
Structural Properties and Biological Prediction of ({[(1E)-3-(1H-Imidazol-1-yl)-1-phenylpropylidene] amino}oxy)(4-methylphenyl)methanone: An In Silico Approach

 ,
,
Abstract
:1. Introduction
2. Experimental
2.1. General
2.2. Synthesis
2.3. Spectroscopic Measurements
2.4. Quantum Chemical Calculations
3. Results and Discussion
3.1. Synthesis
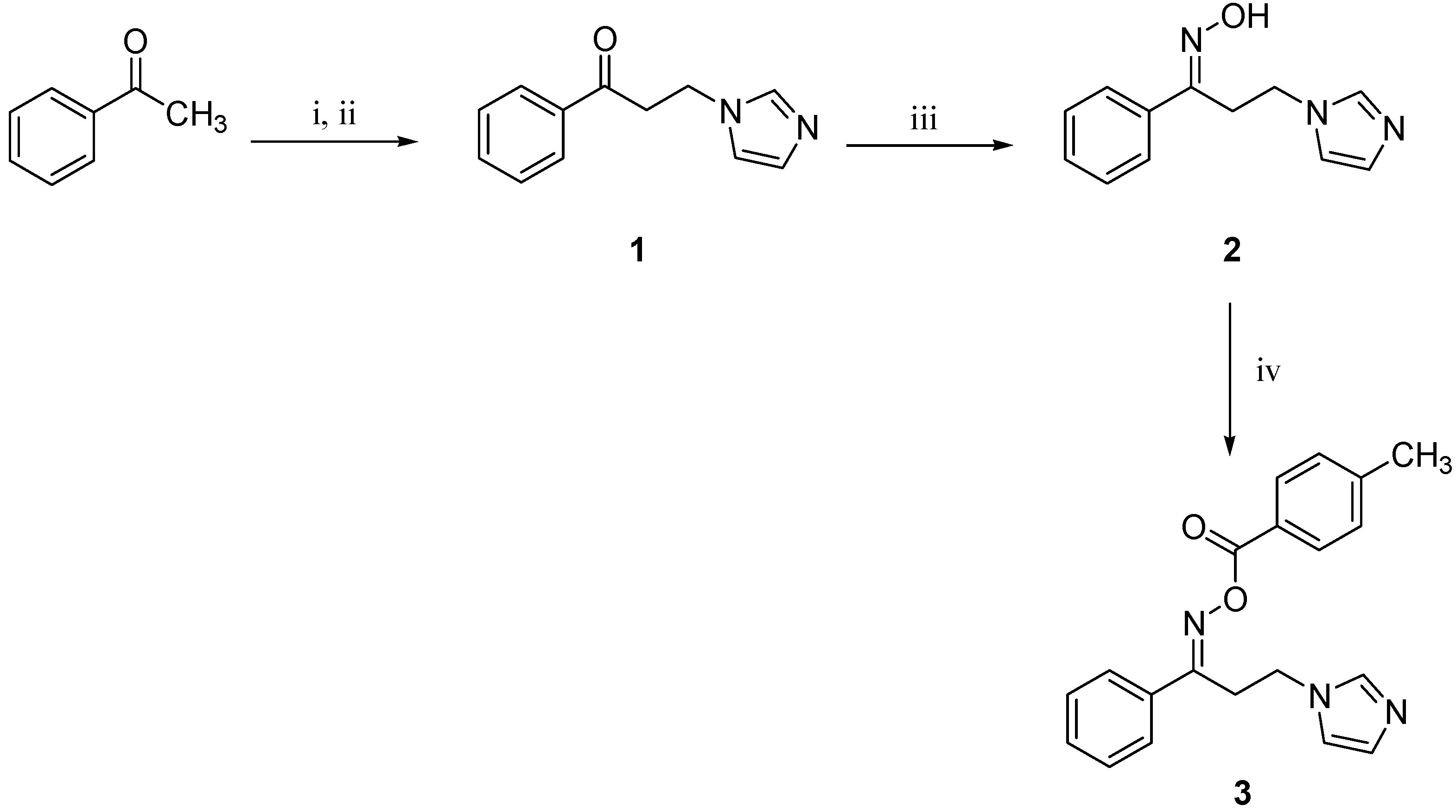
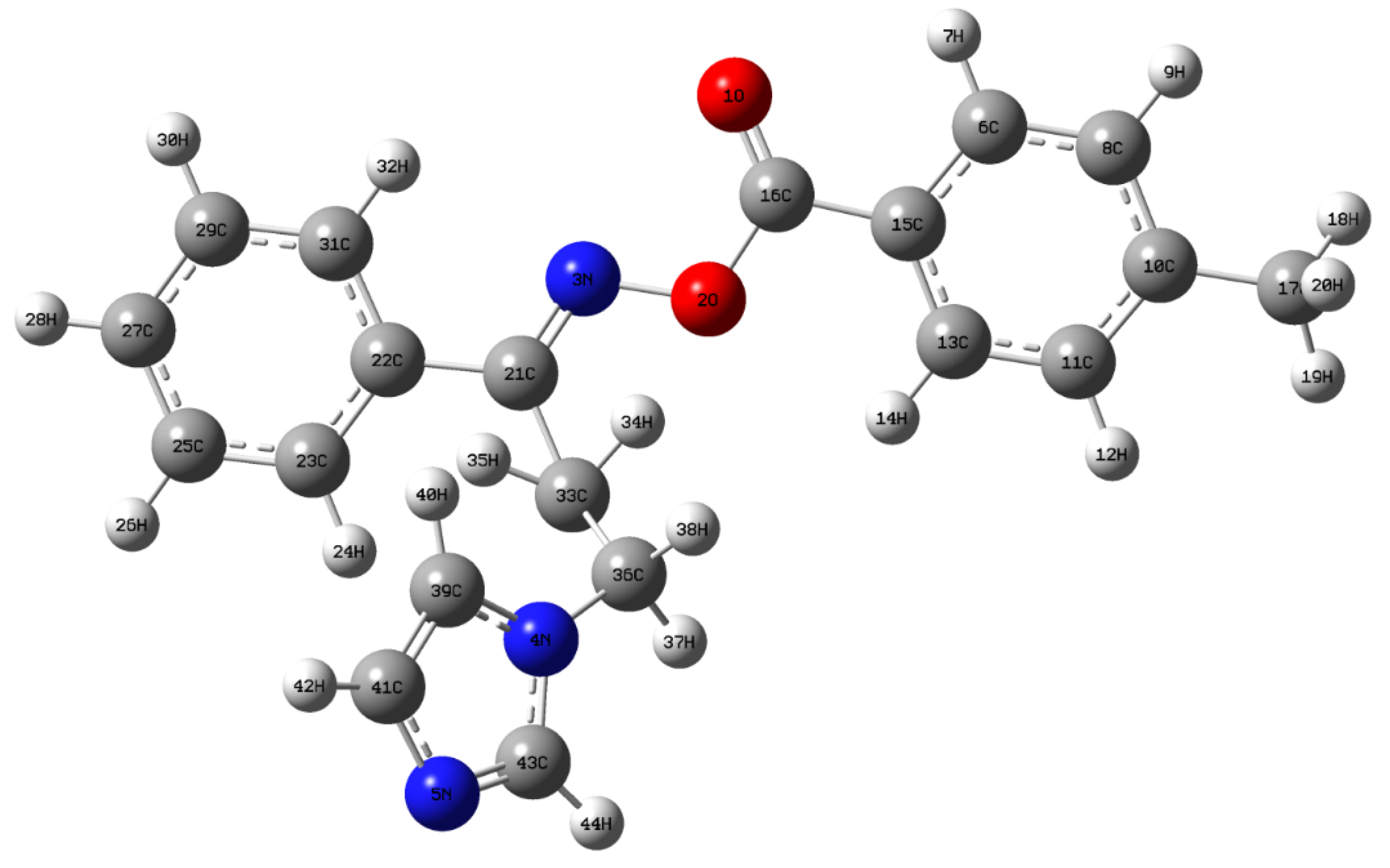
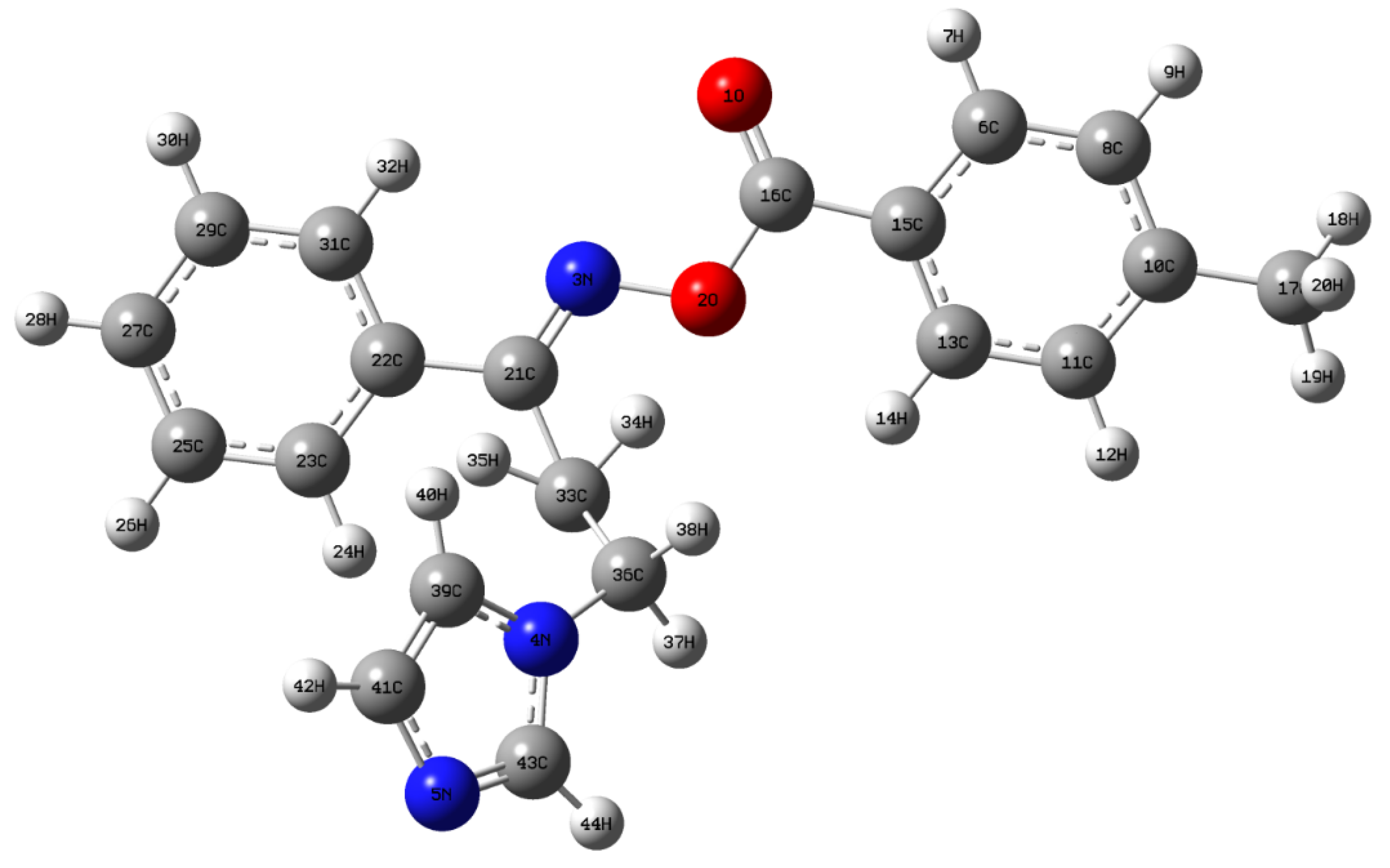
3.2. Structural Geometry Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond Length (Å) | Bond Angle (°) | Dihedral Angle (°) | ||||||
|---|---|---|---|---|---|---|---|---|
| Parameters | Calculated | Experimental | Parameters | Calculated | Experimental | Parameters | Calculated | Experimental |
| O1–C16 | 1.1993 | 1.1960 | N3–O2–C16 | 113.03 | 112.71 | C16–O2–N3–C21 | −169.72 | −174.61 |
| O2–C16 | 1.3828 | 1.360 | O2–N3–C21 | 111.26 | 108.99 | N3–O2–C16–O1 | 9.74 | 4.71 |
| O2–N3 | 1.4179 | 1.438 | C36–N4–C39 | 127.25 | 127.11 | N3–O2–C16–C15 | −171.56 | −175.29 |
| N3–C21 | 1.2859 | 1.283 | C36–N4–C43 | 126.44 | 126.39 | O2–N3–C21–C22 | −179.53 | 178.96 |
| N4–C36 | 1.4568 | 1.456 | C39–N4–C43 | 106.29 | 106.49 | O2–N3–C21–C33 | 0.84 | −0.30 |
| N4–C39 | 1.3817 | 1.360 | C41–N5–C43 | 105.23 | 104.26 | C39–N4–C36–C33 | −87.08 | −87.21 |
| N4–C43 | 1.3692 | 1.345 | H7–C6–C8 | 120.88 | 119.87 | C39–N4–C36–H37 | 152.42 | 151.25 |
| N5–C41 | 1.3757 | 1.365 | H7–C6–C15 | 118.76 | 119.79 | C39–N4–C36–H38 | 36.49 | 34.35 |
| N5–C43 | 1.3131 | 1.310 | C8–C6–C15 | 120.36 | 120.35 | C43–N4–C36–C33 | 90.66 | 94.49 |
| C6–H7 | 1.0831 | 0.930 | C6–C8–H9 | 119.42 | 119.36 | C43–N4–C36–H37 | −29.84 | −27.05 |
| C8–H9 | 1.0852 | 0.930 | C6–C8–C10 | 121.1 | 121.28 | C43–N4–C36–H38 | −145.77 | −143.95 |
| C11–H12 | 1.0851 | 0.930 | H9–C8–C10 | 119.48 | 119.36 | C36–N4–C39–H40 | −1.95 | 1.61 |
| C13–H14 | 1.0818 | 0.929 | C8–C10–C11 | 118.14 | 117.89 | C36–N4–C39–C41 | 178.18 | −178.44 |
| C17–H18 | 1.0926 | 0.960 | C8–C10–C17 | 120.78 | 121.19 | C43–N4–C39–H40 | 179.94 | −179.82 |
| C17–H19 | 1.0918 | 0.959 | C11–C10–C17 | 121.08 | 120.92 | C43–N4–C39–C41 | 0.07 | 0.13 |
| C17–H20 | 1.0955 | 0.960 | C10–C11–H12 | 119.44 | 119.22 | C36–N4–C43–N5 | −178.27 | 179.09 |
| C23–H24 | 1.0825 | 0.930 | C10–C11–C13 | 121.19 | 121.53 | C36–N4–C43–H44 | 2.52 | −0.95 |
| C25–H26 | 1.0839 | 0.930 | H12–C11–C13 | 119.37 | 119.26 | C39–N4–C43–N5 | −0.14 | 0.50 |
| C27–H28 | 1.0841 | 0.930 | C11–C13–H14 | 119.9 | 120.03 | C39–N4–C43–H44 | −179.35 | −179.53 |
| C29–H30 | 1.0841 | 0.930 | C11–C13–C15 | 120.2 | 119.88 | C43–N5–C41–C39 | −0.1 | 0.97 |
| C31–H32 | 1.0823 | 0.930 | H14–C13–C15 | 119.89 | 120.10 | C43–N5–C41–H42 | −179.75 | −179.11 |
| C33–H34 | 1.0926 | 0.970 | C6–C15–C13 | 119.02 | 119.02 | C41–N5–C43–N4 | 0.15 | −0.89 |
| C33–H35 | 1.0887 | 0.970 | C6–C15–C16 | 117.54 | 118.37 | C41–N5–C43–H44 | 179.32 | 179.14 |
| C36–H37 | 1.0924 | 0.970 | C13–C15–C16 | 123.43 | 122.51 | H7–C6–C8–H9 | 0.17 | 1.56 |
| C36–H38 | 1.0894 | 0.970 | O1–C16–O2 | 123.93 | 123.87 | H7–C6–C8–C10 | −179.63 | −178.47 |
| C39–H40 | 1.0774 | 0.930 | O1–C16–C15 | 125.36 | 125.55 | C15–C6–C8–H9 | 179.86 | −178.43 |
| C41–H42 | 1.0788 | 0.931 | O2–C16–C15 | 110.69 | 110.58 | C15–C6–C8–C10 | 0.05 | 1.55 |
