Formation of Hydrocarbons in the Presence of Native Iron under Upper Mantle Conditions: Experimental Constraints



Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. High-Pressure Apparatus
2.3. Analytical Methods
3. Results
3.1. Phase Relations
3.2. Species in Quenched Fluids
4. Discussion
4.1. Formation of Hydrocarbons in the Presence of a Metal Phase
4.2. Interaction of Slab-Derived Fluids with Metal-Saturated Mantle
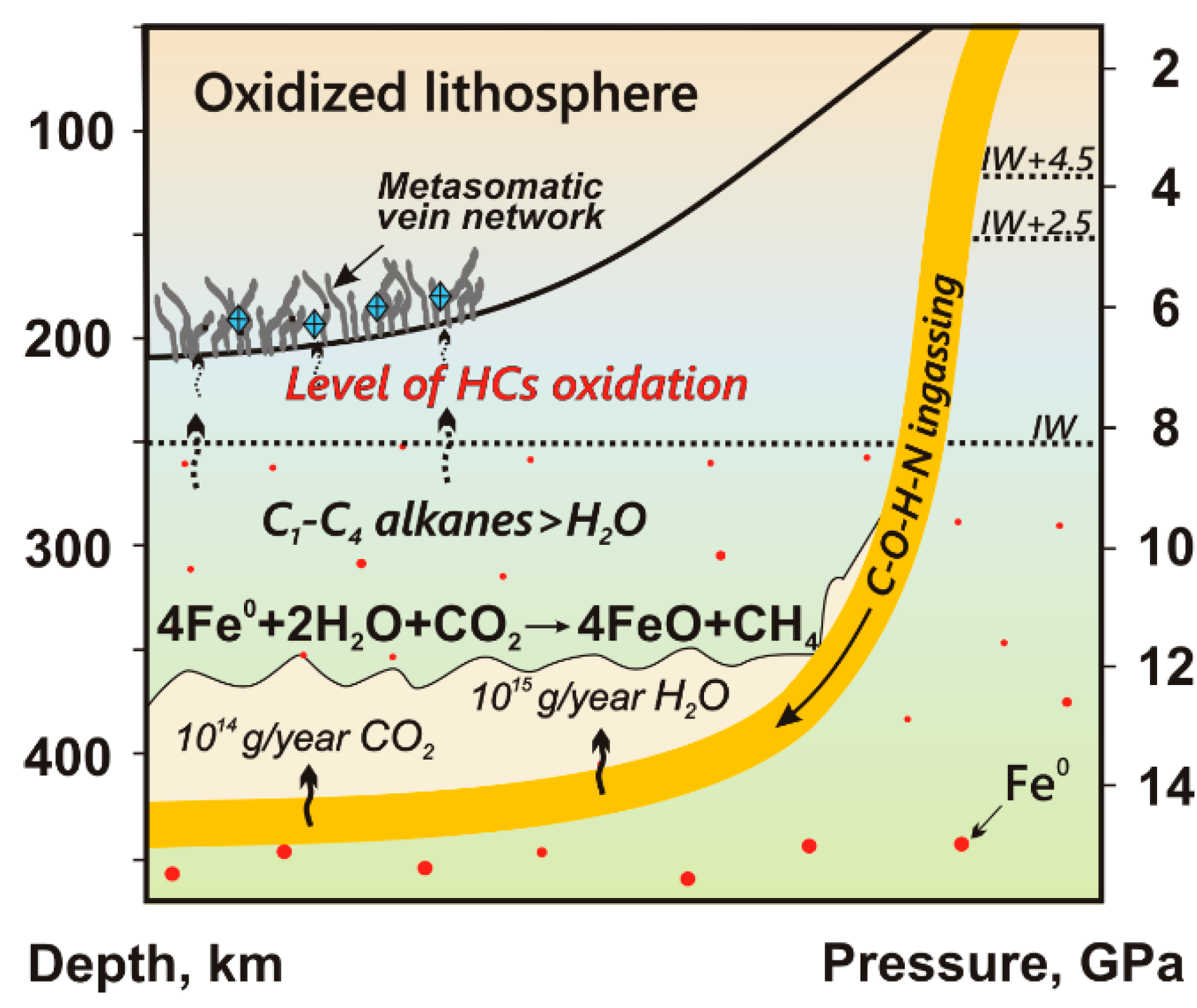
4.3. Fate of HCs in Shallow Mantle
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Horita, J.; Berndt, M.E. Abiogenic methane formation and isotopic fractionation under hydrothermal conditions. Science 1999, 285, 1055–1057. [Google Scholar] [CrossRef]
- Etiope, G.; Schoell, M.; Hosgormez, H. Abiotic methane flux from the Chimaera seep and Tekirova ophiolites (Turkey): Understanding gas exhalation from low temperature serpentinization and implications for Mars. Earth Planet. Sci. Lett. 2011, 310, 96–104. [Google Scholar] [CrossRef]
- Chukanov, N.V.; Pekov, I.V.; Sokolov, S.V.; Nekrasov, A.N.; Chukanova, V.N.; Naumova, I.S. On the problem of the formation and geochemical role of bituminous matter in pegmatites of the Khibiny and Lovozero alkaline massifs, Kola Peninsula, Russia. Geochem. Int. 2006, 44, 715–728. [Google Scholar] [CrossRef]
- Sobolev, N.V.; Sobolev, A.V.; Tomilenko, A.A.; Kuz’min, D.V.; Grakhanov, S.A.; Batanova, V.G.; Logvinova, A.M.; Bul’bak, T.A.; Kostrovitskii, S.I.; Yakovlev, D.A.; et al. Prospects of search for diamondiferous kimberlites in the northeastern Siberian. Platform. Russ. Geol. Geophys. 2018, 59, 1365–1379. [Google Scholar] [CrossRef]
- Sobolev, N.V.; Tomilenko, A.A.; Bul’bak, T.A.; Logvinova, A.M. Composition of volatile components in the diamonds, associated garnet and olivine from diamondiferous peridotites from the Udachnaya pipe, Yakutia, Russia (by coupled gas chromatographic-mass spectrometric analysis). Engineering 2019, 5. [Google Scholar] [CrossRef]
- Sobolev, N.V.; Logvinova, A.M.; Tomilenko, A.A.; Wirth, R.; Bul’bak, T.A.; Luk’yanova, L.I.; Fedorova, E.N.; Reutsky, V.N.; Efimova, E.S. Mineral and fluid inclusions in diamonds from the Urals placers, Russia: Evidence for solid molecular N2 and hydrocarbons in fluid inclusions. Geochim. Cosmochim. Acta 2019, 266, 197–219. [Google Scholar] [CrossRef]
- Smith, E.M.; Shirey, S.B.; Nestola, F.; Bullock, E.S.; Wang, J.; Richardson, S.H.; Wang, W. Large gem diamonds from metallic liquid in Earth’s deep mantle. Science 2016, 354, 1403–1405. [Google Scholar] [CrossRef] [Green Version]
- Yuen, G.; Blair, N.; Des Marais, D.J.; Chang, S. Carbon isotope composition of low molecular weight hydrocarbons and monocarboxylic acids from Murchison meteorite. Nature 1984, 307, 252–254. [Google Scholar] [CrossRef]
