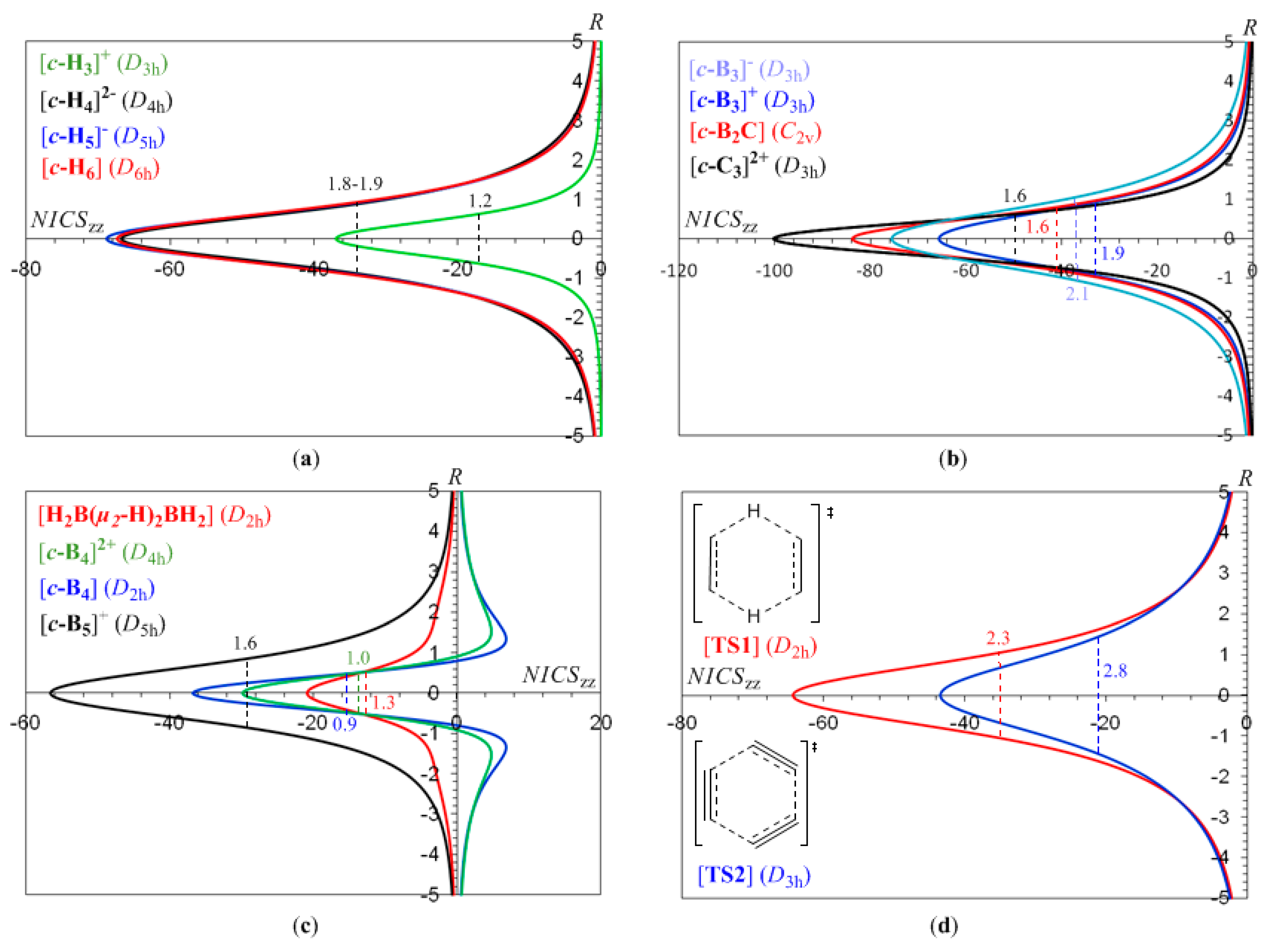
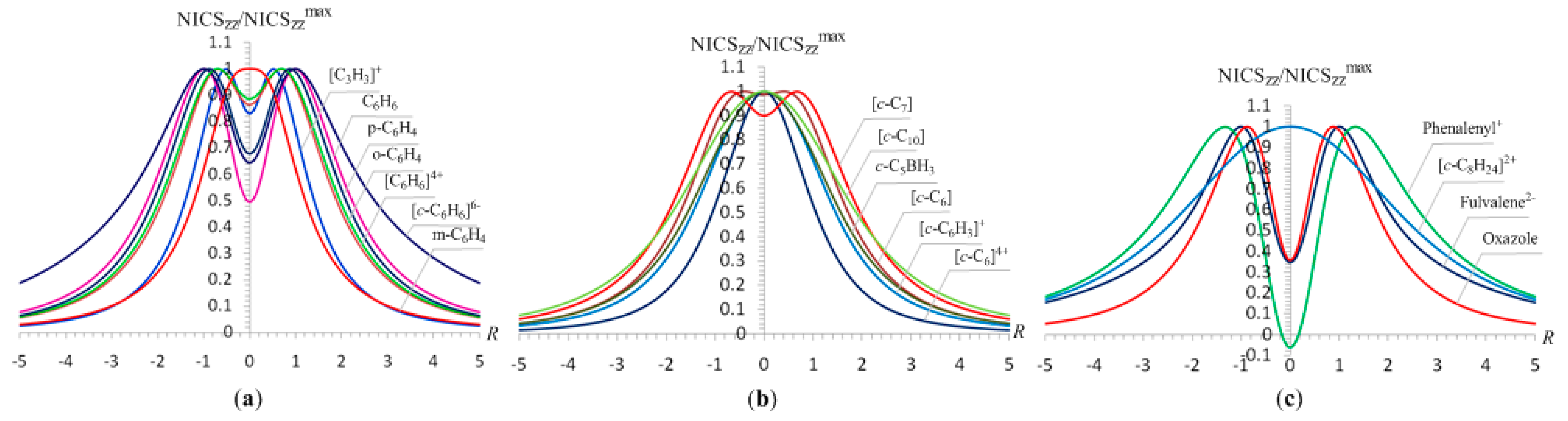
3.1. The NICSzz-scan profiles of 2e- and 6e-σ-aromatics
Let us first examine the shape and most salient features of the NICS
zz-scan profiles of 2e- and 6e-
σ-aromatics, the [
c-H
3]
+ and c-H
6 species being the prototypes [
52,
53,
54,
55]. It should be noted that the optimized structures of the [
c-H
4]
2-, [
c-H
5]
- and [
c-H
6] clusters were obtained under symmetry constrains and correspond to third, second and first order saddle points, respectively. The optimized
Dnh symmetric [
c-H
x]
q aromatic rings are similar to the optimized structures reported previously [
52,
53,
54,
55]. The NICS
zz-scan profiles of selected 2e- and 6e-
σ-aromatics are visualized in
Figure 1, while the main features of the NICS
zz-scan curves along with the
Rav values, e.g. the average distance of the ring current to the center of the ring, are compiled in
Table 1.
The half-band width, e.g., the width of the symmetric curve at half the NICSzz value, of the NICSzz-scan curves depends on the ring radius, Rav, evaluated as , where n is the number of the nuclei of the ring, and Ri is the distance from the ring center to the nucleus i. For the rings where the 2p orbitals (radial 2p orbitals) participate also in the bonding, forming molecular orbitals contributing to the diatropic ring current of the σ-aromatics, Rav was evaluated as , where Zi is the atomic number of the i-atom, and a0 is the Bohr radius. The term 4a0/Zi corresponds to the maximum of the probability of the radial function 4πr2Rn,l2(r) for a 2p atomic orbital.