| C43–H44 | 1.0806 | 0.930 | C10–C17–H18 | 111.36 | 109.45 | H7–C6–C15–C13 | 179.4 | −179.42 |
| C6–C8 | 1.3879 | 1.377 | C10–C17–H19 | 111.49 | 109.47 | H7–C6–C15–C16 | 0.28 | 3.90 |
| C6–C15 | 1.4004 | 1.388 | C10–C17–H20 | 110.72 | 109.44 | C8–C6–C15–C13 | −0.29 | 0.56 |
| C8–C10 | 1.4013 | 1.388 | H18–C17–H19 | 108.26 | 109.44 | C8–C6–C15–C16 | −179.41 | −176.12 |
| C10–C11 | 1.3993 | 1.388 | H18–C17–H20 | 107.27 | 109.53 | C6–C8–C10–C11 | 0.3 | −1.94 |
| C10–C17 | 1.5079 | 1.508 | H19–C17–H20 | 107.55 | 109.50 | C6–C8–C10–C17 | −178.79 | 178.36 |
| C11–C13 | 1.3911 | 1.380 | N3–C21–C22 | 114.73 | 114.19 | H9–C8–C10–C11 | −179.51 | 178.03 |
| C13–C15 | 1.3993 | 1.391 | N3–C21–C33 | 123.56 | 124.71 | H9–C8–C10–C17 | 1.41 | −1.67 |
| C15–C16 | 1.4884 | 1.481 | C22–C21–C33 | 121.72 | 121.09 | C8–C10–C11–H12 | 179.25 | −179.71 |
| C21–C22 | 1.4883 | 1.487 | C21–C22–C23 | 121.7 | 121.76 | C8–C10–C11–C13 | −0.42 | 0.26 |
| C21–C33 | 1.5146 | 1.503 | C21–C22–C31 | 119.81 | 119.94 | C17–C10–C11–H12 | −1.66 | −0.01 |
| C22–C23 | 1.4012 | 1.390 | C23–C22–C31 | 118.48 | 118.29 | C17–C10–C11–C13 | 178.67 | 179.96 |
| C22–C31 | 1.406 | 1.392 | C22–C23–H24 | 120.8 | 119.69 | C8–C10–C17–H18 | −38.01 | −39.32 |
| C23–C25 | 1.3936 | 1.383 | C22–C23–C25 | 120.73 | 120.50 | C8–C10–C17–H19 | −159.05 | −159.26 |
| C25–C27 | 1.3913 | 1.367 | H24–C23–C25 | 118.46 | 119.82 | C8–C10–C17–H20 | 81.25 | 80.72 |
| C27–C29 | 1.396 | 1.377 | C23–C25–H26 | 119.55 | 119.69 | C11–C10–C17–H18 | 142.93 | 140.99 |
| C29–C31 | 1.3883 | 1.378 | C23–C25–C27 | 120.24 | 120.60 | C11–C10–C17–H19 | 21.89 | 21.05 |
| C33–C36 | 1.5455 | 1.520 | H26–C25–C27 | 120.21 | 119.72 | C11–C10–C17–H20 | −97.81 | −98.97 |
| C39–C41 | 1.3711 | 1.348 | C25–C27–H28 | 120.19 | 120.26 | C10–C11–C13–H14 | 179.38 | −178.19 |
| C25–C27–C29 | 119.56 | 119.52 | C10–C11–C13–C15 | 0.18 | 1.81 | |||
| H28–C27–C29 | 120.25 | 120.22 | H12–C11–C13–H14 | −0.29 | 1.78 | |||
| C27–C29–H30 | 120.07 | 119.71 | H12–C11–C13–C15 | −179.49 | −178.22 | |||
| C27–C29–C31 | 120.36 | 120.61 | C11–C13–C15–C6 | 0.17 | −2.21 | |||
| H30–C29–C31 | 119.57 | 119.61 | C11–C13–C15–C16 | 179.24 | 174.32 | |||
| C22–C31–C29 | 120.63 | 120.48 | H14–C13–C15–C6 | −179.02 | 177.8 | |||
| C22–C31–H32 | 118.98 | 119.79 | H14–C13–C15–C16 | 0.05 | −5.67 | |||
| C29–C31–H32 | 120.38 | 119.74 | C6–C15–C16–O1 | 6.16 | 12.52 | |||
| C21–C33–H34 | 108.61 | 108.90 | C6–C15–C16–O2 | −172.53 | −167.47 | |||
| C21–C33–H35 | 110.58 | 108.89 | C13–C15–C16–O1 | −172.92 | −164.03 | |||
| C21–C33–C36 | 113.79 | 113.42 | C13–C15–C16–O2 | 8.39 | 15.97 | |||
| H34–C33–H35 | 107.62 | 107.70 | N3–C21–C22–C23 | 156.34 | 150.54 | |||
| H34–C33–C36 | 107.5 | 108.89 | N3–C21–C22–C31 | −22.63 | −28.46 | |||
| H35–C33–C36 | 108.53 | 108.89 | C33–C21–C22–C23 | −24.03 | −30.17 | |||
| N4–C36–C33 | 114.24 | 114.07 | C33–C21–C22–C31 | 157 | 150.83 | |||
| N4–C36–H37 | 107.49 | 108.76 | N3–C21–C33–H34 | −45.52 | −45.77 | |||
| N4–C36–H38 | 108.62 | 108.77 | N3–C21–C33–H35 | −163.4 | −162.95 | |||
| C33–C36–H37 | 108.55 | 108.73 | N3–C21–C33–C36 | 74.16 | 75.65 | |||
| C33–C36–H38 | 110.28 | 108.73 | C22–C21–C33–H34 | 134.88 | 135.02 | |||
| H37–C36–H38 | 107.42 | 107.59 | C22–C21–C33–H35 | 17 | 17.84 | |||
| N4–C39–H40 | 121.91 | 127.03 | C22–C21–C33–C36 | −105.44 | −103.57 | |||