- Mumma, M.J.; Villanueva, G.L.; Novak, R.E.; Hewagama, T.; Bonev, B.P.; DiSanti, M.A.; Mandell, A.M.; Smith, M.D. Strong release of methane on Mars in northern summer 2003. Science 2009, 323, 1041–1045. [Google Scholar] [CrossRef] [Green Version]
- Etiope, G.; Sherwood Lollar, B. Abiotic methane on Earth. Rev. Geophys. 2013, 51, 276–299. [Google Scholar] [CrossRef]
- Foley, S. A reappraisal of redox melting in the Earth’s mantle as a function of tectonic setting and time. J. Petrol. 2011, 52, 1363–1391. [Google Scholar] [CrossRef]
- Luth, R.W. Volatiles in Earth’s mantle. In Treatise on Geochemistry; Elsevier: Oxford, UK, 2014; Volume 3, pp. 355–391. [Google Scholar]
- Stagno, V.; Ojwang, D.O.; McCammon, C.A.; Frost, D.J. The oxidation state of the mantle and the extraction. Nature 2013, 493, 84. [Google Scholar] [CrossRef]
- Stachel, T.; Luth, R.W. Diamond formation—Where, when and how? Lithos 2015, 220, 200–220. [Google Scholar] [CrossRef]
- Stagno, V.; Cerantola, V.; Aulbach, S.; Lobanov, S.; McCammon, C.A.; Merlini, M. Carbon-bearing phases throughout Earth’s interior—Evolution through space and time. In Deep Carbon: Past to Present; Orcutt, B., Daniel, I., Dasgupta, R., Eds.; Cambridge University Press: Cambridge, UK, 2019; pp. 66–88. [Google Scholar]
- Kenney, J.F.; Kutcherov, V.A.; Bendeliani, N.A.; Alekseev, V.A. The evolution of multicomponent systems at high pressures: The thermodynamic stability of the hydrogen-carbon system: The genesis of hydrocarbons and the origin of petroleum. Proc. Nat. Acad. Sci. USA 2002, 99, 10976–10981. [Google Scholar] [CrossRef] [Green Version]
- Scott, H.P.; Hemley, R.J.; Mao, H.; Herschbach, D.R.; Fried, L.E.; Howard, W.M.; Bastea, S. Generation of methane in the Earth’s mantle: In situ high pressure–temperature measurements of carbonate reduction. Proc. Nat. Acad. Sci. USA 2004, 101, 14023–14026. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Cody, G.D.; Hemley, R.J. In situ diamond-anvil cell observations of methanogenesis at high pressures and temperatures. Energy Fuels 2009, 23, 5571–5579. [Google Scholar] [CrossRef]
- Kolesnikov, A.; Kutcherov, V.G.; Goncharov, A.F. Methane-derived hydrocarbons produced under upper-mantle conditions. Nat. Geosci. 2009, 2, 566–570. [Google Scholar] [CrossRef]
- Palyanov, Y.N.; Borzdov, Y.M.; Kupriyanov, I.N.; Khokhryakov, A.F. Effect of H2O on diamond crystal growth in metal–carbon systems. Cryst. Growth Des. 2010, 12, 5571–5578. [Google Scholar] [CrossRef]
- Kolesnikov, A.Y.; Saul, J.M.; Kutcherov, V.G. Chemistry of hydrocarbons under extreme thermobaric conditions. Chem. Sel. 2017, 2, 1336–1352. [Google Scholar] [CrossRef]
- Mukhina, E.; Kolesnikov, A.; Kutcherov, V. The lower pT limit of deep hydrocarbon synthesis by CaCO3 aqueous reduction. Sci. Rep. 2017, 7, 5749. [Google Scholar] [CrossRef] [Green Version]