It can be seen that the NICS
zz-scan curves are symmetric around the z-axis with the NICS
zz values decaying rapidly and monotically with respect to
R. The half-band widths of the NICS
zz-scan curves for the
σ-aromatics exhibiting
s-orbital aromaticity range from 1.2 to 1.9 Å (
Rav ranging from 0.509 to 0.992 Å) is characteristic of the in-plane pure
σ-aromaticity due to cyclic delocalization of two 1
s electrons over the three-membered ring in [
c-H
3]
+ and six 1
s electrons over the four-, five- and six-membered rings in [
c-H
4]
2-, [
c-H
5]
- and [
c-H
6]. Analogous symmetric sharp NICS
zz-scan curves with half-band widths in the range of 0.9 to 2.1 Å are obtained for the [
c-B
3]
-, [
c-B
3]
+, [
c-B
2C], [
c-C
3]
2+, [
c-B
4], [
c-B
4]
2+, [
c-B
5]
+ (
Figure 1b,c), as well as for the transition states TS1 and TS2 (
Figure 1d) of the concerted transfer of two hydrogen atoms from ethane to ethylene (double group transfer reactions) and the trimerization of acetylene to form benzene respectively
. shown in
Scheme I. The latter two NICS
zz-scan curves have a slightly larger half-band width of 2.3 and 2.8 Å, respectively.
3.2. Magnetotropicity of the 2e- and 6e-σ-aromatics
In the [c-H3]+ species with D3h symmetry, the chief contribution to induced diatropic ring current arises from the Tx,y-allowed HOMO (a1′) → LUMO (e′) transition with excitation energy 18.74 eV. In the [c-H4]2- (D4h) cluster the chief contribution to induced diatropic ring current arises from the Tx,y-allowed HOMO,-1 (eu) → LUMO (b1g) and HOMO,-1 (eu) → LUMO+1 (a1g) transitions having excitation energies 6.33 and 6.77 eV respectively (notice that HOMO,-1 denotes HOMO and HOMO-1, LUMO+1,+2 denotes LUMO+1 and LUMO+2, etc.). In the [c-H5]- (D5h) cluster the chief contribution to induced diatropic ring current arises from the Tx,y-allowed HOMO,-1 (e1′) → LUMO+2 (a1′) with excitation energy 11.02 eV. Finally, in the [c-H6] (D6h) cluster the chief contribution to induced diatropic ring current arises from the Tx,y-allowed HOMO,-1 (e1u) → LUMO+2 (a1g) with excitation energy 14.14 eV. The higher excitation energy of the Tx,y-allowed transition in the [c-H3]+ cluster accounts well for the weaker aromatic character. It should be noted that all orbitals involved in the Tx,y-allowed transitions responsible for the induced diatropic ring current are σ-type MOs.
In the [
c-B
3]
- and [
c-C
3]
2+ species with
D3h symmetry, the T
x,y-allowed HOMO (
a1′) → LUMO+1,+2 (
e′) excitations with excitation energies 3.42 and 4.25 eV respectively are responsible for the induced diatropic ring current. On the other hand, for the [
c-B
3]
+ species the T
x,y-allowed HOMO,-1 (
e′) → LUMO (
a1′) excitation with excitation energy 4.16 eV contributes primarily to the induced diatropic ring current. The lower magnetic aromaticity of the [
c-B
3]
+ species compared to that of the [
c-B
3]
- 2e-
σ-aromatic could be explained by the higher energy difference between the orbitals involved in the respective excitations (4.16
versus 3.42 eV). Both the
e′ and
a1′ MOs participating in the T
x,y-allowed transitions are
σ-type MOs and are primarily responsible for the
σ-aromaticity of the [
c-B
3]
-.and [
c-B
3]
+ species. The 1
a2″ bonding HOMO-1 in [
c-B
3]
- and [
c-C
3]
2+ and HOMO in [
c-B
3]
+ constructed from the overlap of the 2
pz AOs of the ring atoms, being a pure
π-bonding MO similar to the
π-MO in the cyclopropenyl cation ([
c-C
3H
3]
+), is magnetically inactive and therefore cannot contribute to the induced diatropic ring current. The magnetic inactivity of the 1
a2″
π-MO is further confirmed from the NICS
zz-scan profile of the putative [
c-B
3]
3+ species where, due to the removal of two electrons, the 1
a2″
π-MO is vacant. The computed NICS
zz(0) and NICS
zz(1) values for the [
c-B
3]
3+ species are -50.7 and -6.3 ppm, respectively, while the half-band width is 1.0 Å. However, the higher NICS
zz(0) value of the [
c-B
3]
+ species by 15.0 ppm compared to that of the [
c-B
3]
3+ one is probably indicative for a small contribution of the 1
a2″
π-MO to the diatropic ring current as well, supported by the T
x,y-allowed transitions HOMO-1 (
a2″) → LUMO+5,+6 (
e″) in [
c-B
3]
-, HOMO (
a2″) → LUMO+3,+4 (
e″) in [
c-B
3]
+ and HOMO-1 (
a2″) → LUMO+2,+3 (
e″) in [
c-C
3]
2+ having relatively high excitation energies of 5.97, 8.77 and 11.20 eV, respectively. As the contribution of the 1
a2″
π-MO to the diatropic ring current giving rise to
π-aromaticity is small, the NICS
zz-scan curve is still narrow with band width less than 3.0 Å. Boldyrev and co-workers [
56,
57,
58] have identified [
c-B
3]
- as 2+2 doubly aromatic system containing a
π-type (1
a2″) HOMO-1 and radial σ-type (2
a1′) HOMO both molecular orbitals contributing to the induced diatropic ring current. The NICS
zz-scan curve of the [
c-B
3]
- species in conjunction with symmetry-based selection rules are in support of the double aromaticity, but with the induced
π-diatropic ring current much weaker than the dominant induced
σ-diatropic ring current. In effect the canonical orbital contributions to the out-of-plane NICS tensor component (CMO-NICS
zz) for the [
c-B
3]
- species estimated to be -16.1 ppm from the HOMO-4 (1
a1′), -36.0 ppm from the degenerate HOMO-2,-3 (1
e′), -11.5 ppm from the HOMO-1 (1
a2″), the
π-type MO and -11.7 ppm from the HOMO (2
a1′) clearly illustrate the much weaker induced
π-diatropic ring current (-11.5 ppm) than the dominant
σ-diatropic ring current (-63.8 ppm). Therefore, the [
c-B
3]
- species could be considered practically as a
σ-aromatic species. Similarly, the estimated CMO-NICS
zz values for the [
c-B
3]
+ species found to be -16.6 ppm from the HOMO-3 (1
a1′), -37.2 ppm from the degenerate HOMO-1,-2 (1
e′) and only -12.3 ppm from the
π-type HOMO (1
a2″). Compare again the much weaker induced
π-diatropic ring current (-12.3 ppm) than the dominant
σ-diatropic ring current (-53.8 ppm). Finally, the estimated CMO-NICS
zz values for the putative [
c-B
3]
3+ species found to be -18.1 ppm from the HOMO-1 (1
a1′) and -31.2 ppm from the degenerate HOMO,-1 (1
e′) contribute exclusively to the induced
σ-diatropic ring current. Notice that the 1
a1′ and 1
e′ bonding and nonbonding MOs respectively are formed primarily from the overlap of the filled 2
s AOs of the boron atoms. It can be concluded that the CMO-NICS
zz analysis data reinforce the efficiency of the NICS
zz-scan curves and symmetry-based rules model to diagnose the orbital type of aromaticity of the aforementioned 2e- and 6e-
σ-aromatics.
In the [
c-B
4] (
D2h) molecule the T
x,y-allowed transitions contributing to the induced diatropic ring current are the HOMO-2 (
b3u) → LUMO (
ag), HOMO-3 (
b2u) → LUMO (
ag) and HOMO (
ag) → LUMO+4 (
b2u) excitations with excitation energies 4.58, 6.93 and 6.96 eV respectively. In addition the R
z-allowed HOMO (
ag) → LUMO+3 (
b1g) transition with excitation energy 6.27 eV contributes to the long range induced paratropic ring current (
Figure 1c). The same holds true for the [
c-B
4]
2+ (
D4h) dicationic species where the T
x,y-allowed transitions contributing to the induced diatropic ring current are the HOMO-2,-3 (
eu) → LUMO (
a1g) and HOMO (
a2u) → LUMO+2,+3 (
eu) excitations with excitation energies 6.26 and 6.36 eV, respectively, while the R
z-allowed HOMO-1 (
b2g) → LUMO+1 (
b1g) transition with excitation energy 5.25 eV contributes to the long range induced paratropic ring current. Finally, in the [
c-B
5]
+ (
D5h) molecule the T
x,y-allowed transitions contributing to the induced diatropic ring current are the HOMO-1 (
a2″) → LUMO,+1 (
e1″) and HOMO (
a1′) → LUMO+2,+3 (
e1′) excitations with excitation energies 4.86 and 4.96 eV, respectively.