| N4–C39–C41 | 105.71 | 105.95 | C21–C22–C23–H24 | 0.24 | 1.07 | |||
| H40–C39–C41 | 132.38 | 127.02 | C21–C22–C23–C25 | −178.79 | −178.91 | |||
| N5–C41–C39 | 110.5 | 110.84 | C31–C22–C23–H24 | 179.23 | −179.92 | |||
| N5–C41–H42 | 121.55 | 124.57 | C31–C22–C23–C25 | 0.19 | 0.10 | |||
| C39–C41–H42 | 127.94 | 124.59 | C21–C22–C31–C29 | 179.08 | 178.81 | |||
| N4–C43–N5 | 112.27 | 112.45 | C21–C22–C31–H32 | −1.07 | −1.18 | |||
| N4–C43–H44 | 121.89 | 123.80 | C23–C22–C31–C29 | 0.07 | −0.22 | |||
| N5–C43–H44 | 125.84 | 123.75 | C23–C22–C31–H32 | 179.92 | 179.79 | |||
3.3. Natural Bond Orbital (NBO) Analysis
| Donor (i) | ED (e) | Acceptor (j) | ED (e) | E(2) a (kcal/mol) | E(i)–E(j) b (kcal/mol) | F(i,j) c (kcal/mol) |
|---|---|---|---|---|---|---|
| π(N5–C43) | 1.8696 | π*(C39–C41) | 0.2991 | 20.96 | 0.33 | 0.077 |
| π(C6–C8) | 1.6612 | π*(C10–C11) | 0.3353 | 22.79 | 0.29 | 0.072 |
| π(C6–C8) | 1.6612 | π*(C13–C15) | 0.3800 | 18.34 | 0.28 | 0.065 |
| π(C10–C11) | 1.6285 | π*(C6–C8) | 0.2882 | 17.02 | 0.28 | 0.063 |
| π(C10–C11) | 1.6285 | π*(C13–C15) | 0.3800 | 24.38 | 0.28 | 0.074 |
| π(C13–C15) | 1.6473 | π*(O1–C16) | 0.2324 | 20.53 | 0.27 | 0.069 |
| π(C13–C15) | 1.6473 | π*(C6–C8) | 0.2882 | 20.63 | 0.29 | 0.070 |
| π(C13–C15) | 1.6473 | π*(C10–C11) | 0.3353 | 17.40 | 0.29 | 0.064 |
| π(C22–C23) | 1.6487 | π*(N3–C21) | 0.1570 | 15.73 | 0.27 | 0.061 |
| π(C22–C23) | 1.6487 | π*(C25–C27) | 0.3192 | 19.10 | 0.29 | 0.066 |
| π(C22–C23) | 1.6487 | π*(C29–C31) | 0.2875 | 18.74 | 0.29 | 0.067 |
| π(C25–C27) | 1.6565 | π*(C22–C23) | 0.3739 | 21.13 | 0.28 | 0.069 |
| π(C25–C27) | 1.6565 | π*(C29–C31) | 0.2875 | 18.43 | 0.29 | 0.066 |
| π(C29–C31) | 1.6602 | π*(C22–C23) | 0.3739 | 19.73 | 0.28 | 0.067 |
| π(C29–C31) | 1.6602 | π*(C25–C27) | 0.3192 | 20.94 | 0.28 | 0.069 |
| π(C39–C41) | 1.8516 | π*(N5–C43) | 0.3813 | 15.41 | 0.27 | 0.062 |
| LP(2) O1 | 1.8315 | σ*(O2–C16) | 0.1184 | 37.90 | 0.57 | 0.133 |
| LP(2) O1 | 1.8315 | σ*(C15–C16) | 0.0639 | 17.29 | 0.69 | 0.100 |
| LP(2) O2 | 1.7979 | π*(O1–C16) | 0.2324 | 35.59 | 0.36 | 0.102 |
| LP(2) O2 | 1.7979 | π*(N3–C21) | 0.1570 | 12.38 | 0.36 | 0.060 |
| LP(1) N4 | 1.5610 | π*(N5–C43) | 0.3813 | 45.86 | 0.28 | 0.102 |
| LP(1) N4 | 1.5610 | π*(C39–C41) | 0.2991 | 30.46 | 0.29 | 0.088 |
| LP(1) O2 | 1.9728 | π*(C33–H35) | 0.0128 | 0.72 | 1.04 | 0.025 |
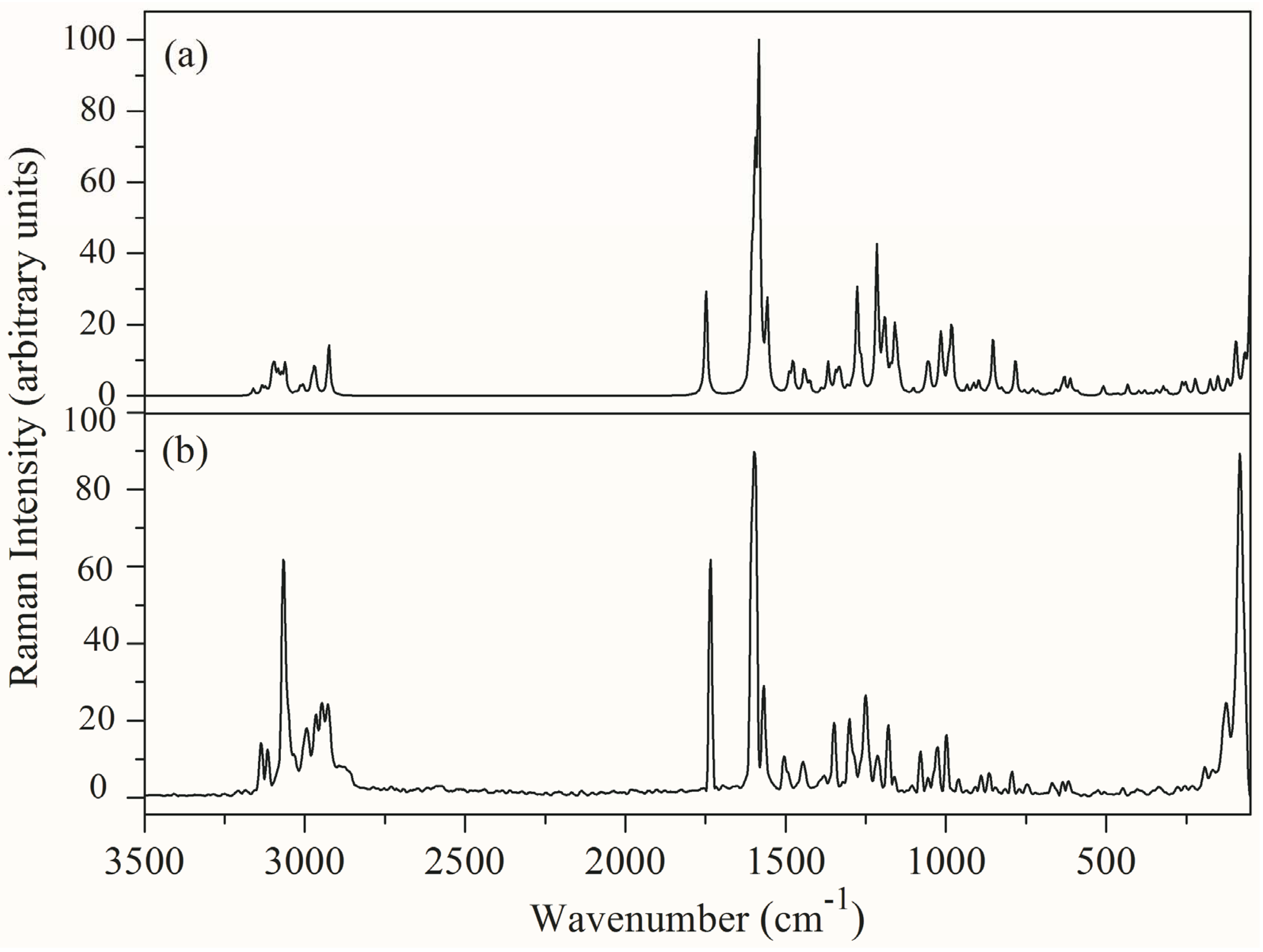
3.4. Vibrational Spectral Analysis
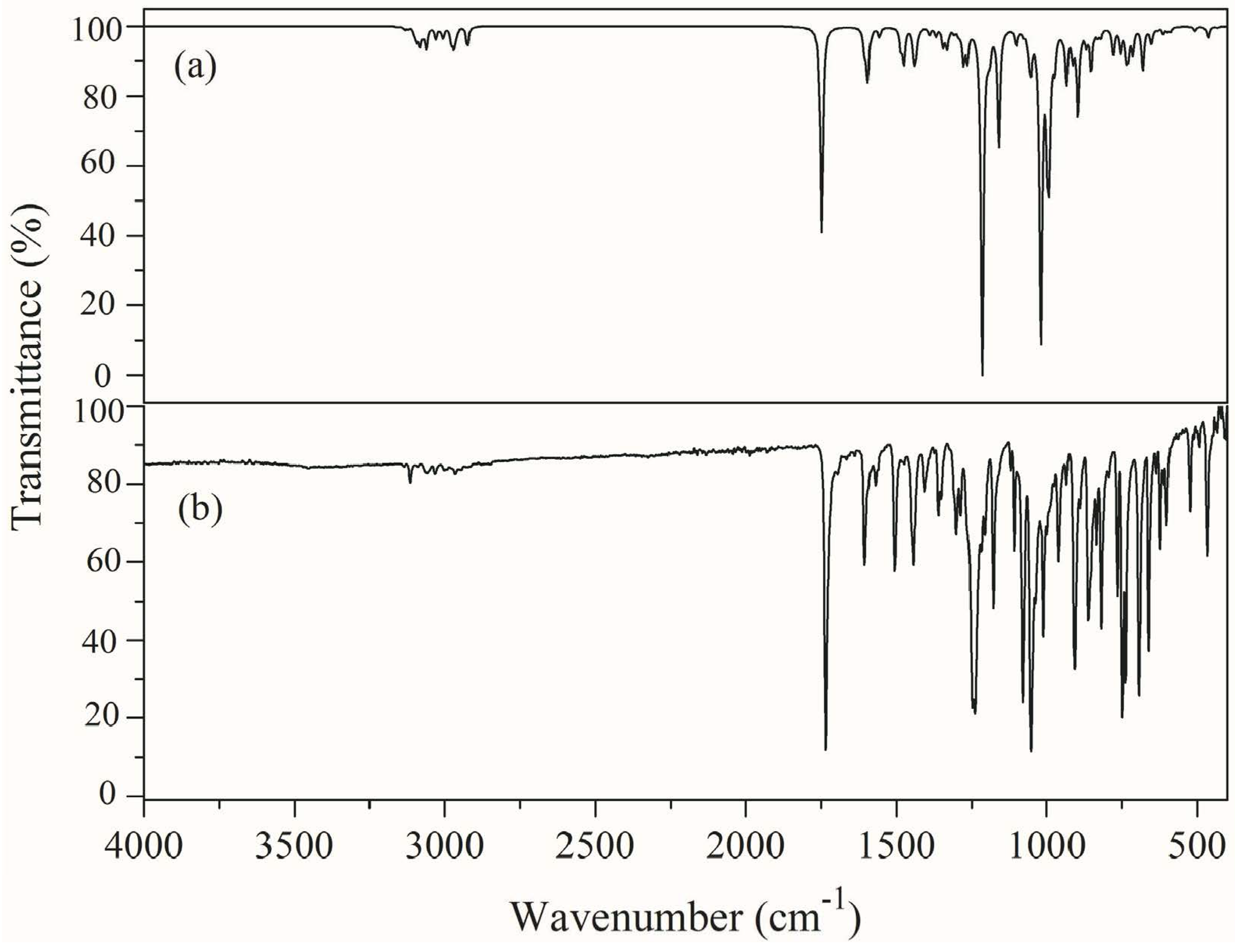
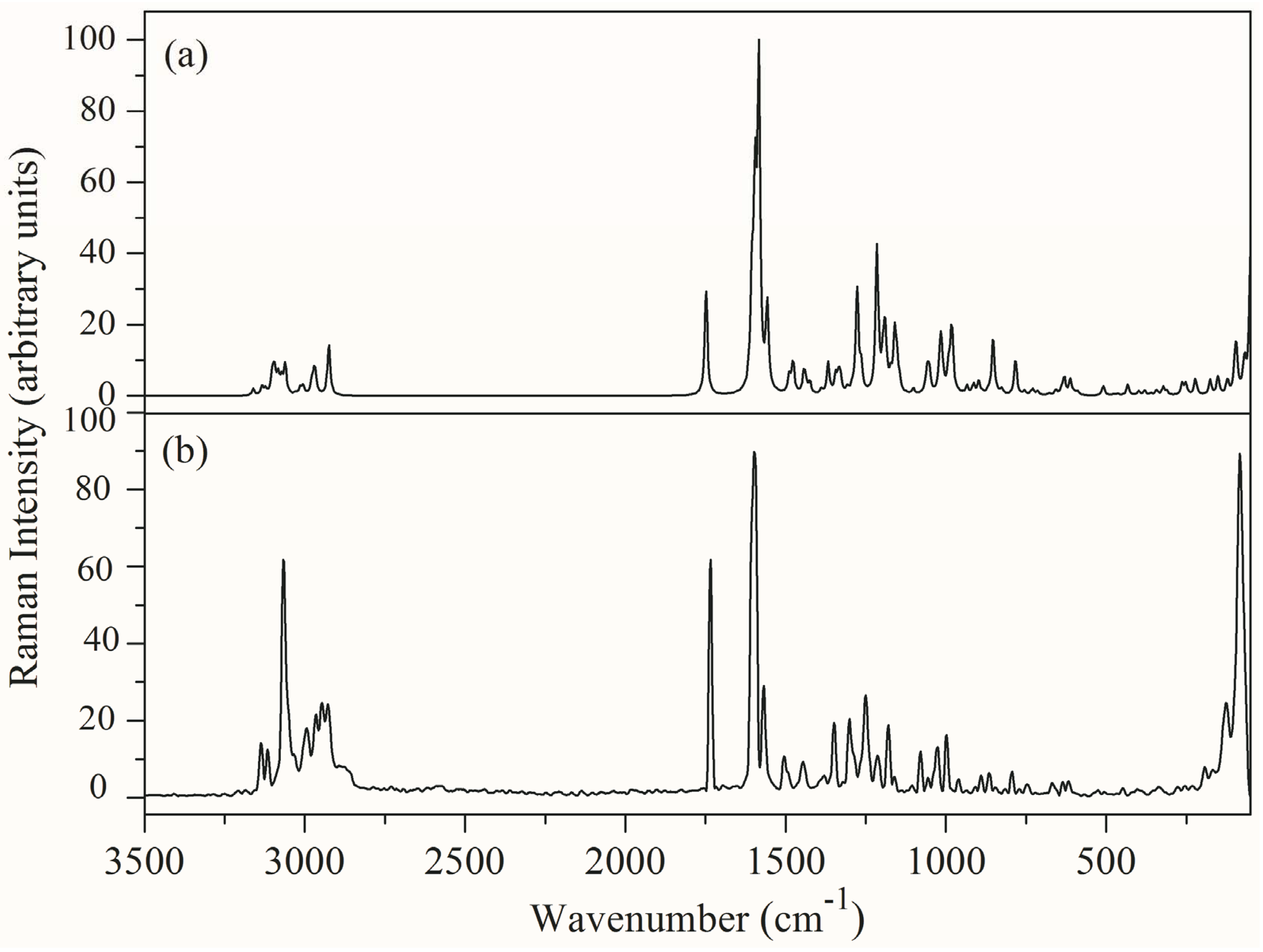
| Calculated Wavenumber (cm−1) | Experimental Wavenumber (cm−1) | IR Intensity | Raman Intensity | Force Constant Mdyne/Å | Assignments with PED% | ||
|---|---|---|---|---|---|---|---|
| Unscaled | Scaled | IR | Raman | ||||
| 3268 | 3161 | – | – | 0.593 | 0.39 | 6.9489 | CH ss (85), CH ss (13) |
| 3239 | 3134 | – | 3136 M | 3.6251 | 0.51 | 6.7536 | CH as (85), CH as (14) |
| 3228 | 3123 | – | – | 2.2934 | 0.35 | 6.7270 | CH v (98) |
| 3208 | 3103 | 3116 VW | 3116 M | 4.7262 | 0.67 | 6.6208 | CH ss (96) |
| 3205 | 3100 | – | – | 2.7163 | 0.64 | 6.6210 | CH ss (88) |
| 3200 | 3095 | – | – | 3.2974 | 0.55 | 6.5918 | CH ss (95) |
| 3198 | 3094 | – | – | 11.246 | 0.8 | 6.5894 | CH ss (69), CH ss (21) |
| 3187 | 3083 | – | – | 21.7901 | 1.12 | 6.5450 | CH as (50), CH as (18) |