- Tao, R.; Zhang, L.; Tian, M.; Zhu, J.; Liu, X.; Liu, J.; Höfer, H.E.; Stagno, V.; Fei, Y. Formation of abiotic hydrocarbon from reduction of carbonate in subduction zones: Constraints from petrological observation and experimental simulation. Geochim. Cosmochim. Acta 2018, 239, 390–408. [Google Scholar] [CrossRef]
- Matveev, S.; Ballhaus, C.; Fricke, K.; Truckenbrodt, J.; Ziegenben, D. Volatiles in the Earth’s mantle: I. Synthesis of CHO fluids at 1273 K and 2.4 GPa. Geochim. Cosmochim. Acta 1997, 61, 3081–3088. [Google Scholar] [CrossRef]
- Sokol, A.G.; Tomilenko, A.A.; Bul’bak, T.A.; Sokol, I.A.; Zaikin, P.A.; Palyanova, G.A.; Palyanov, Y.N. Hydrogenation of carbon at 5.5–7.8 GPa and 1100–1400 °C: Implications to formation of hydrocarbons in reduced mantles of terrestrial planets. Phys. Earth Planet. Inter. 2019, 291, 12–23. [Google Scholar] [CrossRef]
- Lobanov, S.S.; Chen, P.N.; Chen, X.J.; Zha, C.S.; Litasov, K.D.; Mao, H.K.; Goncharov, A.F. Carbon precipitation from heavy hydrocarbon fluid in deep planetary interiors. Nat. Commun. 2013, 4, 2446. [Google Scholar] [CrossRef] [Green Version]
- Frost, D.J.; Liebske, C.; Langenhorst, F.; McCammon, C.A.; Tronnes, R.G.; Rubie, D.C. Experimental evidence for the existence of iron-rich metal in the Earth’s lower mantle. Nature 2004, 248, 409–412. [Google Scholar] [CrossRef]
- Frost, D.J.; McCammon, C.A. The redox state of Earth’s mantle. Annu. Rev. Earth Planet. Sci. 2008, 36, 389–420. [Google Scholar] [CrossRef]
- Ohtani, E. Water in the mantle. Elements 2005, 1, 25–30. [Google Scholar] [CrossRef]
- Manning, C.E. Thermodynamic modeling of fluid-rock interaction at mid-crustal to upper-mantle conditions. Rev. Mineral. Geochem. 2013, 76, 135–164. [Google Scholar] [CrossRef] [Green Version]
- Dasgupta, R. Ingassing, storage, and outgassing of terrestrial carbon through geologic time. Rev. Mineral. Geochem. 2013, 75, 183–229. [Google Scholar] [CrossRef]
- Plank, T.; Langmuir, C.H. The chemical composition of subducting sediment and its consequences for the crust and mantle. Chem. Geol. 1998, 145, 325–394. [Google Scholar] [CrossRef]
- Song, S.; Su, L.; Niu, Y.; Lai, Y.; Zhang, L. CH4 inclusions in orogenic harzburgite: Evidence for reduced slab fluids and implication for redox melting in mantle wedge. Geochim. Cosmochim. Acta 2009, 73, 1737–1754. [Google Scholar] [CrossRef]
- Rea, D.K.; Ruff, L.J. Composition and mass flux of sediment entering the world’s subduction zones: Implications for global sediment budgets, great earthquakes, and volcanism. Earth Planet. Sci. Lett. 1996, 140, 1–12. [Google Scholar] [CrossRef]
- Schmidt, M.; Poli, S. Devolatilization during subduction. In Treatise on Geochemistry, 2nd ed.; Elsevier Science: Oxford, UK, 2014; pp. 669–701. [Google Scholar]
