaces of the molecular orbitals participating in the lowest Tx,y- and Rz-allowed transitions of the corannulene, corannulene dianion and sumanene molecules.
Figure 12.
3D contour surfaces of the molecular orbitals participating in the lowest Tx,y- and Rz-allowed transitions of the corannulene, corannulene dianion and sumanene molecules.


{kind=link}
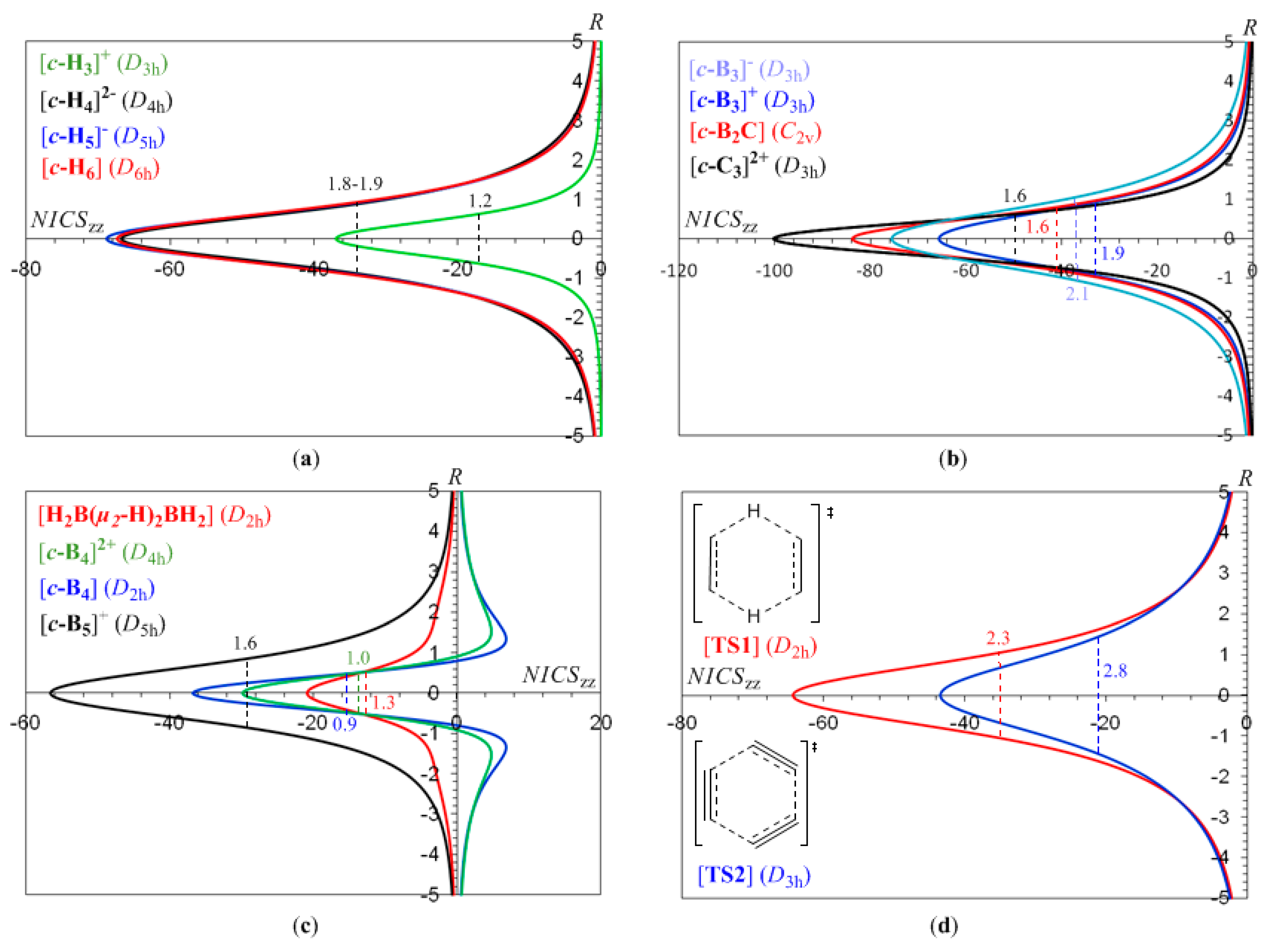{kind=link}