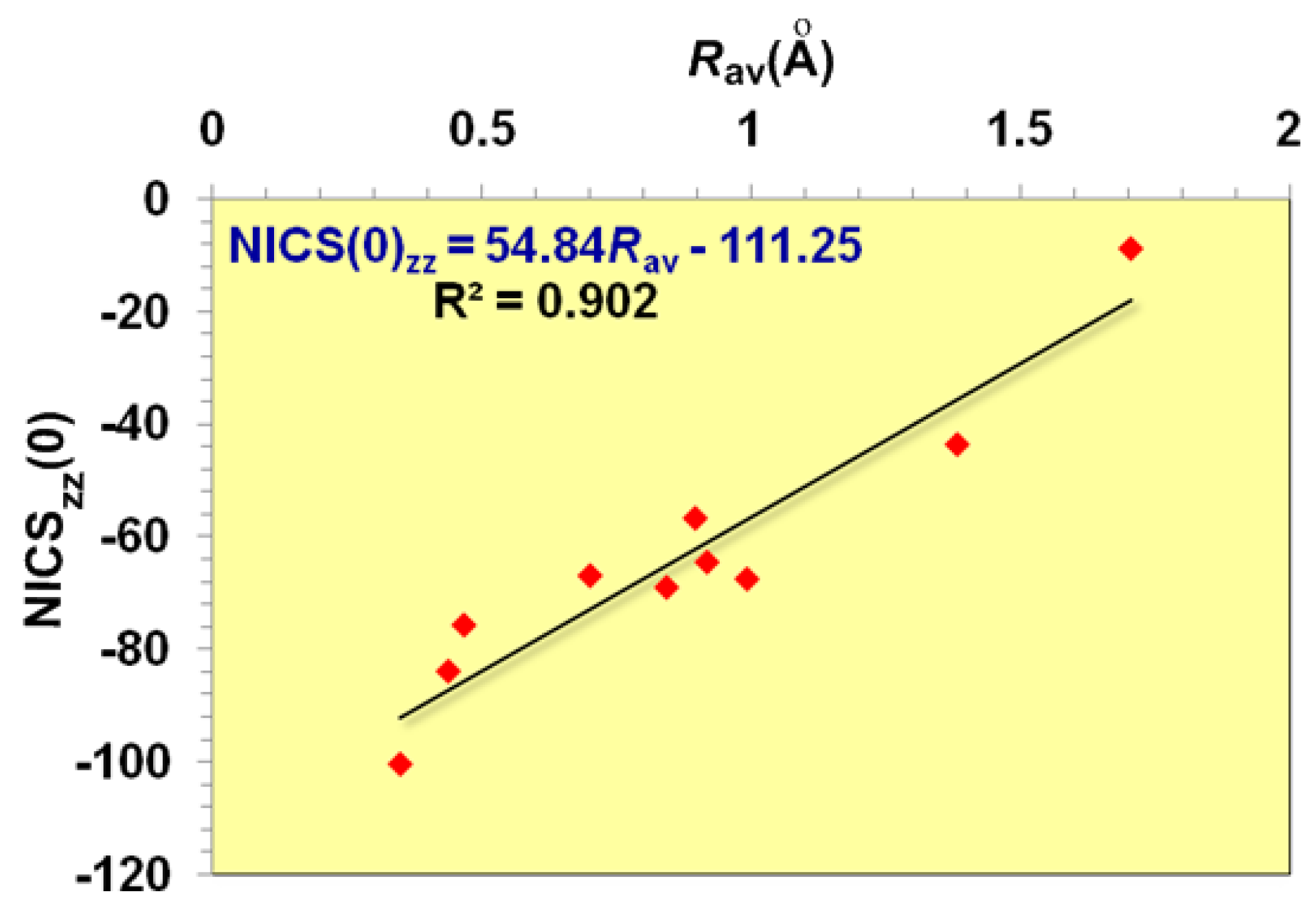
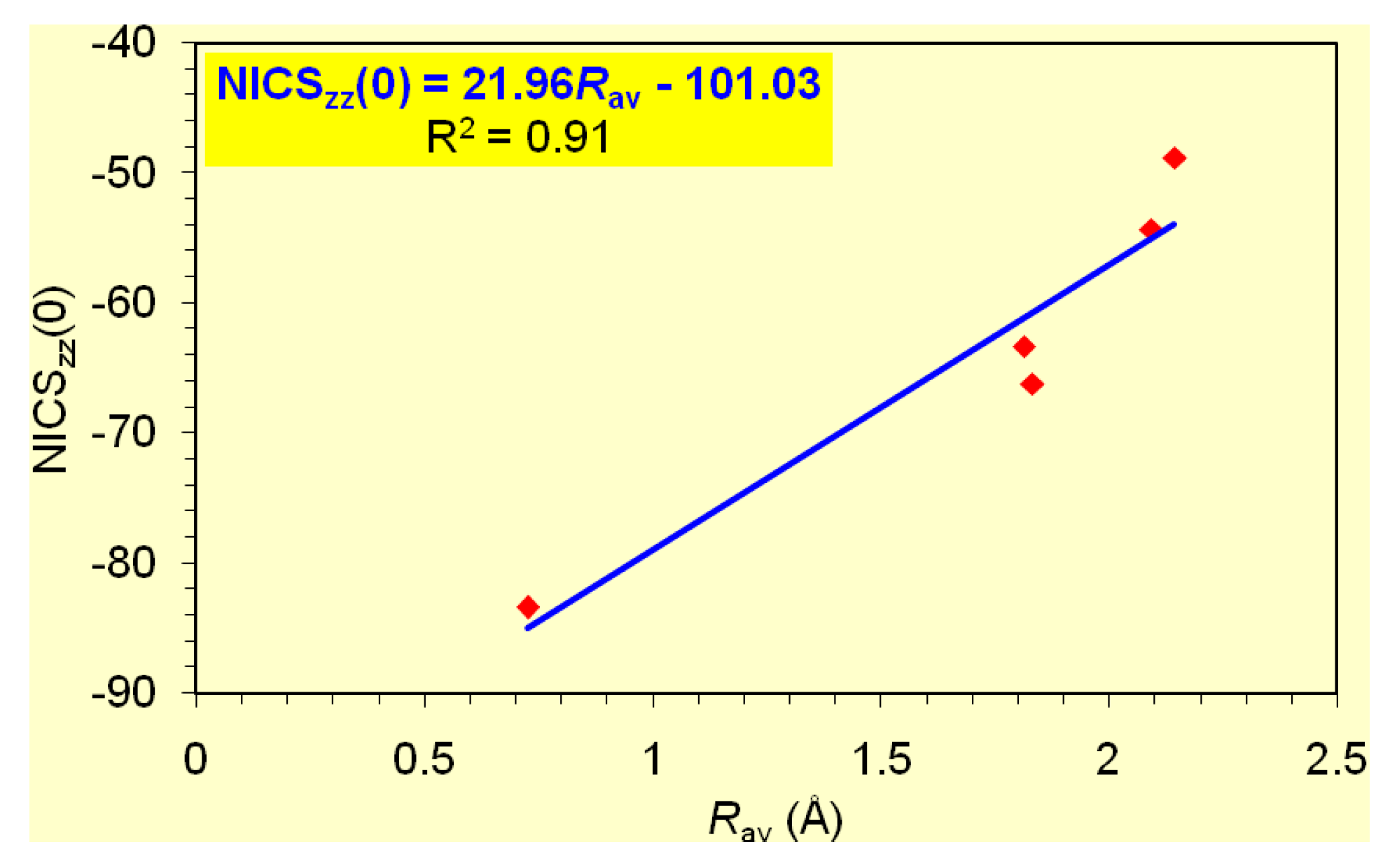
It is important to note that the dominant contribution to aromaticity of the aforementioned
σ-aromatics due to the in-plane
σ-MOs constructed from the overlap of the 2
s and/or 2
p AOs of the ring atoms is nicely reflected on the excellent linear correlation of the NICS
zz(0) with the
Rav of the rings (
Figure 2).