- Stagno, V.; Stopponi, V.; Kono, Y.; Manning, C.; Irifune, T. Experimental determination of the viscosity of Na2CO3 melt between 1.7 and 4.6 GPa at 1200–1700 °C: Implications for the rheology of carbonatite magmas in the Earth’s upper mantle. Chem. Geol. 2018, 501, 19–25. [Google Scholar] [CrossRef]
- Pearson, D.G.; Canil, D.; Shirey, S.B. Mantle samples included in volcanic rocks: Xenoliths and diamonds. Treatise Geochem. 2003, 2, 568. [Google Scholar]
- Pearson, D.G.; Wittig, N. The Formation and Evolution of Cratonic Mantle Lithosphere—Evidence from Mantle Xenoliths. In Treatise on Geochemistry, 2nd ed.; Elsevier Science: Oxford, UK, 2014; Volume 3, pp. 255–292. [Google Scholar]
- Rohrbach, A.; Schmidt, M.W. Redox freezing and melting in the Earth’s deep mantle resulting from carbon-iron redox coupling. Nature 2011, 472, 209. [Google Scholar] [CrossRef]
- Rohrbach, A.; Ghosh, S.; Schmidt, M.W.; Wijbrans, C.H.; Klemme, S. The stability of Fe–Ni carbides in the Earth’s mantle: Evidence for a low Fe-Ni-C melt fraction in the deep mantle. Earth Planet. Sci. Lett. 2014, 388, 211–221. [Google Scholar] [CrossRef]
- Sobolev, N.V.; Efimova, E.S.; Pospelova, L.N. Native iron in Yakutian diamonds and its mineral assemblage. Sov. Geol. Geophys. 1981, 22, 25–28. [Google Scholar]
- Stachel, T.; Harris, J.W.; Brey, G.P. Rare and unusual mineral inclusions in diamonds from Mwadui, Tanzania. Contrib. Mineral. Petrol. 1998, 132, 34–47. [Google Scholar] [CrossRef]
- Jacob, D.E.; Kronz, A.; Viljoen, K.S. Cohenite, native iron and troilite inclusions in garnets from polycrystalline diamond aggregates. Contrib. Mineral. Petrol. 2004, 146, 566–576. [Google Scholar] [CrossRef]
- Kaminsky, F.V.; Wirth, R. Iron carbide inclusions in lower-mantle diamond from Juina, Brazil. Canad. Mineral. 2011, 49, 555–572. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, H.S.C.; Wall, V.J. The Olivine-Orthopyroxene-Spinel oxygen geobarometer, the nickel precipitation curve, and the oxygen fugacity of the Earth’s Upper Mantle. J. Petrol. 1987, 28, 1169–1191. [Google Scholar] [CrossRef]
- Sokol, A.G.; Kruk, A.N.; Seryotkin, Y.V.; Korablin, A.A.; Palyanov, Y.N. Phase relations in the Fe–Fe3C–Fe3N system at 7.8 GPa and 1350 °C: Implications for carbon and nitrogen hosts in Fe0-saturated upper mantle. Phys. Earth Planet. Inter. 2017, 265, 43–53. [Google Scholar] [CrossRef]
- Lord, O.T.; Walter, M.J.; Dasgupta, R.; Walker, D.; Clark, S.M. Melting in the Fe–C system to 70 GPa. Earth Planet. Sci. Lett. 2009, 284, 157–167. [Google Scholar] [CrossRef] [Green Version]
- Kutcherov, V.G.; Kolesnikov, A.Y.; Dyuzheva, T.I.; Kulikova, L.F.; Nikolaev, N.N.; Sazanova, O.A.; Braghkin, V.V. Synthesis of Complex Hydrocarbon Systems at Temperatures and Pressures Corresponding to the Earth’s Upper Mantle Conditions. Dokl. Phys. Chem. 2010, 433, 132–135. [Google Scholar] [CrossRef]