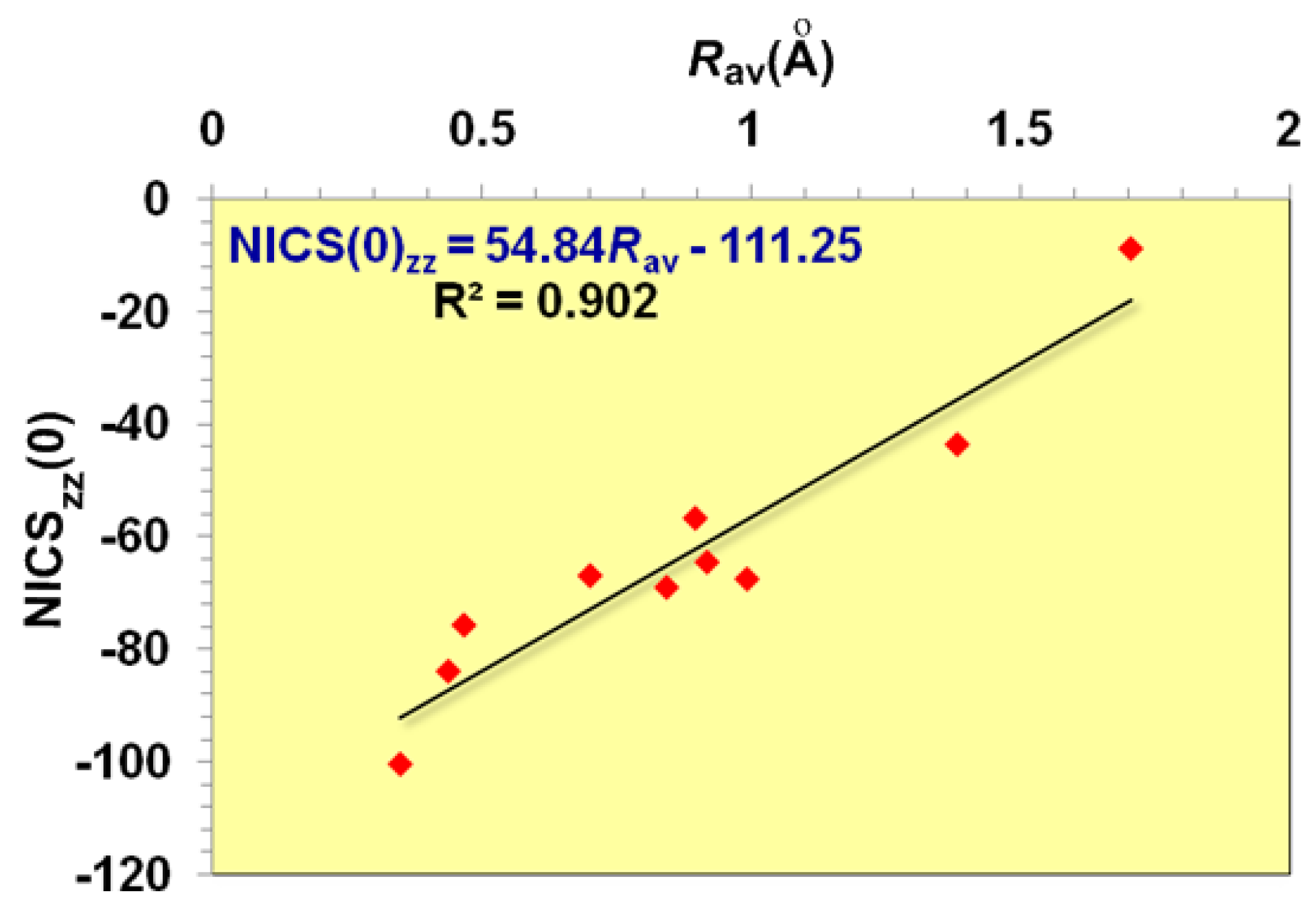{kind=link}
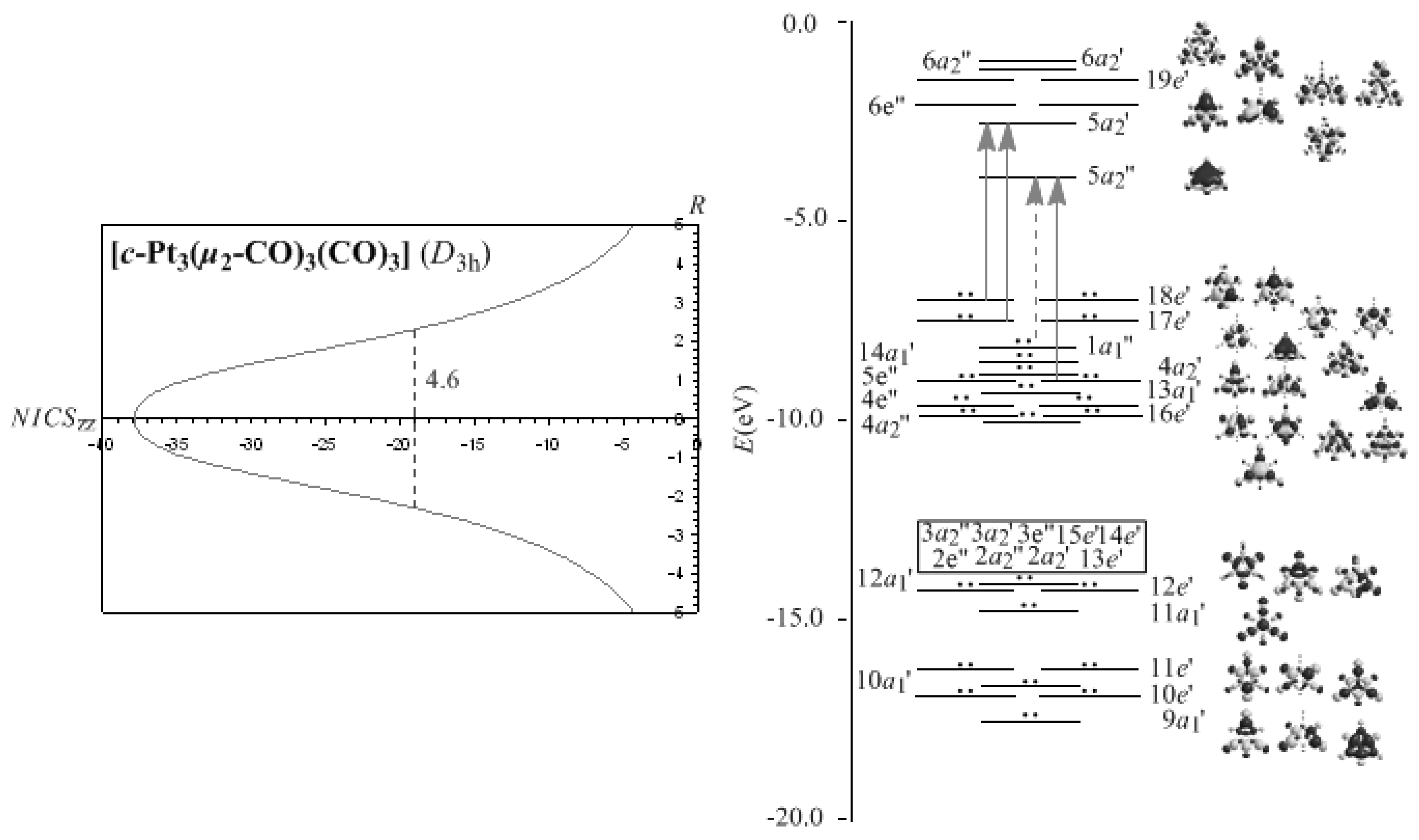{kind=link}
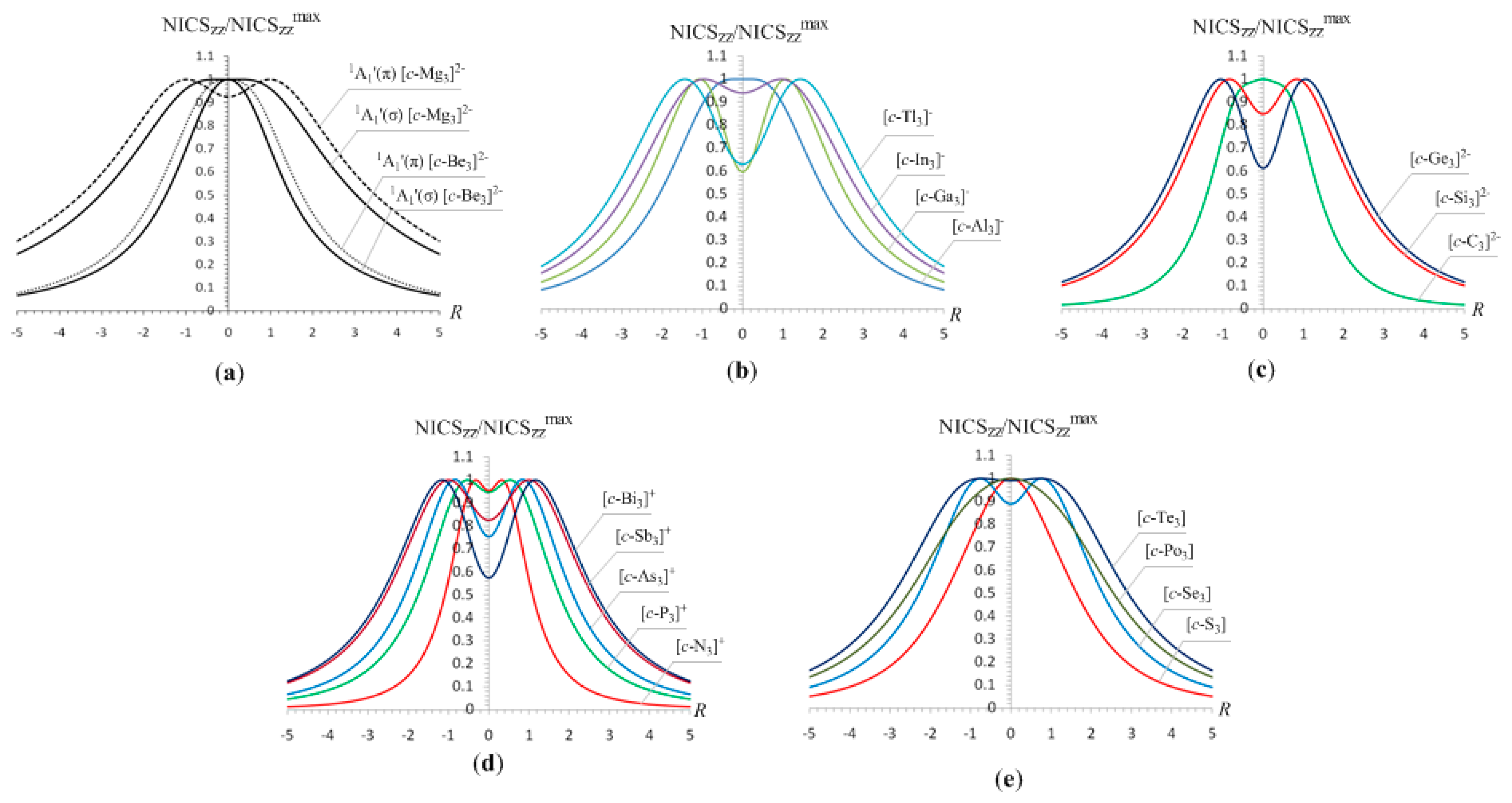{kind=link}
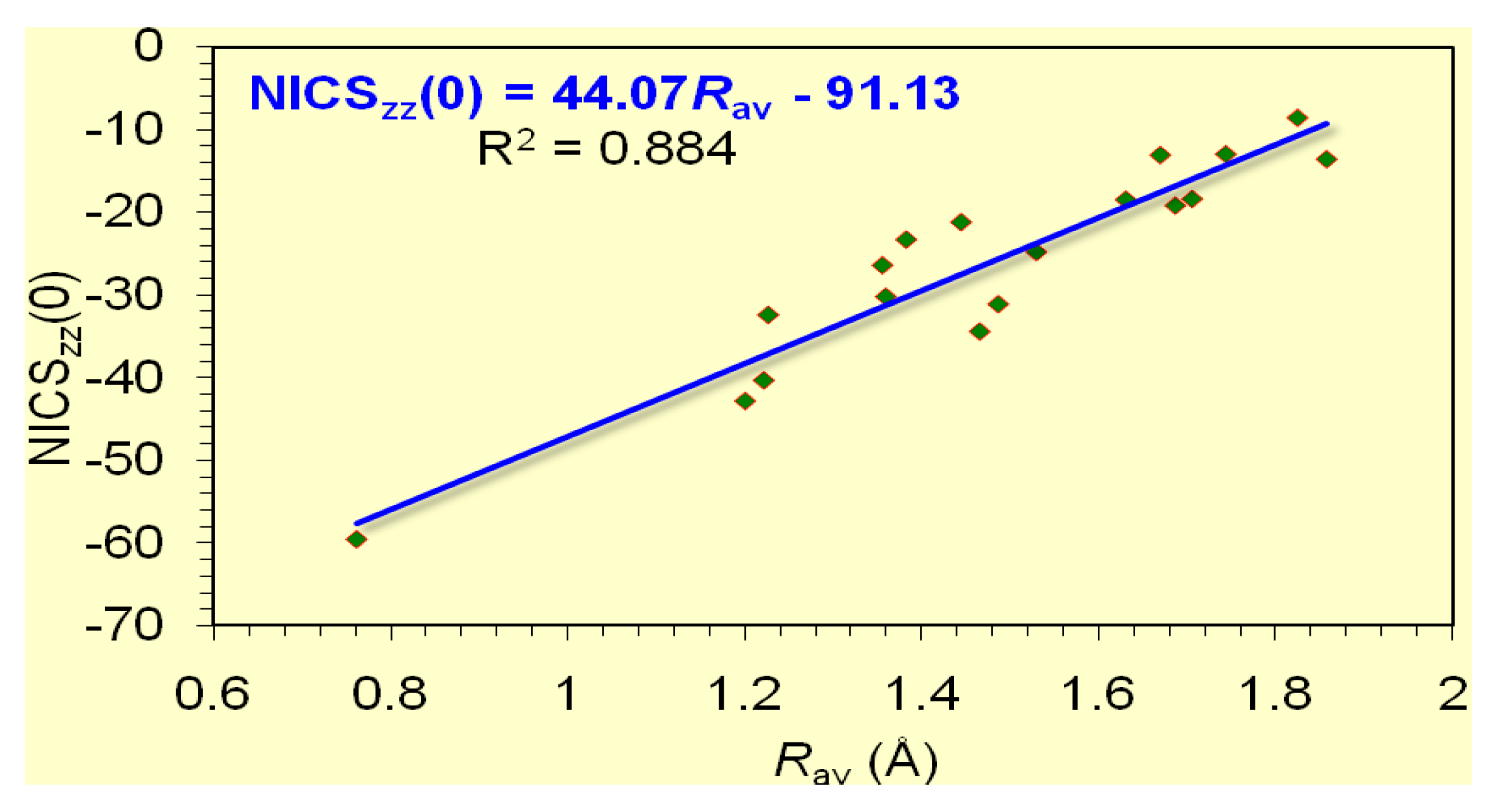{kind=link}