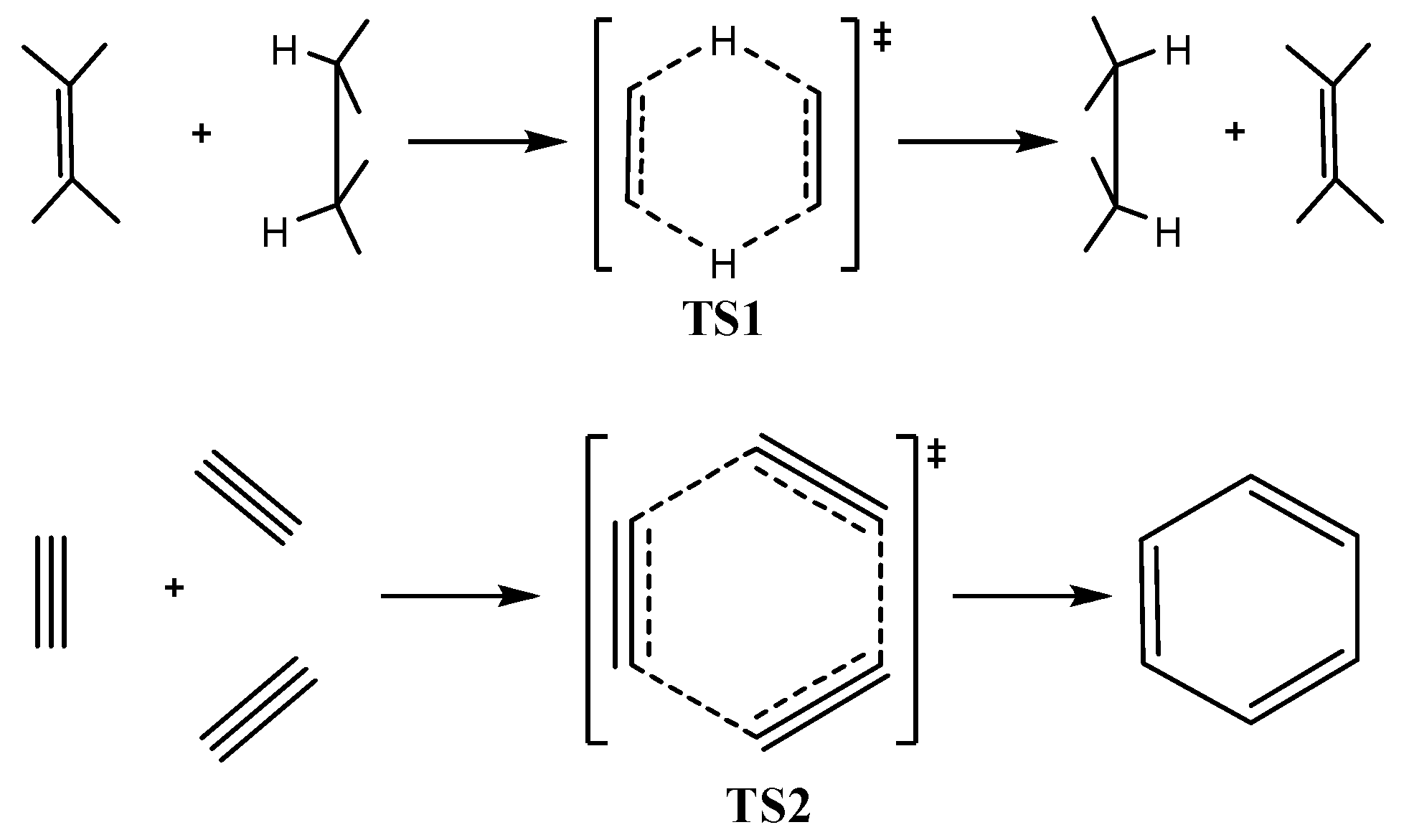
In-plane aromaticity is a general feature of transition states associated with pericyclic reactions [
25,
56,
57,
58]. Cossio
et al. [
25] using a simple ring current model based on the NICS-scan profiles of the transition states was able to characterize the in-plane and
π-aromaticity. For the transition states exhibiting in-plane aromaticity the NICS-scan curves showed a maximum at the molecular plane and decaying rapidly above and below of such plane. In contrast, in
π-aromatic systems the NICS-scan curves showed a maximum at a certain distance above and below the molecular plane, which corresponds to the covalent radii of the involved atoms. The shapes of the NICS
zz-scan curves of the transition states TS1 and TS2 for the concerted transfer of two hydrogen atoms from ethane to ethylene (double group transfer reactions) [
59] and the trimerization of acetylene to form benzene [
60,
61] respectively (
Figure 1d), provide a clear evidence for their in-plane
σ-aromatic character. Indeed the NICS
zz-scan curves are symmetric sharp curves with half-band widths less than 3 Å.
The predictive power of the NICS
zz-scan curves on the orbital type of aromaticity was further corroborated by exploring the orbital type of aromaticity of the [
c-B
3H
7]
2- (
Cs) [
62] and [
c-B
3(
μ2-P)
3]
2- (
D3h)
σ-aromatics, both involving a three-membered B
3 ring. Indeed for both species the NICS
zz-scan curves are symmetric sharp curves with half-band widths of 1.6 and 1.8 Å, respectively. The computed NICS
zz(0), NICS
zz(1) values of the [
c-B
3H
7]
2- and [
c-B
3(
μ2-P)
3]
2- species are -46.1, -18.2 ppm and -38.9, -18.2, respectively. We also computed the NICS
zz-scan curve of diborane, B
2H
6, which showed all the characteristics of the NICS
zz-scan curves of pure
σ-aromatics. It is a sharp symmetric curve with half-band width of 1.3 Å, with the NICS
zz(0) and NICS
zz(1) values being -20.8 and -6.1 ppm, respectively. On the basis of the linear relationship given in
Figure 2 and using the
Rav value of 0.717 Å, one obtains a NICS
zz(0) value of -20.4 ppm, which is in excellent agreement with the computed NICS
zz(0) value of -20.8 ppm.
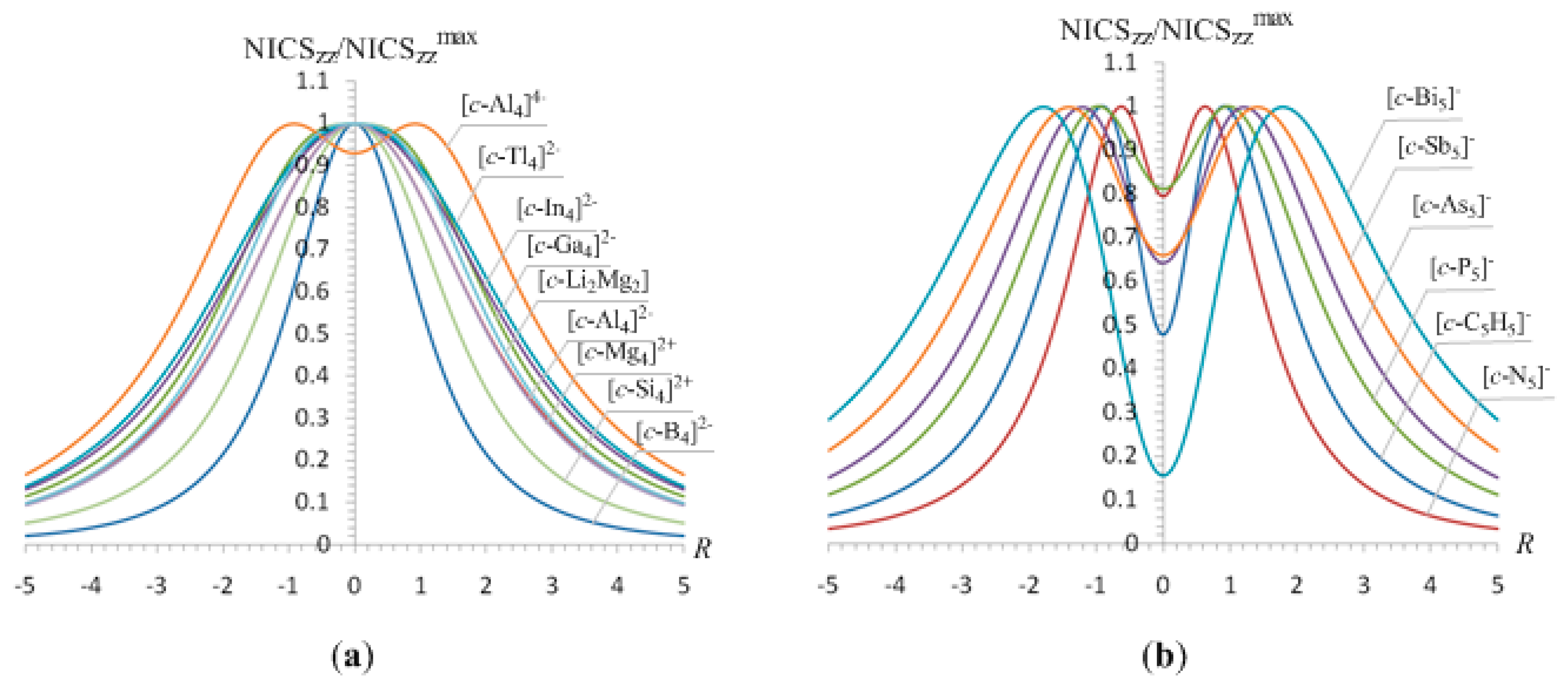
In summary, all 2e- and 6e-σ-aromatics studied herein are characterized by sharp NICSzz-scan curves symmetric around the z-axis (perpendicular to the ring plane) with the NICSzz values decaying rapidly and monotically with respect to R, and having a half-band with approximately less than 3 Å.