- Iizuka-Oku, R.; Yagi, T.; Gotou, H.; Okuchi, T.; Hattori, T.; Sano-Furukawa, A. Hydrogenation of iron in the early stage of Earth’s evolution. Nat. Commun. 2017, 8, 14096. [Google Scholar] [CrossRef] [Green Version]
- Palyanov, Y.N.; Bataleva, Y.V.; Sokol, A.G.; Borzdov, Y.M.; Kupriyanov, I.N.; Reutsky, V.N.; Sobolev, N.V. Mantle–slab interaction and redox mechanism of diamond formation. Proc. Natl. Acad. Sci. USA 2013, 110, 20408–20413. [Google Scholar] [CrossRef] [Green Version]
- Stagno, V. Carbon, carbides, carbonates and carbonatitic melts in the Earth’s interior. J. Geol. Soc. 2019, 176, 375–387. [Google Scholar] [CrossRef]
- Luth, R.W. Natural versus experimental control of oxidation state: Effects on the composition and speciation of C-O-H fluids. Am. Mineral. 1989, 74, 50–57. [Google Scholar]
- Sokol, A.G.; Palyanova, G.A.; Palyanov, Y.N.; Tomilenko, A.A.; Melenevsky, V.N. Fluid regime and diamond formation in the reduced mantle: Experimental constraints. Geochim. Cosmochim. Acta 2009, 73, 5820–5834. [Google Scholar] [CrossRef]
- Palyanov, Y.N.; Borzdov, Y.M.; Khokhryakov, A.F.; Kupriyanov, I.N.; Sokol, A.G. Effect of nitrogen impurity on diamond crystal growth processes. Cryst. Growth Des. 2010, 10, 3169–3175. [Google Scholar] [CrossRef]
- Sokol, A.G.; Borzdov, Y.M.; Palyanov, Y.N.; Khokhryakov, A.F. High-temperature calibration of a multi-anvil high-pressure apparatus. High Press. Res. 2015, 35, 139–147. [Google Scholar] [CrossRef]
- Sokol, A.G.; Tomilenko, A.A.; Bul’bak, T.A.; Palyanova, G.A.; Sokol, I.A.; Palyanov, Y.N. Carbon and Nitrogen Speciation in N-poor C-O-H-N Fluids at 6.3 GPa and 1100–1400 °C. Sci. Rep. 2017, 7, 706. [Google Scholar] [CrossRef] [Green Version]
- Borzdov, Y.; Pal’yanov, Y.; Kupriyanov, I.; Gusev, V.; Khokhryakov, A.; Sokol, A.; Efremov, A. HPHT synthesis of diamond with high nitrogen content from an Fe3N–C system. Diam. Relat. Mater. 2002, 11, 1860–1870. [Google Scholar] [CrossRef]
- Sokol, A.G.; Tomilenko, A.A.; Bul’bak, T.A.; Kruk, A.N.; Zaikin, P.A.; Sokol, I.A.; Seryotkin, Y.V.; Palyanov, Y.N. The Fe–C–O–H–N system at 6.3–7.8 GPa and 1200–1400 °C: Implications for deep carbon and nitrogen cycles. Contrib. Mineral. Petrol. 2018, 173, 47. [Google Scholar] [CrossRef]
- Sokol, A.G.; Palyanov, Y.N.; Tomilenko, A.A.; Bul’bak, T.A.; Palyanova, G.A. Carbon and nitrogen speciation in nitrogen-rich C–O–H–N fluids at 5.5–7.8 GPa. Earth Planet. Sci. Lett. 2017, 460, 234–243. [Google Scholar] [CrossRef]
- Zhang, C.; Duan, Z. A model for C-O-H fluid in the Earth’s mantle. Geochim. Cosmochim. Acta 2009, 73, 2089–2102. [Google Scholar] [CrossRef]