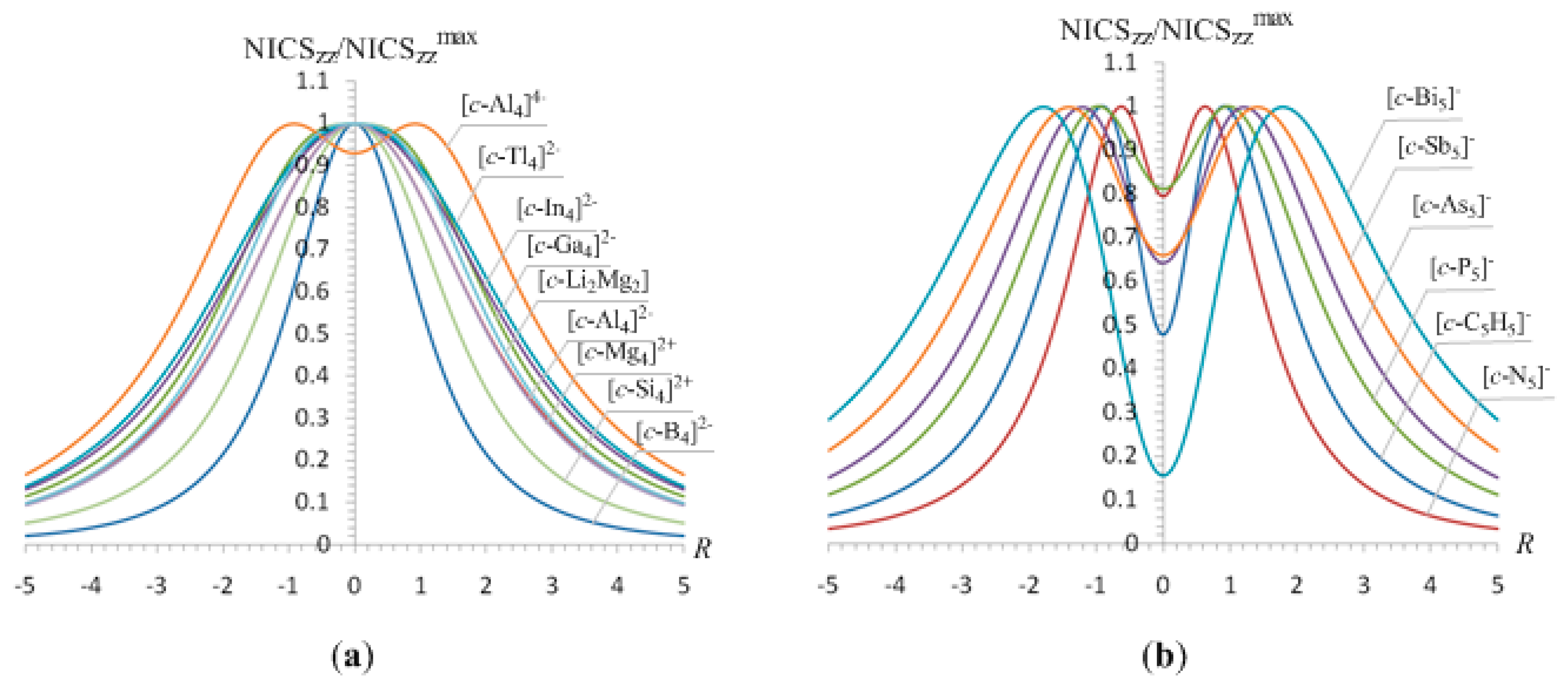{kind=link}
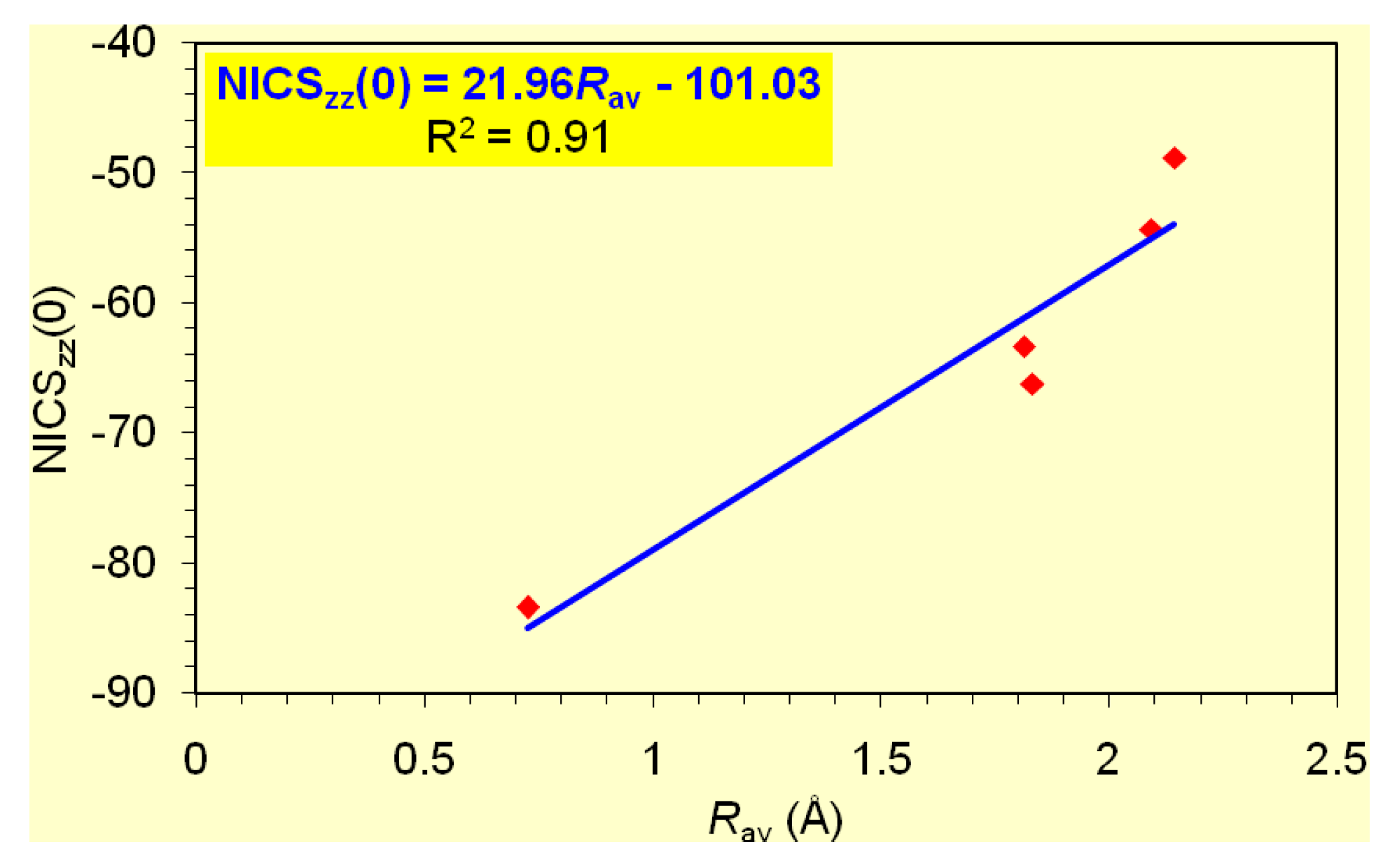{kind=link}
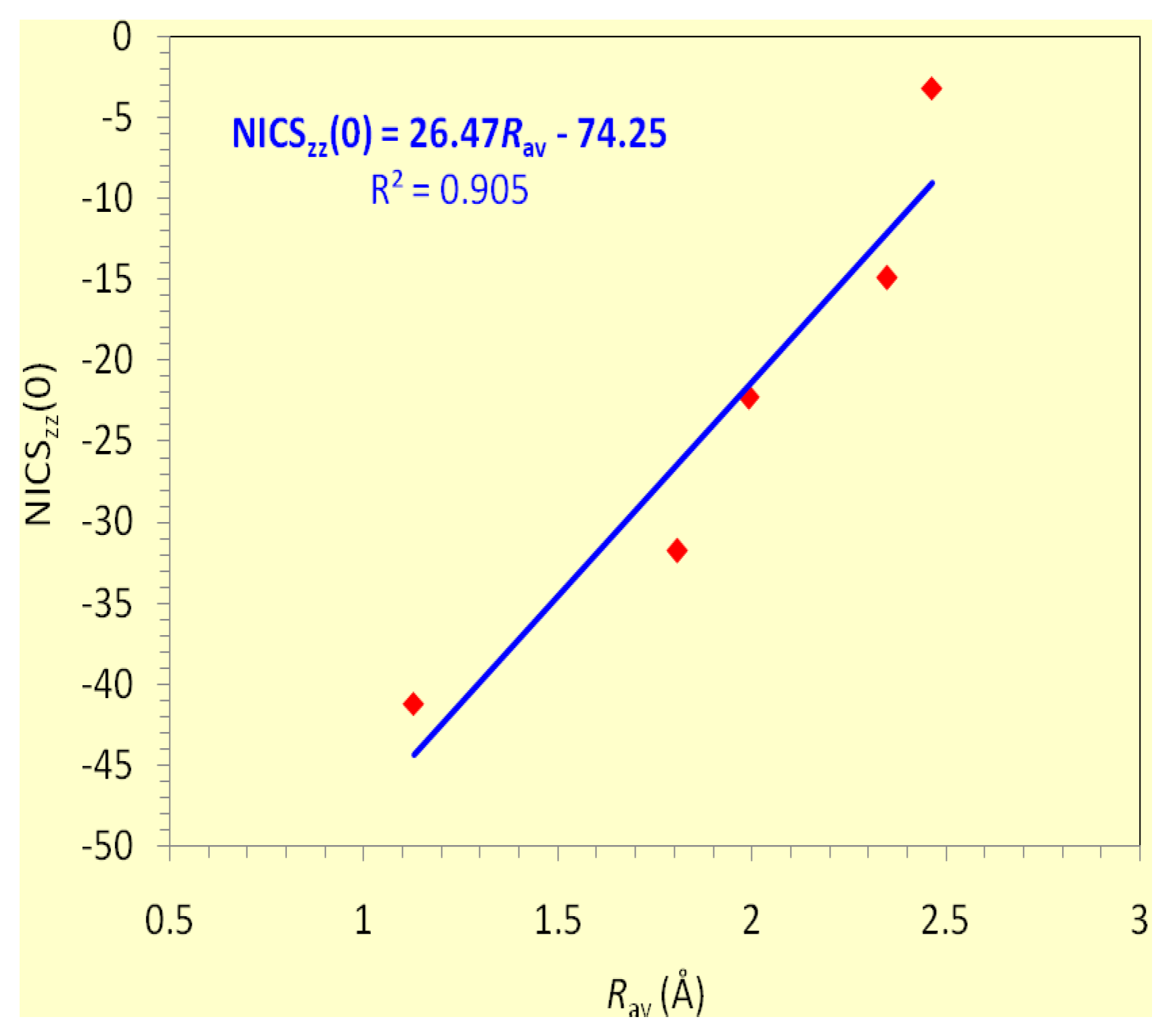{kind=link}
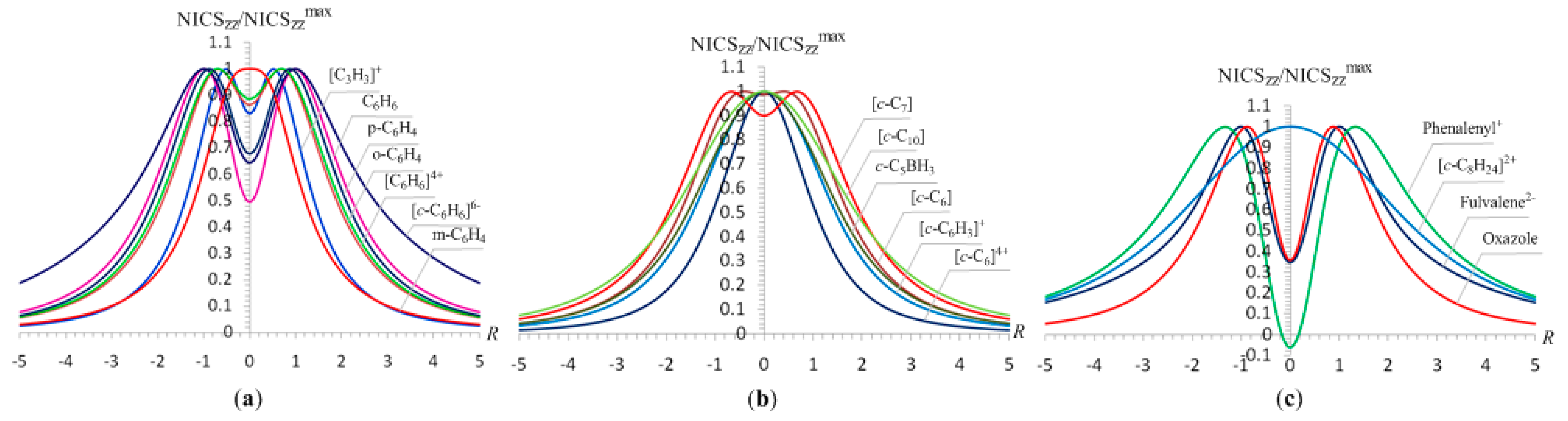{kind=link}