| 3177 | 3073 | – | – | 7.1817 | 0.83 | 6.4757 | CH as (45), CH as (44) |
| 3166 | 3063 | – | 3066 S | 0.2433 | 0.31 | 6.4132 | CH as (44), CH as (28), CH as (25) |
| 3165 | 3061 | – | – | 13.691 | 0.67 | 6.4307 | CH ss (77), CH ss (18) |
| 3164 | 3060 | 3058 VW | – | 12.943 | 0.83 | 6.4245 | CH as (77), CH as (19) |
| 3132 | 3029 | 3032 VW | – | 14.3723 | 0.1 | 6.3489 | CH ss (52), CH ss (33) |
| 3116 | 3014 | – | – | 2.0283 | 0.42 | 6.3011 | CH ss (47), CH ss (31), CH ss (13) |
| 3107 | 3005 | 3001 VW | 2994 M | 13.465 | 0.53 | 6.2686 | CH as (62), CH as (36) |
| 3079 | 2978 | – | – | 16.7718 | 0.78 | 6.1126 | CH as (43), CH as (37), CH as (20) |
| 3070 | 2969 | 2965 VW | 2965 M | 23.7511 | 1.58 | 5.9039 | CH ss (49), CH ss (30), CH ss (11), CH ss (10) |
| 3061 | 2961 | 2945 VW | – | 7.8155 | 0.15 | 5.8801 | CH as (51), CH ss (35) |
| 3024 | 2925 | – | 2928 M | 24.0513 | 3.15 | 5.6052 | CH ss (62), CH ss (20), CH ss (17) |
| 2893 W | Overtones and Combination bands | ||||||
| 2878 W | |||||||
| 2858 W | |||||||
| 1807 | 1748 | 1735 VS | 1735 S | 269.1274 | 6.69 | 25.0634 | O=C ss (88) |
| 1660 | 1605 | 1604 M | – | 26.7859 | 6.42 | 14.4429 | vN=C (60) |
| 1649 | 1595 | – | 1597 VS | 64.4175 | 11.64 | 9.3552 | vCC (22), vCC (17) |
| 1637 | 1584 | – | – | 6.3303 | 20.09 | 8.9281 | vCC (28), vCC (16) |
| 1610 | 1558 | 1569 VW | 1568 M | 8.6839 | 4.96 | 8.1884 | vNC (13), vCC (20), vCC (15), vCC (11) |
| 1608 | 1555 | – | – | 3.9604 | 0.32 | 9.0692 | vCC (32) |
| 1539 | 1489 | 1505 M | 1505 W | 1.8657 | 1.04 | 3.4994 | δCCH (18), δCCH (17), δCCH (16) |
| 1536 | 1486 | – | – | 22.6513 | 0.13 | 5.3525 | vN=C (33), vC=C (33), δCCH (13) |
| 1527 | 1477 | – | – | 2.8843 | 1.4 | 2.9785 | δCCH (14), δCCH (13) |
| 1526 | 1476 | – | – | 42.0124 | 0.54 | 3.0799 | vC=C (13), CH2sci (19) |
| 1493 | 1444 | 1443 M | 1446 W | 13.357 | 0.9 | 1.4623 | CH3sci (41), CH3sci (26) |
| 1489 | 1441 | – | – | 18.9506 | 0.28 | 1.6996 | CH2sci (51), δCH2 (47) |
| 1488 | 1440 | – | – | 10.9672 | 0.46 | 1.3911 | CH3sci(38) |
| 1483 | 1435 | – | – | 23.2028 | 0.29 | 1.5608 | CH2sci (61) |
| 1472 | 1424 | 1407 W | – | 2.639 | 0.7 | 2.5501 | δsCCH (24), vCC (13) |
| 1436 | 1389 | – | 1380 W | 8.6605 | 0.28 | 3.0566 | vCC (20), δCCH (12) |
| 1414 | 1367 | – | – | 7.3558 | 0.25 | 2.0790 | NCHtwi (32) |
| 1414 | 1367 | 1360 W | – | 2.2648 | 1.73 | 1.6400 | δsCH3 (22),δCH2 (13) |
| 1390 | 1344 | – | 1349 M | 21.4422 | 1.09 | 2.4537 | vNC (15), CH2wag (15) |
| 1381 | 1336 | – | – | 3.75 | 0.78 | 2.5704 | vNC (27), CH2wag (19) |
| 1375 | 1330 | – | – | 23.9291 | 1.13 | 1.8331 | δCCH (10) |
| 1351 | 1307 | 1302 W | 1301 M | 5.575 | 0.35 | 1.9302 | vCC (12), δsCCH (20) |
| 1337 | 1293 | – | – | 5.0344 | 0.05 | 6.1367 | vCC (31), vCC (27) |
| 1334 | 1291 | – | – | 2.0028 | 0.24 | 1.4769 | δsCCH (22) |
| 1326 | 1283 | 1286 W | – | 0.9504 | 0.14 | 3.7665 | vCC (24) |
| 1320 | 1277 | – | – | 41.0481 | 6.55 | 2.2416 | vCC (26), δCCH (17) |
| 1307 | 1265 | – | 1250 M | 37.7926 | 1.46 | 1.9430 | δsCCH (35), vNC (12) |
| 1259 | 1218 | 1238 S | – | 65.5138 | 0.34 | 1.5270 | δNCH (55), vNC (10) |
| 1256 | 1215 | 1206 W | – | 394.8687 | 8.95 | 3.0905 | vCC (33), δCCH (12) |
| 1237 | 1197 | – | – | 16.8257 | 1.9 | 1.4384 | δCCH (33) |
| 1230 | 1189 | – | – | 27.0607 | 3.94 | 3.2248 | vCC (39), δCCC (12) |