- Bataleva, Y.V.; Palyanov, Y.N.; Borzdov, Y.M.; Bayukov, O.A.; Zdrokov, E.V. Iron carbide as a source of carbon for graphite and diamond formation under lithospheric mantle P–T parameters. Lithos 2017, 286–287, 151–161. [Google Scholar] [CrossRef]
- Robertson, A.J.B. The Pyrolysis of Methane, Ethane and n-butane on a Platinum Filament. Proc. R. Soc. Lond. Ser. A Math. Phys. Eng. Sci. 1949, 199, 394–411. [Google Scholar]
- Belgued, M.; Amariglio, A.; Paréja, P.; Amariglio, H. Oxygen-Free conversion of methane to higher alkanes through an isothermal two-step reaction on platinum (EUROPT-1): II. hydrogenation of the adspecies resulting from the chemisorption of methane. J. Catal. 1996, 159, 449–457. [Google Scholar] [CrossRef]
- Sverjensky, D.A.; Stagno, V.; Huang, F. Important role for organic carbon in subduction-zone fluids in the deep carbon cycle. Nat. Geosci. 2014, 7, 909. [Google Scholar] [CrossRef]
- Cimino, A.; Boudart, M.; Taylor, H. Ethane hydrogenation-cracking on iron catalysts with and without alkali. J. Phys. Chem. 1954, 58, 796–800. [Google Scholar] [CrossRef]
- Hasterok, D.; Chapman, D.S. Heat production and geotherms for the continental lithosphere. Earth Planet. Sci. Lett. 2011, 307, 59–70. [Google Scholar] [CrossRef]
- Syracuse, E.M.; van Keken, P.E.; Abers, G.A. The global range of subduction zone thermal models. Phys. Earth Planet. Inter. 2010, 183, 73–90. [Google Scholar] [CrossRef]
- Huizenga, J.M.; Crossingham, A.; Viljoen, F. Diamond precipitation from ascending reduced fluids in the Kaapvaal lithosphere: Thermodynamic constraints. C. R. Geosci. 2012, 344, 67–76. [Google Scholar] [CrossRef]
- Sokol, A.G.; Tomilenko, A.A.; Bul’bak, T.A.; Kruk, A.N.; Sokol, I.A.; Palyanov, Y.N. Fate of fluids at the base of subcratonic lithosphere: Experimental constraints at 5.5–7.8 GPa and 1150–1350 °C. Lithos 2018, 318, 419–433. [Google Scholar] [CrossRef]
- Thomassot, E.; Cartigny, P.; Harris, J.W.; (Fanus) Viljoen, K.S. Methane-related diamond crystallization in the Earth’s mantle: Stable isotope evidences from a single diamond-bearing xenolith. Earth Planet. Sci. Lett. 2007, 257, 362–371. [Google Scholar] [CrossRef]
- Shirey, S.B.; Pearson, D.G.; Walter, M.J.; Aulbach, S.; Brenker, F.E.; Bureau, H.; Burnham, A.D.; Cartigny, P.; Chacko, T.; Frost, D.J.; et al. Diamonds and the Mantle Geodynamics of Carbon: Deep Mantle Carbon Evolution from the Diamond Record. In Deep Carbon: Past to Present; Orcutt, B., Daniel, I., Dasgupta, R., Eds.; Cambridge University Press: Cambridge, UK, 2019; pp. 89–128. [Google Scholar]
- Frezzotti, M.L. Diamond growth from organic compounds in hydrous fluids deep within the Earth. Nat. Commun. 2019, 10, 4952. [Google Scholar] [CrossRef]
- Tappe, S.; Foley, S.F.; Kjarsgaard, B.A.; Romer, R.L.; Heaman, L.M.; Stracke, A.; Jenner, G.A. Between carbonatite and lamproite—Diamondiferous Torngat ultramafic lamprophyres formed by carbonate-fluxed melting of cratonic MARID-type metasomes. Geochim. Cosmochim. Acta 2008, 72, 3258–3286. [Google Scholar] [CrossRef] [Green Version]