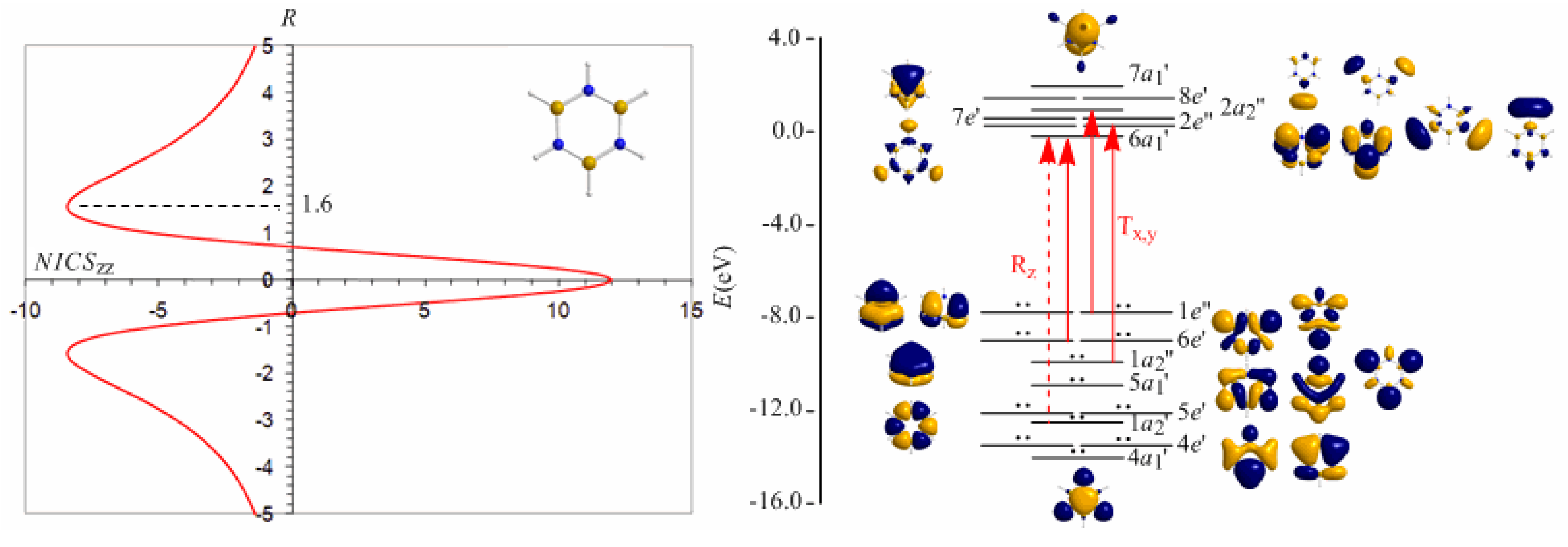{kind=link}
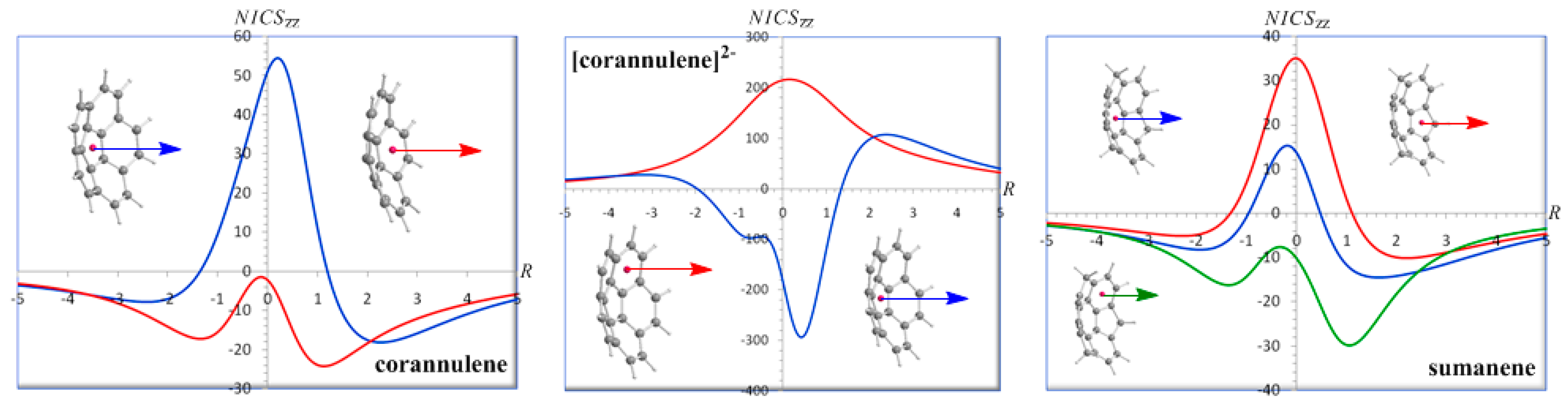{kind=link}
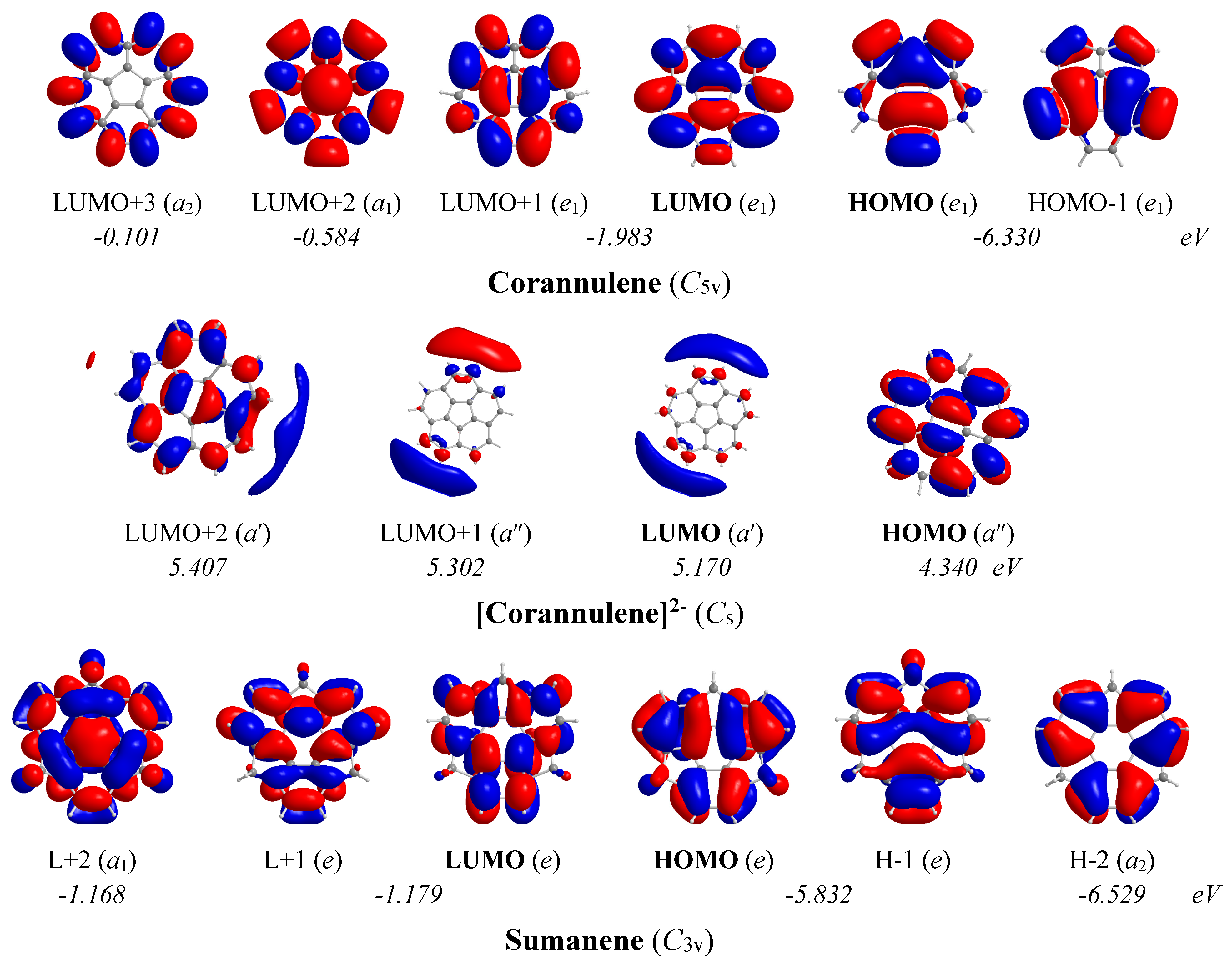{kind=link}
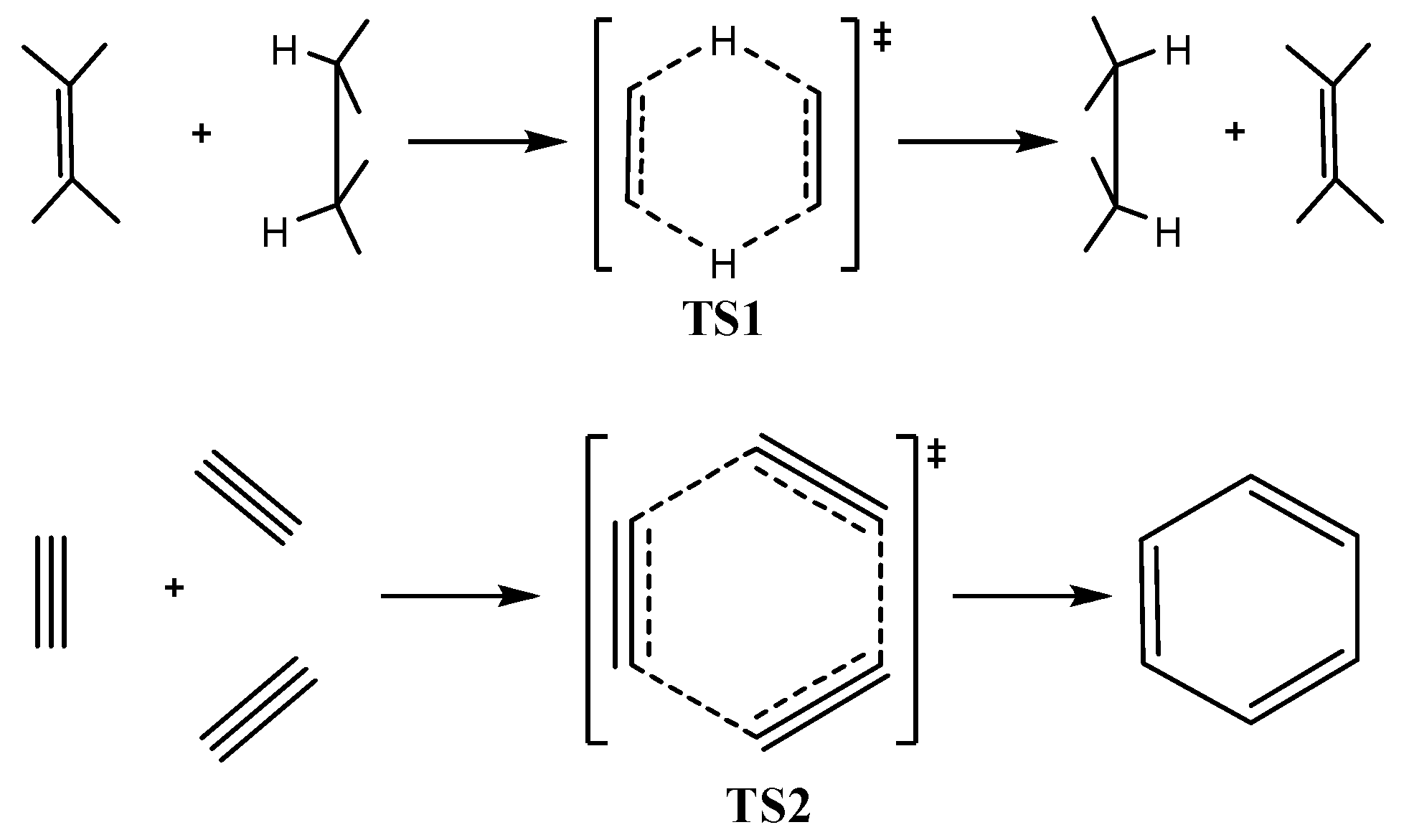{kind=link}
Table 1.