3.3. The case of the [c-Li3]+ and cyclopropane 2e-σ-aromatics
For the [
c-Li
3]
+ (
D3h) cation, another 2e-
σ-aromatic prototype, the NICS
zz-scan curve, has also the features of the NICS
zz-scan curves of the pure
σ-aromatic, but its half-band width is greater than 3 Å (3.7 Å). The broadening of the NICS
zz-scan curve of [
c-Li
3]
+ σ-aromatic is probably due to the bigger size of the three-membered Li
3 ring (
Rav = 1.704 Å) that renders weaker the overlap of the 2
s orbitals forming the 3c-2e bond. This is also mirrored on the estimated interaction energy between the Li
2 and Li
+ fragments (-45.66 kcal/mol), which is much smaller than the interaction energy between the H
2 and H
+ fragments to form the [
c-H
3]
+ species (-154.03 kcal/mol). Another plausible reason for the broadening of the NICS
zz-scan curve of [
c-Li
3]
+ σ-aromatic might be the contribution of the intrinsic local circulation of electrons around the nuclei to the diatropic NICS values. The pure
σ-aromaticity in the [
c-Li
3]
+ cation was previously justified by Alexandrova and Boldyrev [
63].
Considering the ring current model one would expect the
versus z and the
versus z curves to be similar:
According to quantum theory for the
occurring in the molecular plane (z = 0) for the
will have:
For two
σ-aromatics exhibiting
s-orbital aromaticity it can be easily written:
If we use as a reference
σ-aromatic the prototype [
c-H
3]
+ cation we obtain the following relationship:
On the basis of the above relationship we predict for the [c-Li3]+ cation a NICSmax value of -10.0 ppm very close to the estimated NICSzz(0) value of -11.2 ppm.
Cyclopropane being a saturated system was also found to be a strongly
σ-aromatic molecule. Cyclopropane exhibits a ring current density induced by a perpendicular external magnetic field arising in the
σ framework that is intense, diatropic and annular. The hallmarks of the
σ-ring current in cyclopropane is reflected on the shape of the NICS
zz-scan curve, which corresponds to a symmetric curve with a maximum at z = 0 and a half-band width around 3.2 Å. Moreover, the main feature of the strong diatropic circulation in [
c-C
3H
6], which reaches maximal intensity (
jmax = 0.121 a.u.) outside the line of carbon centers [
64,
65], accounts well for the slight broadening of the NICS
zz-scan curve (half-band width > 3 Å). The calculation of the CMO-NICS
zz values for cyclopropane in
D3h symmetry showed that the Walsh orbitals HOMO,-1 (2
e′), being
σ-type MOs, have positive NICS
zz value of 22.2 ppm, the HOMO-2,-3 (1
e″), being
π-type MOs, have also positive NICS
zz value of 8.1 ppm and therefore induce
σ- and
π-type paratropic ring currents, respectively. On the other hand, the HOMO-5 (1
a2″), a
π-type MO, has negative NICS
zz value of -8.7 ppm, which cancels the induced π-paratropic ring current from the 1
e″ MOs. The remaining HOMO-4 (2
a1′), HOMO-6,-7 (1
e′) and HOMO-8 (1
a1′) all being
σ-type MOs, have negative NICS
zz values of -5.4, -18.8 and -21.3 ppm respectively. The CMO-NICS
zz results are nicely mirrored on the shape of the NICS
zz-scan curve of cyclopropane. It is interesting to notice that Pelloni
et al. [