- Hernlund, J.; Leinenweber, K.; Locke, D.; Tyburczy, J. A numerical model for steadystate temperature distributions in solid-medium high-pressure cell assemblies. Am. Mineral. 2006, 91, 295–305. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Run # | Starting Composition (mg) | C (wt.%) | T (°C) | t (hrs) | fH2 Buffer | Capsule | Phase Composition(XRD + EDS) | TIC (Arbitrary Units) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gr | Dm | Fe | Ni | Fe3C | Mgs | OA | ||||||||
| Samples unsaturated with graphite | ||||||||||||||
| 2095_2_2 * | 0.3 | - | 4 | - | - | - | - | 7 | 1200 | 10 | IW | Pt | Fe + Fe3C | 1.23 × 1010 |
| 2095_2_4 * | 0.3 | - | - | - | 4.2 | - | - | 13 | 1200 | 10 | IW | Pt | Fe3C + Fe7C3 | 1.16 × 1010 |
| 688_6_2 * | - | - | - | - | 3.6 | 1.0 | - | 11 | 1200 | 10 | IW | Au | Fe7C3 + Mws + MwsL | 3.39 × 109 |
| 688_6_3 * | - | - | - | - | 2.4 | - | 0.4 | 10 | 1200 | 10 | IW | Au | Fe7C3 + FeO | 3.93 × 1010 |
| Samples saturated with graphite | ||||||||||||||
| 1079_5_1 | 7.3 | - | - | - | - | - | - | 100 | 800 | 10 | MMO | Pt | Gr | 5.65 × 108 |
| 1079_5_2 | 10 | - | 2.4 | - | - | - | 81 | 800 | 10 | MMO | Pt | Gr + Fe3C | 6.28 × 108 | |
| 1001_4_3 | 10 | - | - | 2.3 | - | - | - | 81 | 1000 | 10 | MMO | Pt | Gr + Ni | 4.79 × 1010 |
| 1001_4_4 | 10 | - | 2.1 | - | - | - | - | 83 | 1000 | 10 | MMO | Pt | Gr + Fe3C | 5.24 × 1010 |
| 2095_2_3 * | 1.6 | - | - | - | 4.2 | - | - | 67 | 1200 | 10 | IW | Pt | Gr + Fe7C3 | - |
| 600_8_2 | 10 | - | 3.3 | 1.7 | - | - | - | 67 | 1200 | 15 | IW | Pt | Gr + L | 6.63 × 1010 |
| 600_8_4 | 6.9 | - | - | 4.5 | - | - | - | 61 | 1200 | 15 | IW | Pt | Gr + Ni | 4.43 × 1010 |
| 2100_2_2 | 8.5 | - | - | 8.8 | - | - | 53 | 1200 | 10 | IW | Pt | Gr + Fe7C3 | 7.52 × 1010 | |
| 2100_2_3 | 10.4 | - | - | 1.5 | 9.4 | - | - | 52 | 1200 | 10 | IW | Pt | Gr + L | 3.95 × 1010 |
| 2100_2_4 * | 3 | - | - | - | 7 | - | - | 35 | 1200 | 10 | IW | Pt | Gr + Fe7C3 | 3.39 × 109 |
| 688_6_1 * | - | 0.3 | - | - | 4.0 | - | - | 13 | 1200 | 10 | IW | Au | Dm + Fe3C + Fe7C3 ** | 3.69 × 109 |
| 605_8_5 | 6.8 | - | 4.6 | - | - | - | - | 60 | 1400 | 15 | IW | Pt | Gr + L | 1.4 × 109 |
| 605_8_7 | 6.6 | - | 1.6 | 3.3 | - | - | - | 57 | 1400 | 15 | IW | Pt | Gr + L | 2.09 × 108 |
| 605_8_8 | 6.8 | - | - | 4.5 | - | - | - | 60 | 1400 | 15 | IW | Pt | Gr + L | 1.08 × 109 |
| Run # | n | Phase | Fe | 1σ | Ni | 1σ | O | 1σ | Mg | 1σ | Pt | 1σ | Total |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2095_2_2 | 5 | Fe | 99.0 | 0.4 | - | - | - | - | - | - | - | - | 99.0 |
| 2095_2_2 | 5 | Fe3C | 93.0 | 0.4 | - | - | - | - | - | - | - | - | 93.0 |