The NICS values (in ppm) calculated at the ring center, NICS(0) and 1.0 Å above the ring center, NICS(1), the zz-component of the shielding tensor elements, NICSzz(0) and NICSzz(1) along with the half-band width of the NICSzz-scan curves (in Å) and Rav (in Å) for selected σ-aromatic molecules computed at the B3LYP/6-311+G(d,p) level.
Table 1.
The NICS values (in ppm) calculated at the ring center, NICS(0) and 1.0 Å above the ring center, NICS(1), the zz-component of the shielding tensor elements, NICSzz(0) and NICSzz(1) along with the half-band width of the NICSzz-scan curves (in Å) and Rav (in Å) for selected σ-aromatic molecules computed at the B3LYP/6-311+G(d,p) level.
| Compound | NICS(0) | NICS(1) | NICSzz(0) | NICSzz(1) | Half-band width | Rav |
| [c-H3]+ (D3h) | -33.5 | -1.9 | -36.9 | -7.9 | 1.2 | 0.509 |
| [c-H4]2- (D4h) | -16.0 | -8.9 | -66.8 | -29.7 | 1.8 | 0.701 |
| [c-H5]- (D5h) | -20.1 | -9.4 | -68.9 | -30.5 | 1.8 | 0.843 |
| [c-H6] (D6h) | -24.1 | -10.0 | -67.4 | -31.1 | 1.9 | 0.992 |
| [c-B3]- (D3h) | -73.6 | -28.2 | -75.6 | -39.3 | 2.1 | 0.467 |
| [c-B3]+ (D3h) | -65.4 | -17.3 | -65.7 | -30.3 | 1.9 | 0.480 |
| [c-B2C] (C2v) | -80.1 | -22.6 | -83.8 | -31.6 | 1.6 | 0.438 |
| [c-C3]2+ (D3h) | -95.0 | -16.3 | -100.3 | -25.6 | 1.2 | 0.349 |
| [c-B4] (D2h) | -35.7 | -9.2 | -36.8 | 4.7 | 0.9 | 0.647 |
| [c-B4]2+ (D4h) | -28.8 | -3.4 | -29.9 | 1.5 | 1.0 | 0.698 |
| [H2B(μ2-Η)2ΒΗ2] (D2h) | -26.0 | -3.2 | -20.8 | -6.1 | 1.3 | 0.717 |
| [c-B5]+ (D5h) | -36.2 | -18.8 | -56.6 | -24.3 | 1.6 | 0.896 |
| TS1a (D2h) | -27.8 | -13.3 | -64.4 | -36.7 | 2.3 | 0.918 |
| TS2 b (D3h) | -17.6 | -10.2 | -43.5 | -28.6 | 2.8 | 1.382 |
| [c-Li3]+ (D3h) | -11.1 | -6.8 | -8.7 | -7.3 | 3.7 | 1.704 |
| [c-C3H6] (D3h) | -42.7 | -8.6 | -29.8 | -24.2 | 3.2 | 0.518 |
a TS1 = transition state for the concerted transfer of two hydrogen atoms from ethane to ethylene (double group transfer reactions). b TS2 = transition state for the trimerization of acetylene to form benzene.
Table 2.
The NICS values (ppm) calculated at the ring center, NICS(0) and 1.0 Å above the ring center, NICS(1), the zz-component of the shielding tensor element, NICSzz(0) and NICSzz(1) along with the half-band width (Å) and Rav (Å) for the M3(μ2-CO)3(CO)3 (M = Ni, Pd, Pt) molecules computed at the B3LYP/SDD(Pd,Pt)∪6-311+G(d,p) (C,O) levels.
Table 2.
The NICS values (ppm) calculated at the ring center, NICS(0) and 1.0 Å above the ring center, NICS(1), the zz-component of the shielding tensor element, NICSzz(0) and NICSzz(1) along with the half-band width (Å) and Rav (Å) for the M3(μ2-CO)3(CO)3 (M = Ni, Pd, Pt) molecules computed at the B3LYP/SDD(Pd,Pt)∪6-311+G(d,p) (C,O) levels.
| Compound | NICS(0) | NICS(1) | NICSzz(0) | NICSzz(1) | Half-band width | Rav |
| Ni3(μ2-CO)3(CO)3 (C3v) | -34.2a | -21.0 | -53.9a | -36.6 | 2.7 | 1.427 |
| Pd3(μ2-CO)3(CO)3 (D3h) | -30.2 | -14.0 | -27.7 | -23.6 | 4.6 | 1.602 |
| Pt3(μ2-CO)3(CO)3 (D3h) | -39.3 | -19.3 | -37.9 | -34.4 | 4.6 | 1.602 |
Table 3.
The NICS values (in ppm) calculated at the ring center, NICS(0) and 1.0 Å above the ring center, NICS(1), the zz-component of the shielding tensor element, NICSzz(0) and NICSzz(1), the Rmax (in Å) and the corresponding NICSzzmax along with the Rav (in Å) for selected triangular (D3h) main group double (σ + π)-aromatic clusters computed at the B3LYP/6-311+G(d,p) level.
Table 3.
The NICS values (in ppm) calculated at the ring center, NICS(0) and 1.0 Å above the ring center, NICS(1), the zz-component of the shielding tensor element, NICSzz(0) and NICSzz(1), the Rmax (in Å) and the corresponding NICSzzmax along with the Rav (in Å) for selected triangular (D3h) main group double (σ + π)-aromatic clusters computed at the B3LYP/6-311+G(d,p) level.
| Cluster | NICS(0) | NICS(1) | NICSzz(0) | NICSzz(1) | Rmax (Å) | NICSzzmax | ΔNICSa | Rav (Å) |
| [c-Be3]2- b | 33.6 | 1.7 | -44.1 | -30.9 | 0.0 | -44.1 | 0.0 | 1.245 |
| [c-Be3]2- c | -65.0 | -31.8 | -42.8 | -34.5 | 0.0 | -42.8 | 0.0 | 1.200 |
| [c-Mg3]2- b | -2.8 | -4.0 | -12.7 | -12.2 | 0.4 | -12.7 | 0.0 | 1.948 |
| [c-Mg3]2- c | -36.5 | -24.3 | -13.6 | -14.7 | 1.0 | -14.7 | 1.1 | 1.859 |
| [c-Al3]- | -35.6 | -26.7 | -34.4 | -30.6 | 0.0 | -34.4 | 0.0 | 1.465 |
| [c-Al3]+ | -26.4 | -19.5 | -30.4 | -29.0 | 0.5 | -31.1 | 0.7 | 1.486 |
| [c-Ga3]- | -27.3 | -22.6 | -15.0 | -25.1 | 1.1 | -25.2 | 10.2 | 1.473 |
| [c-Ga3]+ | -14.4 | -14.6 | -11.8 | -24.6 | 1.1 | -24.8 | 13.0 | 1.529 |
| [c-In3]- | -6.1 | -10.6 | -13.0 | -13.8 | 1.0 | -13.8 | 0.8 | 1.743 |
| [c-Tl3]- | 16.6 | 1.7 | -5.4 | -8.1 | 1.4 | -8.6 | 3.2 | 1.824 |
| [c-C3]2- | 13.2 | -10.2 | -15.4 | -11.6 | 0.0 | -15.4 | 0.0 | 0.824 |
| [c-Si3]2- | -3.1 | -12.8 | -26.4 | -30.7 | 0.8 | -31.0 | 4.6 | 1.355 |
| [c-Ge3]2- | -4.2 | -13.4 | -21.2 | -34.6 | 1.1 | -34.6 | 13.4 | 1.444 |
| [c-N3]+ | -8.1 | -20.5 | -59.5 | -35.3 | 0.3 | -62.3 | 2.8 | 0.761 |
| [c-P3]+ | -12.9 | -18.3 | -40.3 | -37.5 | 0.5 | -42.6 | 2.3 | 1.221 |
| [c-As3]+ | -13.3 | -18.4 | -30.2 | -39.4 | 0.8 | -40.1 | 9.9 | 1.359 |
| [c-Sb3]+ | 0.6 | -7.3 | -18.5 | -22.4 | 1.0 | -22.4 | 3.9 | 1.630 |
| [c-Bi3]+ | -11.8 | -15.3 | -19.2 | -33.0 | 1.2 | -33.4 | 14.2 | 1.686 |
| c-S3 | -42.3 | -7.0 | -32.4 | -23.9 | 0.0 | -32.4 | 0.0 | 1.226 |
| c-Se3 | -42.7 | -9.8 | -23.3 | -25.6 | 0.8 | -26.2 | 2.9 | 1.382 |
| c-Te3 | -23.8 | -7.5 | -13.1 | -13.1 | 0.8 | -13.2 | 0.1 | 1.669 |
| c-Po3 | -29.4 | -9.5 | -18.4 | -16.7 | 0.0 | -16.7 | 0.0 | 1.705 |
a ΔNICS = NICSzzmax - NICSzz(0). b 1A1′(σ) ground state. c 1A1′(π) electronic state.