| 1212 | 1173 | 1177 M | 1180 M | 2.3703 | 0.97 | 0.9970 | vCC (11), δsCCH (20) |
| 1199 | 1160 | – | 1161 W | 148.5022 | 3.52 | 1.0149 | δsCCH (20) |
| 1193 | 1154 | – | – | l | 1.51 | 1.1323 | vNC (11), δNCH (14) |
| 1185 | 1146 | – | – | 0.1824 | 0.74 | 0.9300 | δsCCH (33) |
| 1140 | 1103 | 1107 W | 1106 VW | 1.9151 | 0.07 | 1.0086 | vCC (19), δsCCH (20) |
| 1138 | 1101 | – | – | 17.4222 | 0.25 | 1.5829 | vNC (62), δsCCH (20) |
| 1113 | 1077 | 1078 S | – | 5.3066 | 0.03 | 1.1960 | vCC (20), δsCCH (15) |
| 1095 | 1059 | – | – | 27.4394 | 1.54 | 1.0008 | vCC (19), δsCCH (46) |
| 1087 | 1052 | 1051 VS | 1056 M | 42.9985 | 1.43 | 1.8490 | – |
| 1060 | 1025 | – | 1026 M | 10.928 | 0.07 | 1.0692 | δasCCH (62) |
| 1054 | 1020 | – | – | 382.4008 | 1.22 | 1.9040 | vOC (24) |
| 1050 | 1015 | – | – | 16.3115 | 2.41 | 1.7360 | vCC (27) |
| 1046 | 1012 | 1010 M | – | 14.8755 | 1 | 2.0414 | vNC (20), δsCCN (31) |
| 1032 | 998 | – | 998 M | 119.1442 | 0.1 | 2.0580 | δsCCC (24) |
| 1025 | 992 | – | – | 150.1159 | 1.56 | 1.7637 | vCC (15) |
| 1015 | 982 | – | – | 10.5957 | 4.12 | 3.5570 | vCC (14), δsCCC (27) |
| 1010 | 977 | – | – | 0.2509 | 0.09 | 0.7885 | HCCHtwi (47) |
| 1006 | 973 | – | – | 39.6089 | 0.16 | 0.8858 | δsCH3 (55) |
| 1001 | 968 | – | – | 0.7333 | 0.01 | 0.8021 | HCCHtwi (80) |
| 991 | 959 | 960 W | 961 W | 0.145 | 0.08 | 0.7930 | HCCHtwi (40) |
| 980 | 948 | – | – | 3.9185 | 0.04 | 0.7761 | δasCCCH (54) |
| 966 | 934 | 935W | – | 66.3139 | 0.53 | 2.0584 | vCC (36) |
| 945 | 914 | – | – | 33.2026 | 0.63 | 0.9804 | HCCHtwi (24) |
| 928 | 897 | 906 M | – | 111.0668 | 0.83 | 1.0686 | vON (17), HCCHtwi (15) |
| 917 | 887 | – | 890 W | 5.8457 | 0.1 | 3.1525 | δsCNC (70) |
| 899 | 870 | 863 M | 865 W | 18.2761 | 0.18 | 1.8980 | vCC (12), vON (13) |
| 882 | 853 | – | – | 53.555 | 3.69 | 2.6957 | vON (22) |
| 869 | 841 | – | – | 1.649 | 0.15 | 0.6090 | HCCHtwi (80) |
| 861 | 832 | 835 W | – | 8.1045 | 0.01 | 0.6028 | CCCHopb (79) |
| 854 | 826 | – | – | 1.9587 | 0.31 | 0.5388 | HCCHopb (50) |
| 848 | 821 | 819 M | – | 9.9934 | 0.04 | 0.6698 | CCCHopb (50) |
| 810 | 783 | – | 794 W | 3.9159 | 2.27 | 1.8007 | vCC (18), δCCC (30) |
| 806 | 780 | 764 M | – | 31.983 | 0.03 | 0.5309 | NCCNopb (87) |
| 781 | 755 | 749 S | – | 28.9486 | 0.23 | 0.7004 | CCCHopb (53) |
| 761 | 736 | 737 S | – | 33.4661 | 0.13 | 1.0165 | CCCCopb (13), τOCCO (38) |
| 753 | 729 | – | – | 30.0271 | 0.34 | 1.2388 | vCC (15) |
| 738 | 714 | – | – | 30.7013 | 0.22 | 0.3981 | HCNCopb (76) |
| 703 | 680 | 694 S | – | 54.5898 | 0.01 | 0.5922 | CCCCopb (39) |
| 701 | 678 | – | – | 1.2881 | 0.09 | 0.8929 | CCCCopb (19), τOCCO (16) |
| 679 | 657 | – | – | 3.2453 | 0.31 | 1.2364 | δCCC (14) |
| 673 | 651 | – | – | 18.7772 | 0.01 | 0.9418 | NCCNopb (89) |
| 661 | 639 | 636 VW | 636 VW | 0.5431 | 0.36 | 1.0390 | vNC (29), δCCN (13) |
| 652 | 631 | 623 W | 618 VW | 2.8157 | 1.06 | 1.6943 | δCCC (43) |
| 635 | 614 | – | – | 5.4204 | 0.34 | 0.9863 | CCNCopb (44), δCCC (15) |
| 632 | 611 | 612 W | – | 2.7071 | 0.68 | 1.1482 | δCCC (34), τCCNC (25) |
| 620 | 599 | 602 W | – | 5.011 | 0.16 | 1.0360 | δCCC (25), δCC=O (12) |
| 609 | 589 | – | – | 5.5383 | 0.21 | 0.8308 | NCCCopb (13) |
| 526 | 509 | 525 W | – | 5.7184 | 0.59 | 0.8315 | CCNopb (12) |