| 2095_2_4 | 15 | Fe3C | 92.4 | 0.4 | - | - | - | - | - | - | - | - | 92.4 |
| 2095_2_4 | 6 | Fe7C3 | 90.4 | 0.4 | - | - | - | - | - | - | - | - | 90.4 |
| 688_6_2 | 8 | Fe7C3 | 90.3 | 0.4 | - | - | 90.3 | ||||||
| 688_6_2 | 8 | Mws | 62.6 | 0.6 | - | - | 24.7 | 0.5 | 11.7 | 0.3 | - | - | 99.0 |
| 688_6_2 | 9 | MwsL | 63.8 | 2.7 | - | - | 26.0 | 1.5 | 11.2 | 1.1 | - | - | 101.0 |
| 1079_5_2 | 7 | Fe3C | 92.5 | 0.4 | - | - | - | - | - | - | - | - | 92.5 |
| 1001_4_3 | 7 | Ni | - | - | 99.7 | 0.2 | - | - | - | - | - | - | 99.7 |
| 1001_4_4 | 8 | Fe3C | 92.7 | 0.5 | - | - | - | - | - | - | - | - | 92.7 |
| 1001_4_4 | 6 | FeO | 78.9 | 0.6 | - | - | 22.1 | 0.5 | - | - | - | - | 100.9 |
| 2095_2_3 | 6 | Fe7C3 | 90.6 | 0.4 | - | - | - | - | - | - | - | - | 90.6 |
| 600_8_2 | 7 | L | 65.8 | 0.8 | 29.1 | 0.6 | - | - | - | - | 1.8 | 0.5 | 96.7 |
| 600_8_4 | 5 | Ni | - | - | 99.3 | 0.7 | - | - | - | - | - | - | 99.3 |
| 2100_2_2 | 8 | Fe3C | 91.1 | 0.1 | - | - | - | - | - | - | - | - | 91.1 |
| 2100_2_3 | 6 | L | 73.1 | 0.2 | 15.4 | 0.5 | - | - | - | - | 7.2 | 0.4 | 95.7 |
| 2100_2_4 | 6 | Fe7C3 | 90.6 | 0.4 | - | - | - | - | - | - | - | - | 90.6 |
| 688_6_1 | 8 | Fe3C | 92.7 | 0.3 | - | - | - | - | - | - | - | - | 92.7 |
| 688_6_1 | 7 | Fe7C3 | 90.2 | 0.4 | - | - | - | - | - | - | - | - | 90.2 |
| 688_6_3 | 7 | Fe7C3 | 90.3 | 0.3 | - | - | - | - | - | - | 90.3 | ||
| 688_6_3 | 9 | FeO | 77.8 | 0.3 | - | - | 23.2 | 0.3 | - | - | - | - | 101.0 |
| 605_8_5 | 7 | L | 79.4 | 0.2 | - | - | - | - | - | - | 16.3 | 0.8 | 95.6 |
| 605_8_5 | 8 | FeO | 76.6 | 0.2 | - | - | 21.9 | 0.5 | - | - | - | - | 98.5 |
| 605_8_7 | 6 | L | 26.2 | 0.5 | 47.8 | 1.1 | - | - | - | - | 25.9 | 1.6 | 99.9 |
| 605_8_8 | 9 | L | - | - | 49.8 | 0.1 | 0.6 | 0.1 | - | - | 50.1 | 0.7 | 100.5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sokol, A.; Tomilenko, A.; Sokol, I.; Zaikin, P.; Bul’bak, T. Formation of Hydrocarbons in the Presence of Native Iron under Upper Mantle Conditions: Experimental Constraints. Minerals 2020, 10, 88. https://doi.org/10.3390/min10020088
Sokol A, Tomilenko A, Sokol I, Zaikin P, Bul’bak T. Formation of Hydrocarbons in the Presence of Native Iron under Upper Mantle Conditions: Experimental Constraints. Minerals. 2020; 10(2):88. https://doi.org/10.3390/min10020088
Chicago/Turabian StyleSokol, Alexander, Anatoly Tomilenko, Ivan Sokol, Pavel Zaikin, and Taras Bul’bak. 2020. "Formation of Hydrocarbons in the Presence of Native Iron under Upper Mantle Conditions: Experimental Constraints" Minerals 10, no. 2: 88. https://doi.org/10.3390/min10020088
APA StyleSokol, A., Tomilenko, A., Sokol, I., Zaikin, P., & Bul’bak, T. (2020). Formation of Hydrocarbons in the Presence of Native Iron under Upper Mantle Conditions: Experimental Constraints. Minerals, 10(2), 88. https://doi.org/10.3390/min10020088