Table 4.
Estimated excitation energies (in eV) of the lowest Tx,y- and Rz-allowed transitions of the three-membered rings of main group atoms.
Table 4.
Estimated excitation energies (in eV) of the lowest Tx,y- and Rz-allowed transitions of the three-membered rings of main group atoms.
| Cluster | Excitations | |
| a1′ → e′ (σ-type) | a2″ → e″ (π-type) | |
| [c-Al3]- | 1.58 | 3.16 |
| [c-Ga3]- | 1.50 | 2.46 |
| [c-In3]- | 1.11 | 2.62 |
| [c-Tl3]- | 1.12 | 2.45 |
| [c-C3]2- | 5.18 | 7.39 |
| [c-Si3]2- | 3.41 | 3.61 |
| [c-Ge3]2- | 2.58 | 2.08 |
| [c-N3]+ | 18.04 | 11.11 |
| [c-P3]+ | 9.47 | 5.60 |
| [c-As3]+ | 8.29 | 4.63 |
| [c-Sb3]+ | 6.56 | 3.75 |
| [c-Bi3]+ | 6.27 | 3.23 |
| [c-S3] | 9.29 | 8.00 |
| [c-Se3] | 7.98 | 7.29 |
| [c-Te3] | 6.13 | 7.30a |
| [c-Po3] | 5.11 | 6.18a |
a e″ → a2″ transition
Table 5.
The NICS values (ppm) calculated at the ring center, NICS(0) and 1.0 Å above the ring center, NICS(1), the zz-component of the shielding tensor element, NICSzz(0) and NICSzz(1), the Rmax (in Å) and the corresponding NICSzzmax along with the Rav (in Å) for selected four- and five-membered σ-, π- and double (σ + π)-aromatic molecules.
Table 5.
The NICS values (ppm) calculated at the ring center, NICS(0) and 1.0 Å above the ring center, NICS(1), the zz-component of the shielding tensor element, NICSzz(0) and NICSzz(1), the Rmax (in Å) and the corresponding NICSzzmax along with the Rav (in Å) for selected four- and five-membered σ-, π- and double (σ + π)-aromatic molecules.
| Compound | NICS(0) | NICS(1) | NICSzz(0) | NICSzz(1) | Rmax (Å) | NICSzzmax | ΔNICSa | Rav |
| [c-B4]2- (D4h) | -44.8 | -24.3 | -83.4 | -47.8 | 0.0 | -83.4 | 0.0 | 0.727 |
| [c-Al4]2- (D4h) | -34.4 | -27.7 | -66.2 | -54.9 | 0.0 | -66.2 | 0.0 | 1.832 |
| [c-Ga4]2- (D4h) | -38.6 | -29.7 | -63.3 | -57.6 | 0.0 | -63.3 | 0.0 | 1.814 |
| [c-In4]2- (D4h) | -22.6 | -20.9 | -54.3 | -47.9 | 0.0 | -54.3 | 0.0 | 2.094 |
| [c-Tl4]2- (D4h) | -18.7 | -18.7 | -48.8 | -43.8 | 0.0 | -43.8 | 0.0 | 2.144 |
| [c-Al4]4- (D2h) | -6.0 | -12.1 | -30.0 | -32.2 | 0.9 | -32.3 | 2.3 | 1.885 |
| [c-Si4]2+ (D4h) | -42.0 | -28.8 | -78.1 | -58.0 | 0.0 | -78.1 | 0.0 | 1.633 |
| [c-Mg4]2+ (D4h) | -10.7 | -8.6 | -31.0 | -25.7 | 0.0 | -31.0 | 0.0 | 2.213 |
| c-Li2Mg2 (D2h) | 4.0 | -1.0 | -26.6 | -23.6 | 0.0 | -26.6 | 0.0 | 2.066 |
| [C5H5]- (D5h) | -12.6 | -9.6 | -16.2 | -33.7 | 0.9 | -34.0 | 17.8 | 1.204 |
| [c-N5]- (D5h) | -16.5 | -16.5 | -41.2 | -45.2 | 0.6 | -51.9 | 10.7 | 1.128 |
| [c-P5]- (D5h) | -16.7 | -15.8 | -31.8 | -39.3 | 1.0 | -39.3 | 7.5 | 1.808 |
| [c-As5]- (D5h) | -15.3 | -14.7 | -22.3 | -33.9 | 1.2 | -34.6 | 12.3 | 1.993 |
| [c-Sb5]- (D5h) | -9.0 | -9.5 | -14.9 | -21.2 | 1.4 | -22.5 | 7.6 | 2.347 |
| [c-Bi5]- (D5h) | -9.3 | -9.5 | -3.2 | -15.2 | 1.8 | -20.8 | 17.7 | 2.464 |
a ΔNICS = NICSzzmax - NICSzz(0)
Table 6.
Estimated excitation energies (in eV) of the main translational σ- and π-type transitions of double (σ + π)-aromatic four-membered [c-Al4]2-, [c-Ga4]2-, [c-In4]2-, [c-Tl4]2- and [c-Si4]2+ clusters in D4h symmetry.
Table 6.
Estimated excitation energies (in eV) of the main translational σ- and π-type transitions of double (σ + π)-aromatic four-membered [c-Al4]2-, [c-Ga4]2-, [c-In4]2-, [c-Tl4]2- and [c-Si4]2+ clusters in D4h symmetry.
| excitation | type | Excitation energy | ||||
| [c-Al4]2- | [c-Ga4]2- | [c-In4]2- | [c-Tl4]2- | [c-Si4]2+ | ||
| a1g → eu | σ- | 2.00 | 0.56 | 1.92 | 1.96 | 3.14 |
| a2u → eg | π- | 2.30 | 1.17 | 2.07 | 2.03 | 3.68 |
| b2g → eu | σ- | 4.20 | ||||
| b1g → eu | σ- | 4.85 | ||||
Table 7.
Estimated excitation energies (in eV) of the main Tx,y-allowed σ- and π-type transitions of double (σ + π)-aromatic five-membered pnictogen [c-Pn5]- (Pn = N, P, As, Sb, Bi) rings and the [c-C5H5]- ligand in D5h symmetry.
Table 7.
Estimated excitation energies (in eV) of the main Tx,y-allowed σ- and π-type transitions of double (σ + π)-aromatic five-membered pnictogen [c-Pn5]- (Pn = N, P, As, Sb, Bi) rings and the [c-C5H5]- ligand in D5h symmetry.
| excitation | Type | Excitation energy | |||||
| [c-N5]- | [c-P5]- | [c-As5]- | [c-Sb5]- | [c-Bi5]- | [c-C5H5]- | ||
| e1′ → a1′ | σ- | 9.24 | |||||
| e1′ → e2′ | σ- | 9.82 | |||||
| e2′ → e2′ | σ- | 9.84 | |||||
| e1″ → e2″ | π- | 8.20 | |||||
| e1′ → a1′ | σ- | 5.86 | |||||
| e1′ → e2′ | σ- | 5.99 | |||||
| e1″ → e2″ | π- | 4.28 | 3.58 | 2.95 | 2.05 | ||
| e1′ → a1′ | σ- | 5.23 | 4.72 | ||||
| e1′ → e2′ | σ- | 5.10 | 5.10 | 3.84 | |||
| a1′ → e1′ | σ- | 5.996 | 4.99 | ||||
| e1″ → e2″ | π- | 6.31 | |||||
| e1′ → a1′ | σ- | 8.88 | |||||
| e2′ → e1′ | σ- | 8.59 | |||||
Table 8.
The NICS values (in ppm) calculated at the ring center, NICS(0) and 1.0 Å above the ring center, NICS(1), the zz-component of the shielding tensor element, NICSzz(0) and NICSzz(1), the Rmax (in Å) and the corresponding NICSzzmax (ppm) along with Rav (Å) for selected aromatic organic molecules computed at the B3LYP/6-311+G(d,p) level.