| 493 | 477 | 493 VW | – | 1.2364 | 0.06 | 0.4729 | CCCCopb (28) |
| 479 | 464 | 466 W | – | 13.7513 | 0.1 | 0.4369 | CCCCopb (36) |
| 448 VW | |||||||
| 447 | 433 | 434 VW | – | 1.5492 | 0.7 | 0.5503 | δCCC (20), δCCN (17) |
| 416 | 403 | 404 VW | 403 VW | 0.0615 | 0 | 0.2964 | CCCCopb (47) |
| 412 | 398 | – | – | 0.1288 | 0.26 | 0.2902 | CCCCopb (48) |
| 393 | 380 | – | – | 2.3925 | 0.3 | 0.3513 | δCCN (31) |
| 372 | 360 | – | – | 1.3636 | 0.11 | 0.5099 | vCC (14) |
| 355 | 344 | – | – | 2.0459 | 0.21 | 0.1924 | CH2opb (14), δCNC (29) |
| 354 | 342 | – | 335 VW | 2.8112 | 0.11 | 0.1949 | CH2opb (37), δCNC (16) |
| 333 | 322 | – | – | 1.1973 | 0.51 | 0.4237 | CCCCopb (11), τCCNO (35) |
| 322 | 311 | – | – | 2.7272 | 0.25 | 0.2073 | CH2opb (28), δCNC (20) |
| 272 | 263 | – | – | 3.6921 | 0.75 | 0.2031 | CCCCopb (46) |
| 260 | 252 | – | – | 2.0039 | 0.71 | 0.2151 | CCCopb (31), δCON (22) |
| 229 | 222 | – | – | 0.4991 | 1.01 | 0.0980 | vOC (10), δCCC (10) |
| 182 | 176 | – | 192 W | 1.7133 | 0.95 | 0.0980 | CNC=Nopb (19), δCCC (30) |
| 157 | 152 | – | – | 0.9648 | 1.13 | 0.0804 | CCCCopb (34) |
| 126 | 122 | – | 125 M | 1.9862 | 0.87 | 0.0521 | δCCC (22), δCCO (17) |
| 103 | 99 | – | – | 1.3736 | 0.72 | 0.0298 | CCCCopb (26), δCNC=N (27) |
| 98 | 95 | – | 83 VS | 0.972 | 2.92 | 0.0174 | δCNC=N (25), τCCCC (23) |
| 71 | 69 | – | – | 1.0649 | 1.91 | 0.0174 | CCNCopb (14), τCCCO (29) |
| 52 | 50 | – | – | 1.6541 | 6.9 | 0.0070 | CCNCopb (40), τCCCO (25) |
| 46 | 45 | – | – | 0.1833 | 1.85 | 0.0064 | δCON (25), δCCO (16) |
| 38 | 37 | – | – | 0.9354 | 6.12 | 0.0034 | τCCCO (28), τCCCN (15) |
| 33 | 32 | – | – | 0.2588 | 2.81 | 0.0007 | C-CH3opb (89) |
| 27 | 26 | – | – | 1.8136 | 9.93 | 0.0023 | CCCNopb (21) |
| 26 | 25 | – | – | 0.0469 | 2.53 | 0.0017 | (29), τCONC (29) |
| 17 | 17 | – | – | 0.7348 | 35 | 0.0009 | τCCCN (25),τCCON (18) |
| 12 | 12 | – | – | 0.9577 | 41.04 | 0.0005 | τCCON (44) |
3.4.1. Phenyl Ring Vibrations
3.4.2. Methylene Vibrations
3.4.3. Methyl Vibrations
3.4.4. Imidazole Ring Vibrations
3.4.5. Skeletal Mode Vibrations
3.5. HOMO–LUMO Energy Analysis

3.6. NMR Spectral Analysis
| 13C | 1H | ||||
|---|---|---|---|---|---|
| Atom | δexp. | δcal. | Atom | δexp. | δcal. |
| C6 | 131.23 | 131.08 | H7 | 7.85 | 7.85 |
| C8 | 129.01 | 128.71 | H9 | 7.23 | 7.58 |
| C10 | 144.64 | 146.91 | H12 | 7.23 | 6.94 |
| C11 | 129.01 | 127.70 | H14 | 7.85 | 7.60 |
| C13 | 129.54 | 128.47 | H18 | 2.37 | 1.12 |
| C15 | 125.80 | 125.88 | H19 | 2.37 | 1.11 |
| C16 | 163.17 | 162.04 | H20 | 2.37 | 2.37 |
| C17 | 21.79 | 19.86 | H24 | 7.59 | 7.35 |
| C21 | 163.48 | 164.41 | H26 | 7.35 | 7.33 |
| C22 | 136.91 | 135.25 | H28 | 7.41 | 7.39 |
| C23 | 129.61 | 126.13 | H30 | 7.35 | 7.41 |
| C25 | 130.09 | 127.30 | H32 | 7.59 | 7.86 |
| C27 | 131.23 | 130.22 | H34 | 3.36 | 3.24 |
| C29 | 130.09 | 128.05 | H35 | 3.36 | 2.82 |
| C31 | 129.61 | 126.99 | H37 | 4.21 | 3.37 |
| C33 | 31.04 | 30.68 | H38 | 4.21 | 4.21 |
| C36 | 43.65 | 44.04 | H40 | 6.84 | 6.94 |
| C39 | 118.73 | 119.84 | H42 | 6.94 | 6.84 |
| C41 | 127.20 | 128.46 | H44 | 7.37 | 7.36 |
| C43 | 133.07 | 135.83 | – | – | – |
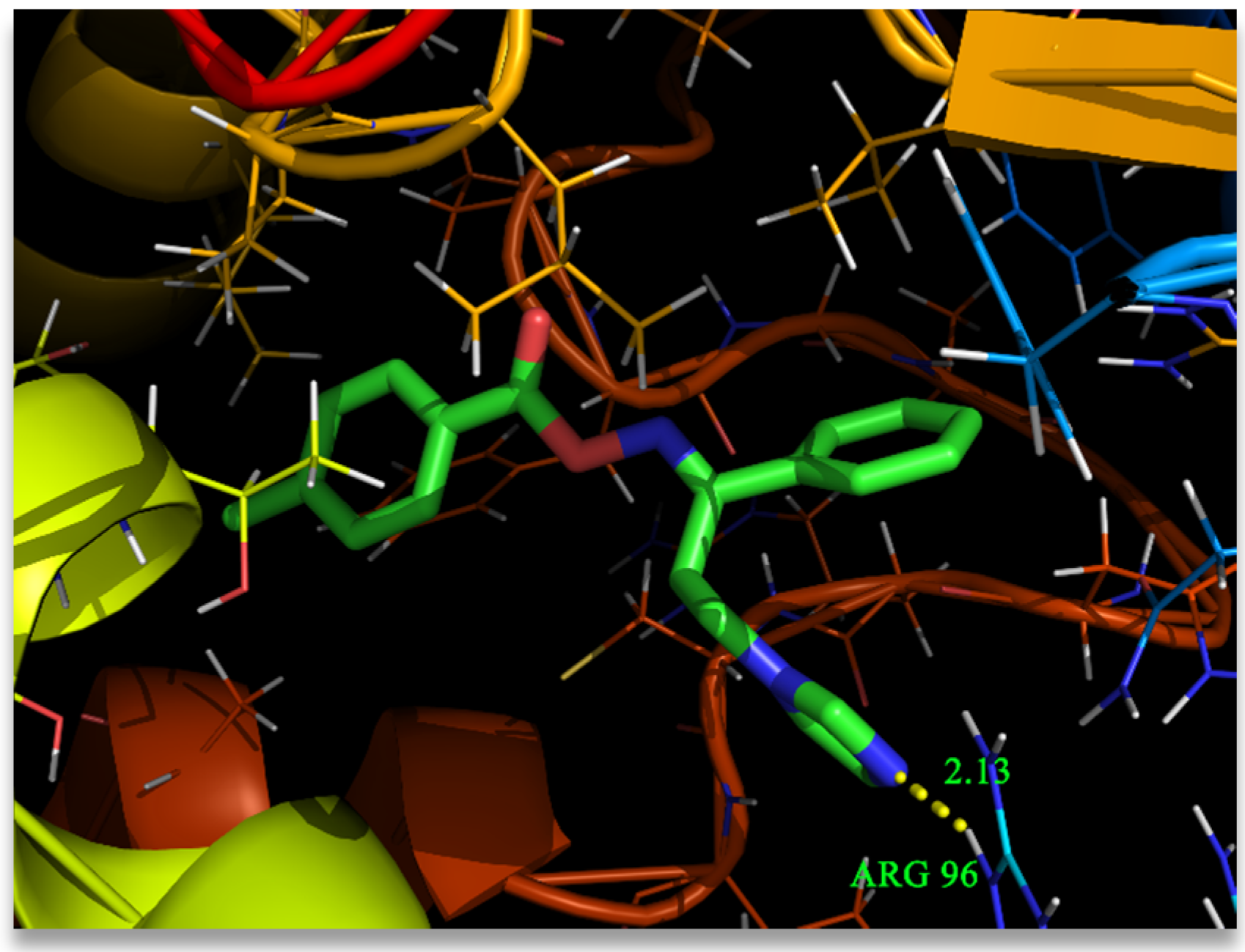
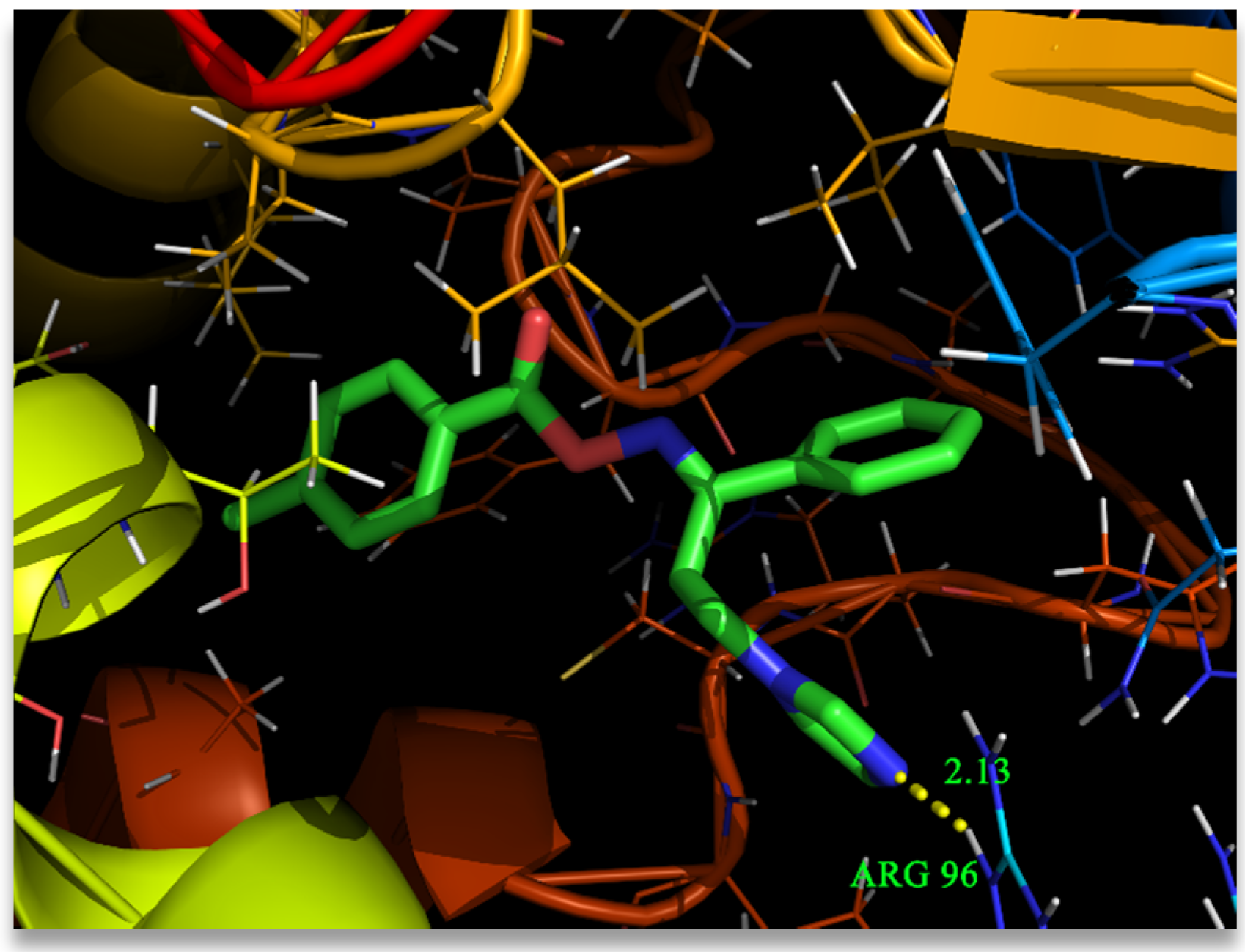
3.7. Molecular Docking Analysis
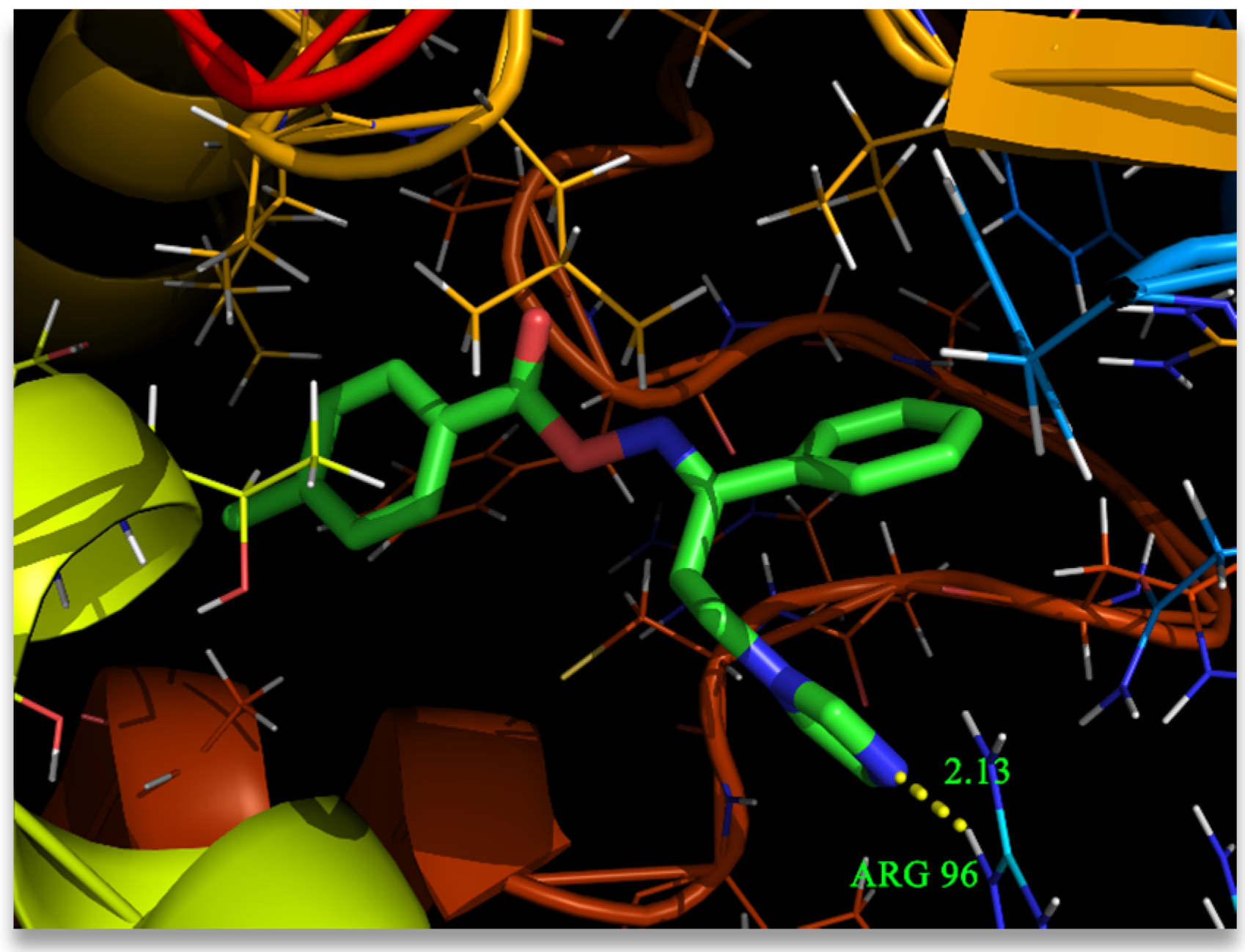
4. Conclusions
Acknowledgments