65] demonstrated that both the large negative NICS value and anisotropies of cyclopropane are mainly determined by the in-plane components rather than the more relevant out-of-plane contribution. The existence of
σ-aromaticity in cyclopropane has recently been challenged by
ab initio valence bond (VBSCF/cc-PVTZ) computations which revealed directly that the
σ-aromatic stabilization energy of cyclopropane is, at most, 3.5 kcal/mol relative to propane [
66]. However, such small energy difference raised the question whether the cyclopropane is really the
σ-aromatic paradigm.
3.4. The NICSzz-scan curves of the M3(μ2-CO)3(CO)3 (M = Ni, Pd, Pt) molecules predict their σ-aromaticity
Let us now explore the efficiency of the NICS
zz-scan profiles to predict the pure
σ-aromaticity characterizing the M
3(
μ2-CO)
3(CO)
3 (M = Ni, Pd, Pt) molecules. Recently, R. B. King [
67] applied the
σ-aromaticity model used for cyclopropane to Group 8 trinuclear metal-carbonyl complexes, [M
3(CO)
12] (M = Fe, Ru, Os). The
σ-aromaticity of these molecules was further verified by a structural and NICS analysis reported recently [
68]. According to this model the bonding in the M
3 triangles is composed by a core 3c-2e bond of Hückel topology formed by overlap of inward pointing radial hybrid orbitals of M and a surface (in-plane) 3c-4e bond of Möbius topology formed by tangential
p-orbitals of M. Notice that for the M
3(
μ2-CO)
3(CO)
3 (M = Ni, Pd, Pt) molecules the presence of the
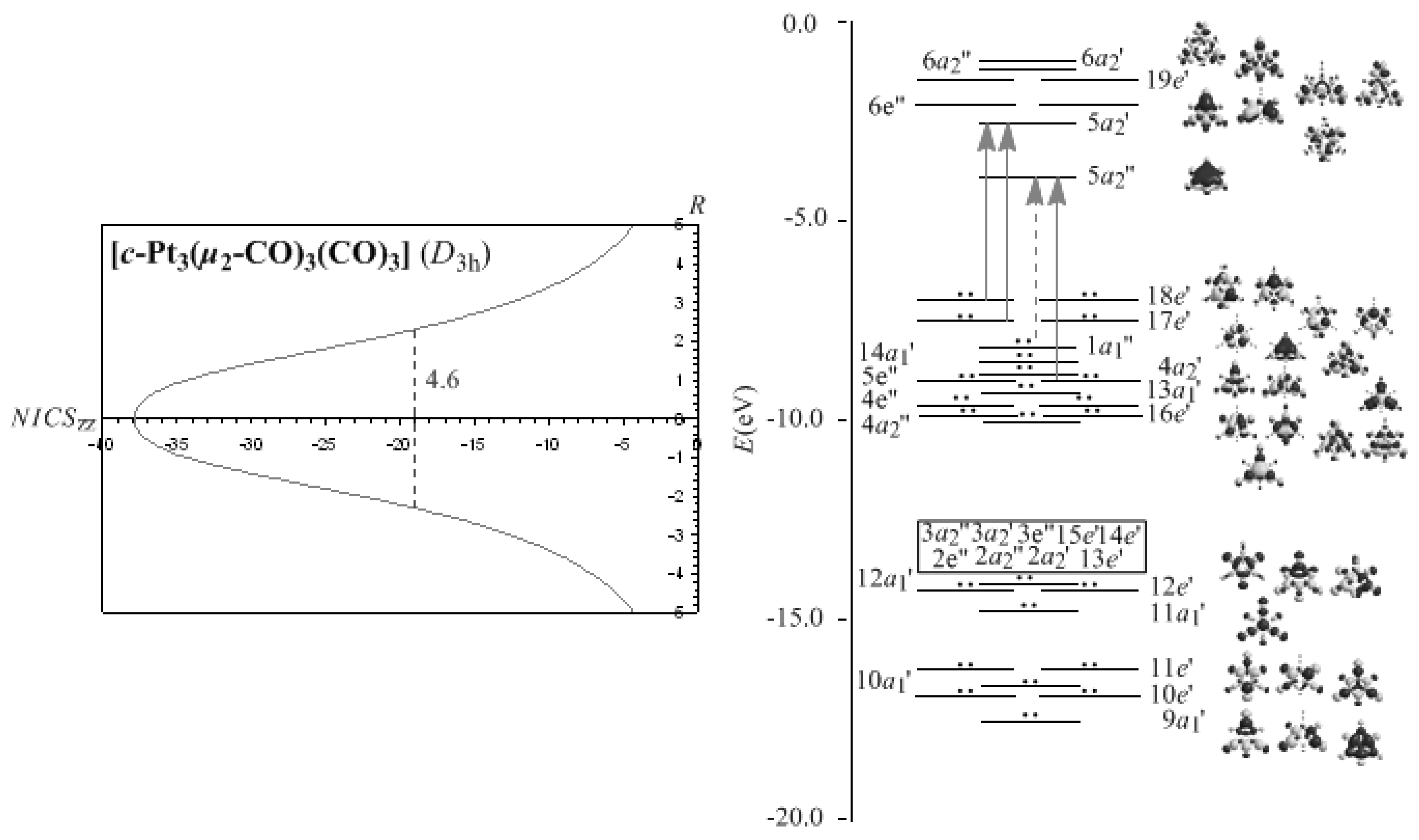
μ2-CO ligands precludes the 3c-4e Möbius surface bonding orbitals. The NICS
zz-scan curves of the Pt
3(
μ2-CO)
3(CO)
3 molecule along with the molecular orbital energy level diagram, symmetries, 3D molecular orbital pictures and lowest T
x,y-allowed transitions contributing to the diamagnetic ring current are visualized in
Figure 3, while the most salient features of the NICS
zz-scan curves along with the half-band widths and
Rav values of the rings of the M
3(
μ2-CO)
3(CO)
3 (M = Ni, Pd, Pt) molecules are compiled in
Table 2.
It can be seen that the NICSzz-scan curves of the M3 (M = Ni, Pd, Pt) clusters are sharp symmetric curves with half-band width less than 2 Å, illustrating their pure σ-aromatic character. The broadening of the NICSzz-scan curves of the M3(μ2-CO)3(CO)3 (M = Ni, Pd, Pt) molecules (half-band width of 2.7-4.6 Å), could be attributed to the contribution of the diatropic NICS values of the strongly σ-aromatic c-M2C rings to the diatropic NICS values of the M3 core rings.
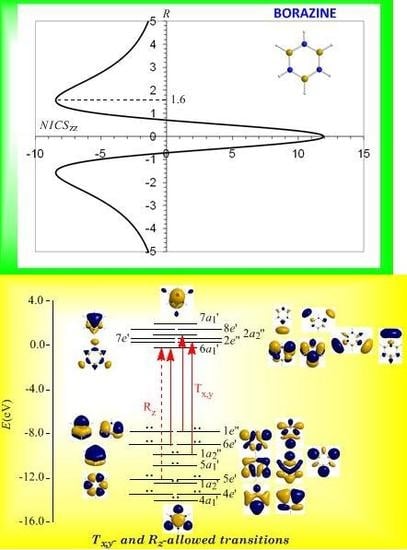
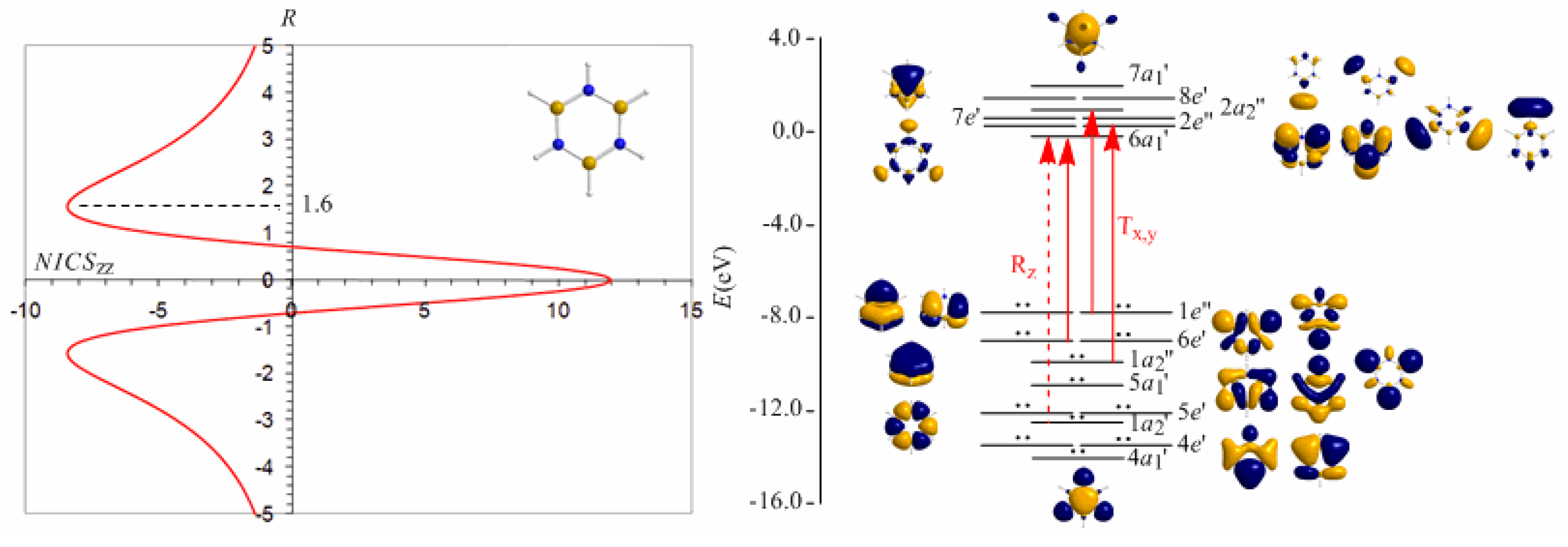
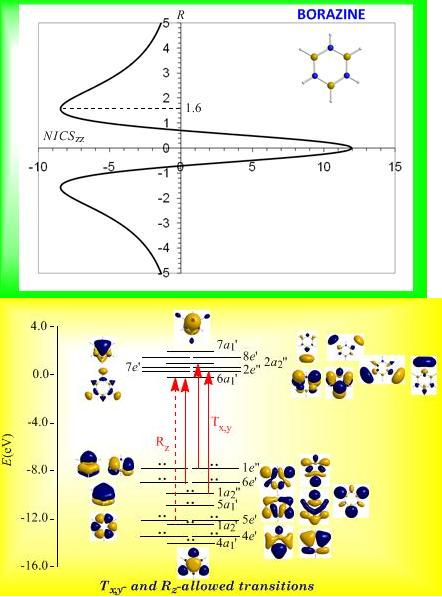
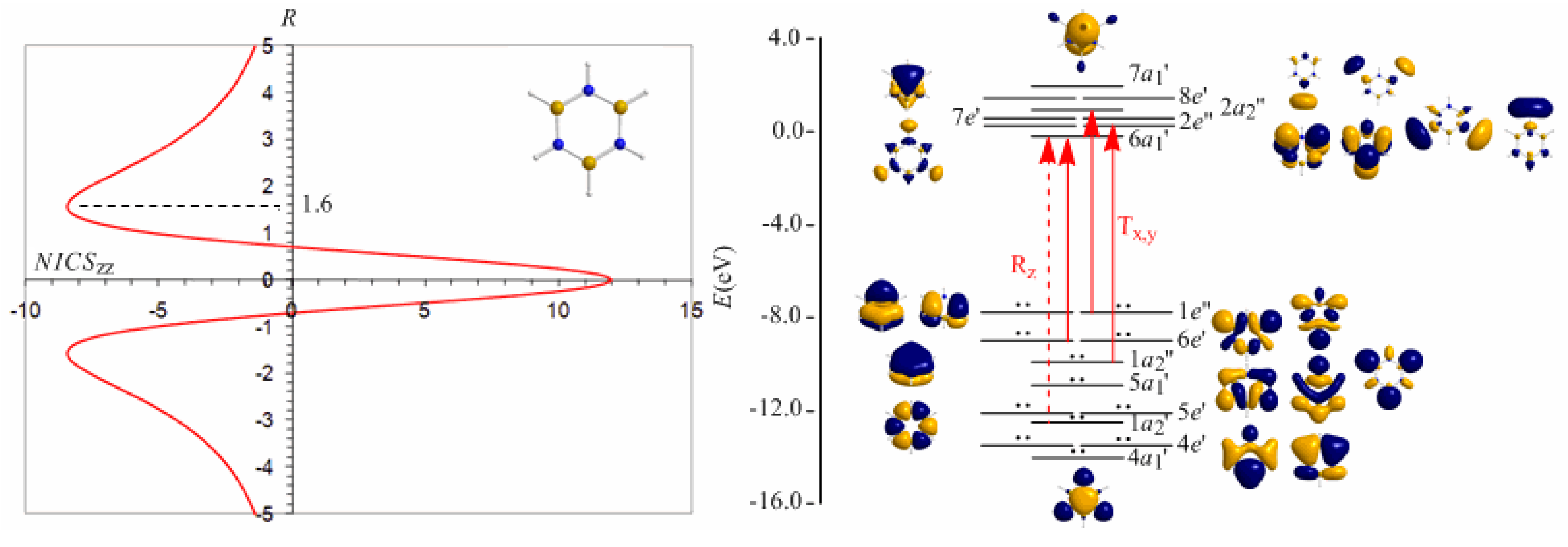
3.8. Predicting the peculiar aromaticity of borazine
Borazine,
c-B
3N
3H
6 - the boron-nitrogen six-membered ring compound often referred to as “inorganic benzene - has been subject of controversial discussions with respect to the aromaticity of this molecule. Very recently Islas
et al. [
94] in their publication entitled
Borazine: to be or not to be aromatic based on a detailed analysis of two molecular fields, the induced magnetic field (
Bind) and the electron localization function (ELF) indicated that borazine can be described as a