Table 8.
The NICS values (in ppm) calculated at the ring center, NICS(0) and 1.0 Å above the ring center, NICS(1), the zz-component of the shielding tensor element, NICSzz(0) and NICSzz(1), the Rmax (in Å) and the corresponding NICSzzmax (ppm) along with Rav (Å) for selected aromatic organic molecules computed at the B3LYP/6-311+G(d,p) level.
| Compound | NICS(0) | NICS(1) | NICSzz(0) | NICSzz(1) | Rmax | NICSzzmax | ΔNICSa | Rav |
| C3H3+ (D3h) | -22.9 | -14.7 | -31.1 | -28.8 | 0.5 | -37.4 | 6.3 | 0.787 |
| C6H6 (D6h) | -8.0 | -10.2 | -14.5 | -29.2 | 1.0 | -29.2 | 14.7 | 1.395 |
| C6H64- (D6h) | -12.9 | -14.1 | -22.6 | -37.9 | 1.0 | -37.9 | 15.3 | 1.404 |
| C6H66- (D6h) | -16.0 | -16.9 | -26.7 | -41.5 | 1.0 | -41.5 | 14.8 | 1.395 |
| o-C6H4 (C2v) (S0) | -18.0 | -12.7 | -30.7 | -32.9 | 0.7 | -34.7 | 4.0 | 1.369 |
| m-C6H4 (C2v) (S0) | -42.2 | -16.3 | -64.4 | -39.9 | 0.0 | -64.4 | 0.0 | 1.225 |
| p-C6H4 (D2h) (S0) | -29.5 | -18.7 | -63.6 | -47.3 | 0.0 | -63.6 | 0.0 | 1.388 |
| C6 (D3h) | -24.8 | -12.0 | -32.4 | -28.3 | 0.4 | -32.8 | 0.4 | 1.284 |
| C64+ (D3h) | -61.4 | -18.5 | -69.0 | -36.7 | 0.0 | -69.0 | 0.0 | 1.356 |
| C6H3+ (D6h) | -42.7 | -19.9 | -72.4 | -51.9 | 0.0 | -72.4 | 0.0 | 1.313 |
| C5BH3 (C2v) | -34.5 | -16.8 | -58.3 | -43.2 | 0.0 | -58.3 | 0.0 | 1.336 |
| Fulvalene2- (D2) | -12.1 | -16.6 | -9.9 | -28.5 | 1.0 | -28.5 | 18.6 | 1.224 |
| Phenalenyl+ (D3h) | -3.6 | -7.2 | 1.1 | -17.3 | 1.3 | -19.0 | 20.1 | 1.409 |
| Oxazole (Cs) | -11.4 | -9.7 | -10.2 | -27.9 | 0.9 | -28.3 | 18.1 | 1.150 |
| c-C7 (C2v) | -16.9 | -14.2 | -32.3 | -33.7 | 0.7 | -35.9 | 3.6 | 1.483 |
| c-C8H242+ (D4h) | -16.1 | -13.7 | -43.8 | -39.0 | 0.0 | -43.8 | 0.0 | 3.072 |
| c-C10 (D5h) | -29.9 | -22.3 | -73.0 | -59.0 | 0.0 | -73.0 | 0.0 | 2.064 |
a ΔNICS = NICSzzmax - NICSzz(0)
Table 9.
Estimated excitation energies (in eV) of the main Tx,y- and Rz-allowed σ- and π-type transitions of selected aromatic organic molecules.
Table 9.
Estimated excitation energies (in eV) of the main Tx,y- and Rz-allowed σ- and π-type transitions of selected aromatic organic molecules.
| excitation | type | Excitation energies | ||||||
| C3H3+ | C6H6 | C6H64- | C6H66- | o-C6H4 | m-C6H4 | p-C6H4 | ||
| a2″ → e″ | π- | 10.03 | ||||||
| e′ → a1′ | σ- | 13.50 | ||||||
| e′ → a1′ | σ- | 13.79 | ||||||
| e1g → e2u | π- | 6.59 | 6.32 | |||||
| e1g → e2u | π- | 8.78 | 7.07 | |||||
| e2g → e2g | σ- | 10.78 | 7.82 | 6.66 | ||||
| a1g → e1u | π- | 2.80 | ||||||
| e1u → a1g | π- | 3.93 | ||||||
| a2 → b2 | π- | 5.12 | ||||||
| a1 → b2 | π- | 5.15 | ||||||
| b2 → b2 | σ- | 8.71 | ||||||
| a1 → a2 | π-Rz | 7.35 | ||||||
| b1 → b2 | π-Rz | 5.11 | ||||||
| a2 → b1 | π- | 5.41 | ||||||
| a1 → b1 | σ- | 5.65 | ||||||
| a1 → b2 | σ- | 7.37 | ||||||
| a1 → a2 | π-Rz | 6.75 | ||||||
| b1 → b2 | π-Rz | 8.46 | ||||||
| b2g → au | π- | 6.29 | ||||||
| b3u → ag | π- | 6.93 | ||||||
| b2u → ag | σ- | 8.00 | ||||||
| b1g → ag | π-Rz | 3.64 | ||||||
| b1u → au | π-Rz | 4.76 | ||||||
Table 10.
Estimated excitation energies (in eV) of the lowest Tx,y- and Rz-allowed σ- and π-type transitions of selected aromatic organic molecules.
Table 10.
Estimated excitation energies (in eV) of the lowest Tx,y- and Rz-allowed σ- and π-type transitions of selected aromatic organic molecules.
| excitation | type | Excitation energies | |||||
| c-C6 | [c-C6]4+ | [c-C6H3]+ | c-C5BH3 | c-C7 | c-C10 | ||
| e′ → a1′ | σ- | 3.96 | |||||
| e″→ a2″ | π- | 9.89 | |||||
| a1′ → a2′ | σ-Rz | 9.14 | |||||
| a1g → e1u | σ- | 4.66 | |||||
| e1g → e2u | π- | 7.60 | |||||
| a1′ → e′ | σ- | 6.51 | |||||
| e″→ a2″ | π- | 10.59 | |||||
| a1 → b2 | σ- | 5.99 | |||||
| b1 → a1 | π- | 6.55 | |||||
| b1 → b2 | π-Rz | 5.98 | |||||
| a1 → b2 | σ- | 3.40 | |||||
| a1 → b1 | π- | 3.03 | |||||
| a1 → a2 | σ-Rz | 6.36 | |||||
| e2′ → e1′ | σ- | 6.53 | |||||
| e1′ → e2′ | σ- | 8.35 | |||||
| e1″ → e2″ | π- | 8.57 | |||||
Table 11.
Estimated excitation energies (in eV) of the lowest Tx,y- and Rz-allowed σ- and π-type transitions of selected aromatic organic molecules.
Table 11.
Estimated excitation energies (in eV) of the lowest Tx,y- and Rz-allowed σ- and π-type transitions of selected aromatic organic molecules.
| Excitation energies | |||||
| excitation | Type | Fulvalene2- | Phenalenyl+ | Oxazole | [c-C8H24]2+ |
| a → b2 | π- | 1.96 | |||
| b2 → b1 | π- | 2.00 | |||
| a → b3 | π- | 2.69 | |||
| b2 → b3 | π-Rz | 2.10 | |||
| e″ → a1″ | π- | 3.68 | |||
| e″ → a2″ | π- | 6.51 | |||
| a2″ → a1″ | π-Rz | 3.58 | |||
| e1′ → a1′ | σ- | 10.51 | |||
| a2′ → a1′ | σ-Rz | 11.75 | |||
| a″ → a″ | π- | 6.64 | |||
| a′ → a′ | σ- | 8.52 | |||
| eu→ b1g | σ- | 3.00 | |||
| eu→ b2g | σ- | 3.42 | |||
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Tsipis, A.C.; Depastas, I.G.; Tsipis, C.A. Diagnosis of the σ-, π- and (σ+π)-Aromaticity by the Shape of the NICSzz-Scan Curves and Symmetry-Based Selection Rules. Symmetry 2010, 2, 284-319. https://doi.org/10.3390/sym2010284
AMA Style
Tsipis AC, Depastas IG, Tsipis CA. Diagnosis of the σ-, π- and (σ+π)-Aromaticity by the Shape of the NICSzz-Scan Curves and Symmetry-Based Selection Rules. Symmetry. 2010; 2(1):284-319. https://doi.org/10.3390/sym2010284
Chicago/Turabian StyleTsipis, Athanassios C., Ioannis G. Depastas, and Constantinos A. Tsipis. 2010. "Diagnosis of the σ-, π- and (σ+π)-Aromaticity by the Shape of the NICSzz-Scan Curves and Symmetry-Based Selection Rules" Symmetry 2, no. 1: 284-319. https://doi.org/10.3390/sym2010284