Author Contributions
Conflict of Interests
References
- Vanden-Bossche, H.; Dromer, F.; Improvisi, I.; Lozano-Chiu, M.; Rex, J.H.; Sanglards, D. Antifungal drug resistance in pathogenic fungi. Med. Mycol. 1998, 36, 119–128. [Google Scholar] [PubMed]
- Odds, F.; Brown, A.J.P.; Gow, N.A.R. Antifungal agents: Mechanisms of action. Trends Microbiol. 2003, 11, 272–279. [Google Scholar] [CrossRef]
- Hamdan, J.S.; Hahn, R.C. Antifungal drugs for systemic mycosis: An overview of mechanism of action and resistance. Anti-Infect. Agents Med. Chem. 2006, 5, 403–412. [Google Scholar] [CrossRef]
- Almutairi, M.S.; Attia, M.I.; Ghabbour, H.A.; Ghoneim, S.W.; Abdel-Aziz, H.A.; Fun, H.-K. Crystal structure of ({(E)-[3-(1H-imidazol-1-yl)-1-phenylpropylidene]amino}oxy)(4-methylphenyl)-methanone, C20H19N3O2. Z. Kristallogr. New Cryst. Struct. 2014, 229, 307–308. [Google Scholar] [CrossRef]
- Attia, M.I.; Zakaria, A.S.; Almutairi, M.S.; Ghoneim, S.W. In Vitro anti-Candida activity of certain new 3-(1H-imidazol-1-yl)propan-1-one oxime esters. Molecules 2013, 18, 12208–12221. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Scott, A.P.; Radom, L. Harmonic vibrational frequencies: An evaluation of Hartree-Fock, Møller-Plesset, quadratic configuration interaction, density functional theory, and semiempirical scale factors. J. Phys. Chem. 1996, 100, 16502–16513. [Google Scholar] [CrossRef]
- Foresman, J.B.; Frisch, A. Exploring Chemistry with Electronic Structure Methods; Gaussian Inc.: Pittsburgh, PA, USA, 1996. [Google Scholar]
- Keresztury, G.; Chalmers, J.M.; Griffith, P.R. Raman Spectroscopy: Theory in Handbook of Vibrational Spectroscopy; John Wiley & Sons: New York, NY, USA, 2002. [Google Scholar]
- Glendening, E.D.; Reed, A.E.; Carpenter, J.E.; Weinhold, F. NBOi, version 3.1; TCI, University of Wisconsin: Madison, WI, USA, 1998. [Google Scholar]
- Glendening, E.D.; Reed, A.E.; Carpenter, J.E.; Weinhold, F. NBO, version 3.1; Theoretical Chemistry Institute and Department of Chemistry, University of Wisconsin: Madison, WI, USA, 1998. [Google Scholar]
- Varsanyi, G. Vibrational Spectra of Benzene Derivatives; Academic Press: New York, NY, USA, 1969. [Google Scholar]
- Jamroz, M.H. Vibrational Energy Distribution Analysis VEDA 4. Hindawi: Warsaw, Poland, 2004. [Google Scholar]
- Socrates, G. Infrared Characteristic Group Frequencies; John Wiley & Sons: New York, NY, USA, 1980. [Google Scholar]
- Smith, B.C. Infrared Spectral Interpretation: A Systematic Approach; CRC Press: New York, NY, USA, 1999. [Google Scholar]
- Lin-Vein, D.; Colthup, N.B.; Fateley, W.G.; Grasselli, J.G. The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules; Academic Press: New York, NY, USA, 1991. [Google Scholar]
- Casado, J.; Hernandez, V.; Hotta, S.; Lopez Navarrete, J.T. Vibrational spectra of charged defects in a series of α,α′-dimethyl end-capped oligothiophenes induced by chemical doping with iodine. J. Chem. Phys. 1998, 109, 10419–10429. [Google Scholar] [CrossRef]
- Gussoni, M. Infrared intensities: A new tool in chemistry. J. Mol. Struct. 1986, 141, 63–92. [Google Scholar] [CrossRef]
- Bellamy, L.J. The Infra-Red Spectra of Complex Molecules; Chapman & Hall: London, UK, 1975. [Google Scholar]