π-aromatic compound but it is not a globally aromatic species, since the delocalized system is not as homogeneous as that of benzene. The induced magnetic field in borazine is weaker than that of benzene but nevertheless pronounced and long-ranged. The NICS
zz-scan curve of borazine shown in
Figure 10 clearly demonstrates the long-range
π-aromaticity and the in-plane
σ-antiaromaticity of borazine. It shows a maximum negative (diatropic) NICS
zz value of -8.4 ppm at z = 1.6 Å above and below the ring and a maximum positive (paratropic) NICS
zz value of 12.0 ppm at z = 0.
The estimated excitation energies of the lowest T
x,y-allowed transitions are for the
π-type
e″ →
a2″ and
a2″ →
e″ excitations 8.57 eV and 10.19 eV respectively and for the
σ-type
e′ →
a1′ excitation 9.05 eV. The latter is outweighed by the T
x,y-allowed
a2′ →
a1′ transition with excitation energy 12.04 eV. The relatively high excitation energies lead to weak induced
π-diatropic ring currents, which nicely explain the substantial decrease of the
π-electron delocalization relative to benzene (compare the NICS
zzmax values of -8.4 and -29.2 ppm, for the borazine and benzene molecules, respectively). It is interesting to notice that the local contributions of the
σ electrons in borazine according to the calculations carried out by Islas
et al. [
94] generate a short-range response and a paratropic (antiaromatic) region in the center of the ring, while the
π-orbital contribution to
Bind shows the typical response of an aromatic system. This is exactly the topology of the total ring currents represented by the NICS
zz-scan curve of borazine (
Figure 10).
3.9. Applying the magnetic NICSzz-scan criterion to diagnose aromaticity/antiaromaticity in the non-planar corannulene, corannulene dianion and sumarene molecules
The bowl-shaped corannulene, C
20H
10, corannulene dianion, [C
20H
10]
2- and sumarene, C
21H
12, polycyclic hydrocarbons, which share a fullerene substructure, have been studied extensively in recent years [
95,
96]. Such studies have emphasized the description of their aromaticity and electron interactions on curved surfaces from both the experimental and theoretical perspectives.
The counter-rotating ring currents in coronene and corannulene have been investigated by Jenneskens
et al. [
97] Very recently Monaco and Zanasi [
98] calculated the
π current of aromatic polycyclic hydrocarbons placed in a uniform magnetic field and found substantial localization on subunits in some cases. Moreover, Zanasi
et al. [
99] in their recent publication entitled
Magnetic Euripi in Corannulene computed
ab initio current densities for corannulene dianion, dication and tetraanion and found large
π-ring currents with respect to benzene which undergo remarkable changes in response to variation in the oxidation state. The three corannulene ions along with the neutral species constitute a full set that spans all of the possible patterns of rim and hub circulations. Thus, for the neutral corannulene the magnetotropicity corresponds to paratropic/hub-diatropic/rim pattern, while for the corannulene dianion to diatropic/hub-paratropic/rim pattern.
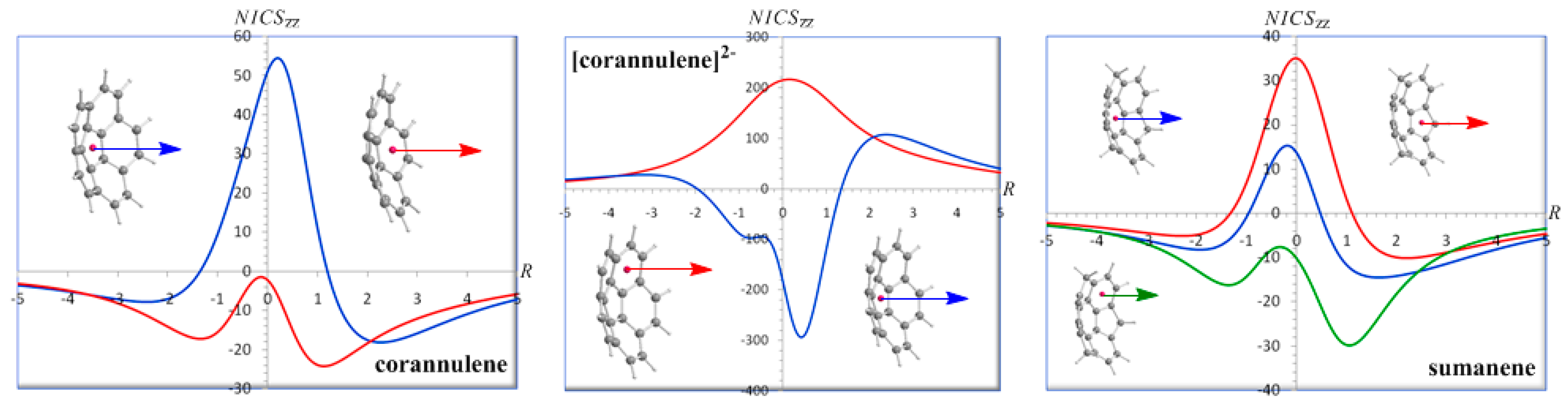
The NICS
zz-scan curves of corannulene, corannulene dianion and sumanene computed for both the inner (hub) and outer (rim) rings are shown in
Figure 11.
The NICS
zz-scan curve of the inner (hub) five-membered ring of corannulene clearly demonstrates the antiaromatic character of the ring (maximum NICS
zz(0.2) value of 54.5 ppm) in line with the computed current-density maps for the
σ,
π, and total (
σ +
π) electrons at 1 a
0 above the molecular planes showing a paramagnetic counter-circulation on the inner hub [
97,
98,
99]. The NICS
zz-scan curve of the inner (hub) five-membered ring of corannulene is similar to the NICS
zz-scan curve of the highly antiaromatic cross-conjugated annelated five-membered ring system, acepentalene, [
45] corresponding to relatively narrow curve with half-band width of 1.3 Å. On the other hand, the NICS
zz-scan curve of the outer (rim) six-membered rings of corannulene exhibiting two maxima at a certain distance above and below the molecular plane and having NICS
zz(0) value close to zero indicates their pure
π-type aromatic character (minimum NICS
zz(1.1) and NICS
zz(-1.3) values of -24.1 and -17.1 ppm respectively), which is also consistent with the computed current-density maps showing a clear diamagnetic circulation around the outer rim [
97,
98,
99]. It is important to be noticed that both the central paramagnetic (paratropic) and the outer diamagnetic (diatropic) induced ring currents are enhanced inside the bowl but depleted on the outside as a result of ring current superpositions.
The NICS
zz-scan curve of the inner (hub) five-membered ring exhibiting two maxima at a certain distance above and below the molecular plane and having NICS
zz(0) value close to zero clearly demonstrate the high
π-aromaticity (diatropicity) of the hub (minimum NICS
zz(0.4) value of -293.8 ppm). The NICS
zz-scan curve of the outer (rim) six-membered rings of corannulene dianion is typical of highly in-plane antiaromatic systems [
45] (maximum NICS
zz(0.2) value of 216.6 ppm). Notice again that the inner (hub) diamagnetic (diatropic) induced ring current is strongly enhanced inside the bowl but depleted on the outside. It is important to be noted that the NICS
zz-scan curves of corannulene and corannulene dianion correctly predict the paratropic/hub-diatropic/rim and diatropic/hub-paratropic/rim patterns respectively obtained by the
ab initio computed probability current densities [
99].
The NICS
zz-scan curve of the inner six-membered ring of sumanene indicates paratropicity at the ring center with a maximum NICS
zz(-0.2) value of 15.3 ppm and a long range diatropicity with minimum NICS
zz(1.7) and NICS
zz(-1.9) values of -14.5 and -8.2 ppm respectively. Therefore the inner six-membered ring of sumanene can be considered practically as non aromatic. Notice again the enhancement of the induced diatropic ring currents inside the bowl. The NICS
zz-scan curve of the outer five-membered ring of sumanene (a typical curve for in-plane antiaromatic systems) [
45] clearly illustrates the antiaromatic character of the ring, while the NICS
zz-scan curve of the outer six-membered ring of sumanene exhibiting two maxima at a certain distance above and below the molecular plane and having NICS
zz(0) value close to 0 is characteristic of the pure
π-aromaticity enhanced inside the bowl (NICS
zz(1.1) = -29.8 ppm), but depleted on the outside (NICS
zz(-1.4) = -16.2 ppm).
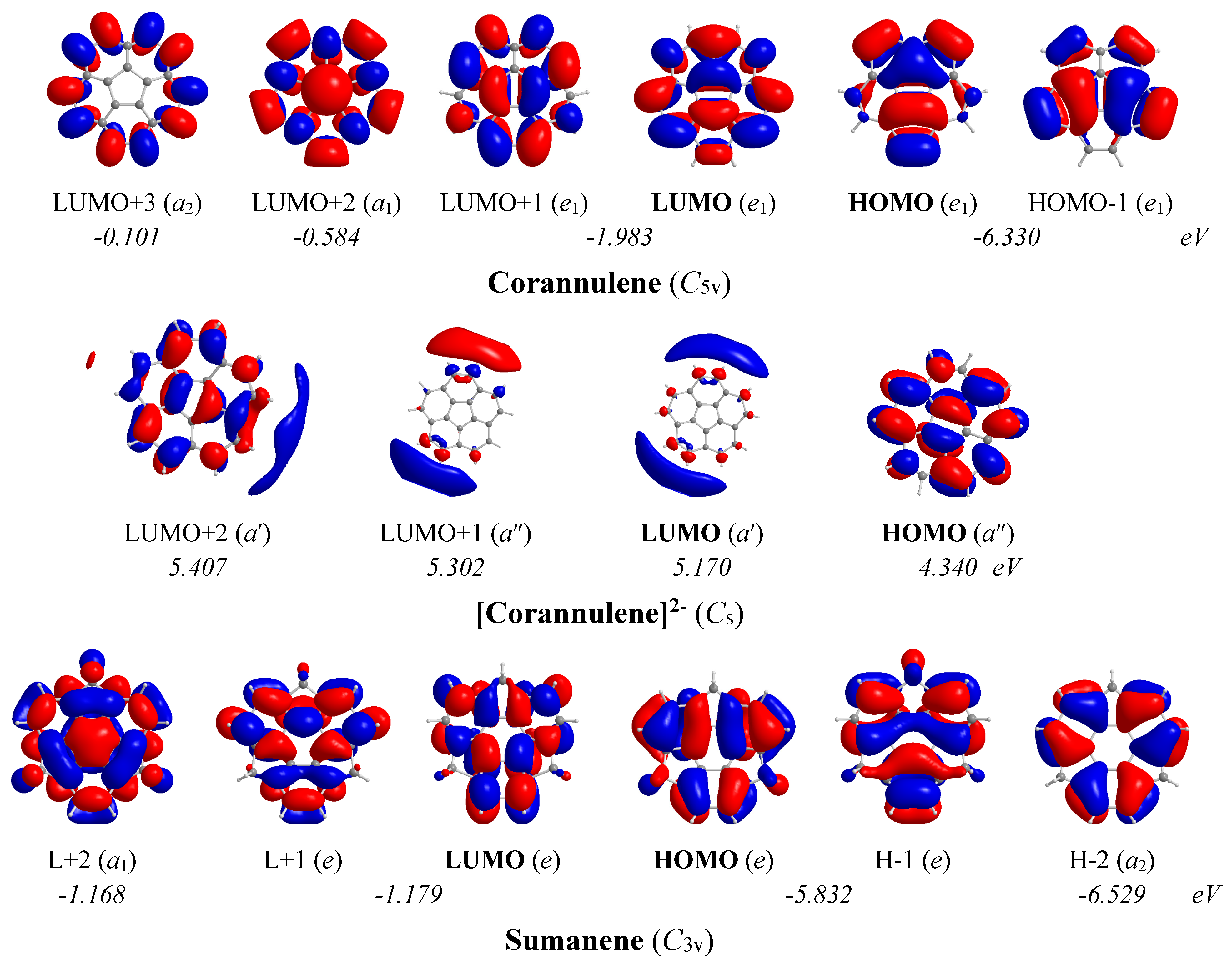
The 3D molecular orbital pictures of the MOs participating in the lowest T
x,y- and R
z-allowed transitions in corannulene, corannulene dianion and sumanene are shown in
Figure 12.
In the corannulene molecule with C5v symmetry, the chief contribution to induced diatropic ring current arises from the Tx,y-allowed HOMO,-1 (e1) → LUMO+2 (a1) and HOMO,-1 (e1) → LUMO+3 (a2) transitions with excitation energies 5.75 and 6.23 eV respectively. On the other hand, the chief contribution to induced paratropic ring current arises from the Rz-allowed HOMO,-1 (e1) → LUMO,+1 (e1) transition with excitation energy 4.35 eV. For the corannulene dianion with Cs symmetry, the chief mixed contribution to induced paratropic and diatropic ring currents arises from the lowest Rz- and Tx,y-allowed HOMO (a″) → LUMO (a′), HOMO (a″) → LUMO+1 (a″) and HOMO (a″) → LUMO+2 (a′) transitions with excitation energies 0.83, 0.96 and 1.07 eV respectively.
In the sumanene molecule with C3v symmetry, the chief contribution to induced diatropic ring current arises from the lowest Tx,y-allowed HOMO,-1 (e) → LUMO+2 (a1) transition with excitation energy 4.66 eV. Furthermore, the chief contribution to induced paratropic ring current arises from the lowest Rz-allowed HOMO,-1 (e) → LUMO,+1 (e) and HOMO-2 (a2) → LUMO+2 (a1) transitions with excitation energies 4.65 and 5.36 eV, respectively.
The estimated excitation energies of the lowest Tx,y- and Rz-allowed transitions of the corannulene, corannulene dianion and sumanene molecules in conjunction with the 3D molecular orbital pictures of the relevant MOs account well for the magnetotropicity patterns derived from the shapes of the NICSzz-scan curves.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}