- Forsyth, D.A.; Sebag, A.B. Computed 13C·NMR chemical shifts via empirically scaled GIAO shieldings and molecular mechanics geometries. Conformation and configuration from 13C shifts. J. Am. Chem. Soc. 1997, 119, 9483–9494. [Google Scholar] [CrossRef]
- Rablen, P.R.; Pearlman, S.A.; Finkbiner, J. A comparison of density functional methods for the estimation of proton chemical shifts with chemical accuracy. J. Phys. Chem. A 1999, 103, 7357–7363. [Google Scholar] [CrossRef]
- Costa, L.P.A.; de Albuquerque, C.F.; dos Santos, F.M.; de Amorim, M.B., Jr. GIAO-HDFT scaling factor for 13C·NMR chemical shifts calculation. J. Phys. Org. Chem. 2010, 23, 972–977. [Google Scholar]
- Van Eikema-Hommes, N.J.R.; Clark, T. Regression formulae for ab initio and density functional calculated chemical shifts. J. Mol. Model. 2005, 11, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Bally, T.; Rablen, P.R. Calculating accurate proton chemical shifts of organic molecules with density functional methods and modest basis sets. J. Org. Chem. 2009, 74, 4017–4023. [Google Scholar] [CrossRef] [PubMed]
- Giesen, D.; Zumbulyadis, N. A hybrid quantum mechanical and empirical model for the prediction of isotropic 13C shielding constants of organic molecules. Phys. Chem. Chem. Phys. 2002, 4, 5498–5507. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 16, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Podust, L.M.; Poulos, T.L.; Waterman, M.R. Crystal structure of cytochrome P450 14alpha-sterol demethylase (CYP51) from Mycobacterium tuberculosis in complex with azole inhibitors. Proc. Natl. Acad. Sci. USA 2001, 98, 3068–3073. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, F.C.; Koetzle, T.F.; Williams, G.J.; Meyer, E.E., Jr.; Brice, M.D.; Rodgers, J.R.; Kennard, O.; Shimanouchi, T.; Tasumi, M. The Protein Data Bank: A computer-based archival file for macromolecular structures. J. Mol. Biol. 1977, 112, 535–542. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Huey, R.; Morris, G.M.; Olson, A.J.; Goodsell, D.S. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007, 28, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almutairi, M.S.; Manimaran, D.; Joe, I.H.; Saleh, O.A.; Attia, M.I. Structural Properties and Biological Prediction of ({[(1E)-3-(1H-Imidazol-1-yl)-1-phenylpropylidene] amino}oxy)(4-methylphenyl)methanone: An In Silico Approach. Symmetry 2016, 8, 1. https://doi.org/10.3390/sym8010001
Almutairi MS, Manimaran D, Joe IH, Saleh OA, Attia MI. Structural Properties and Biological Prediction of ({[(1E)-3-(1H-Imidazol-1-yl)-1-phenylpropylidene] amino}oxy)(4-methylphenyl)methanone: An In Silico Approach. Symmetry. 2016; 8(1):1. https://doi.org/10.3390/sym8010001
Chicago/Turabian StyleAlmutairi, Maha S., Devarasu Manimaran, Issac Hubert Joe, Ola A. Saleh, and Mohamed I. Attia. 2016. "Structural Properties and Biological Prediction of ({[(1E)-3-(1H-Imidazol-1-yl)-1-phenylpropylidene] amino}oxy)(4-methylphenyl)methanone: An In Silico Approach" Symmetry 8, no. 1: 1. https://doi.org/10.3390/sym8010001
APA StyleAlmutairi, M. S., Manimaran, D., Joe, I. H., Saleh, O. A., & Attia, M. I. (2016). Structural Properties and Biological Prediction of ({[(1E)-3-(1H-Imidazol-1-yl)-1-phenylpropylidene] amino}oxy)(4-methylphenyl)methanone: An In Silico Approach. Symmetry, 8(1), 1. https://doi.org/10.3390/sym8010001