High-Pressure Raman and Infrared Spectroscopic Study of Prehnite


Abstract
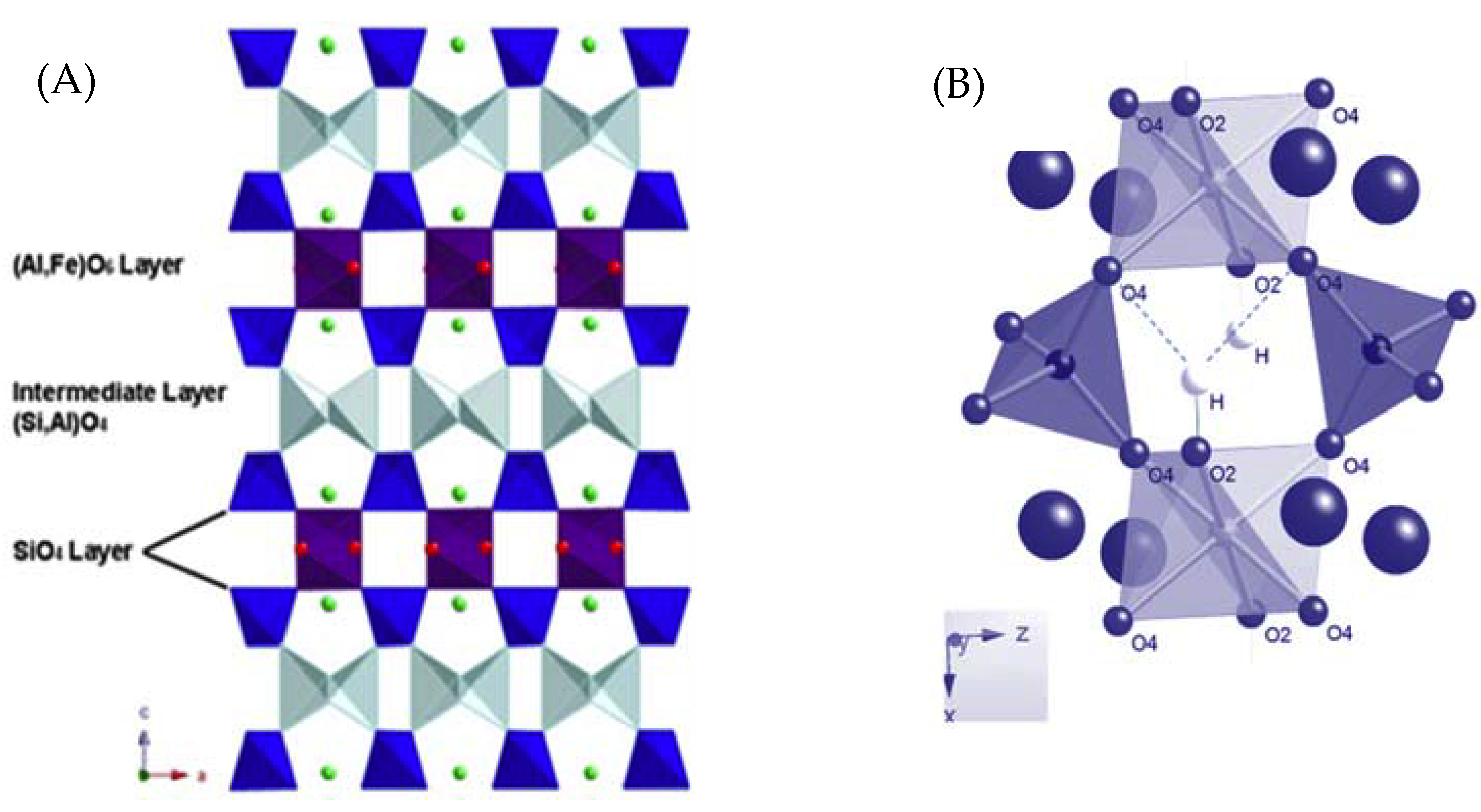
1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. IR and Raman Spectra of Prehnite Measured under Ambient Conditions
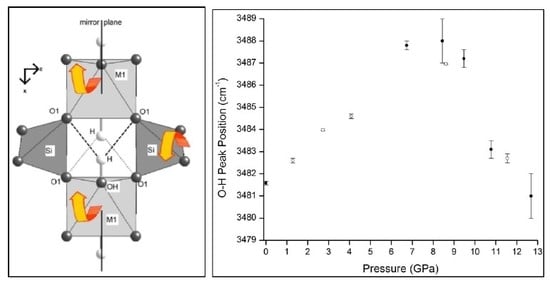
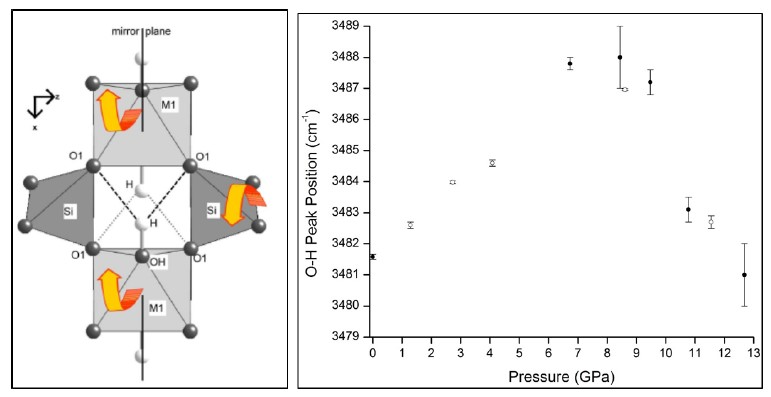
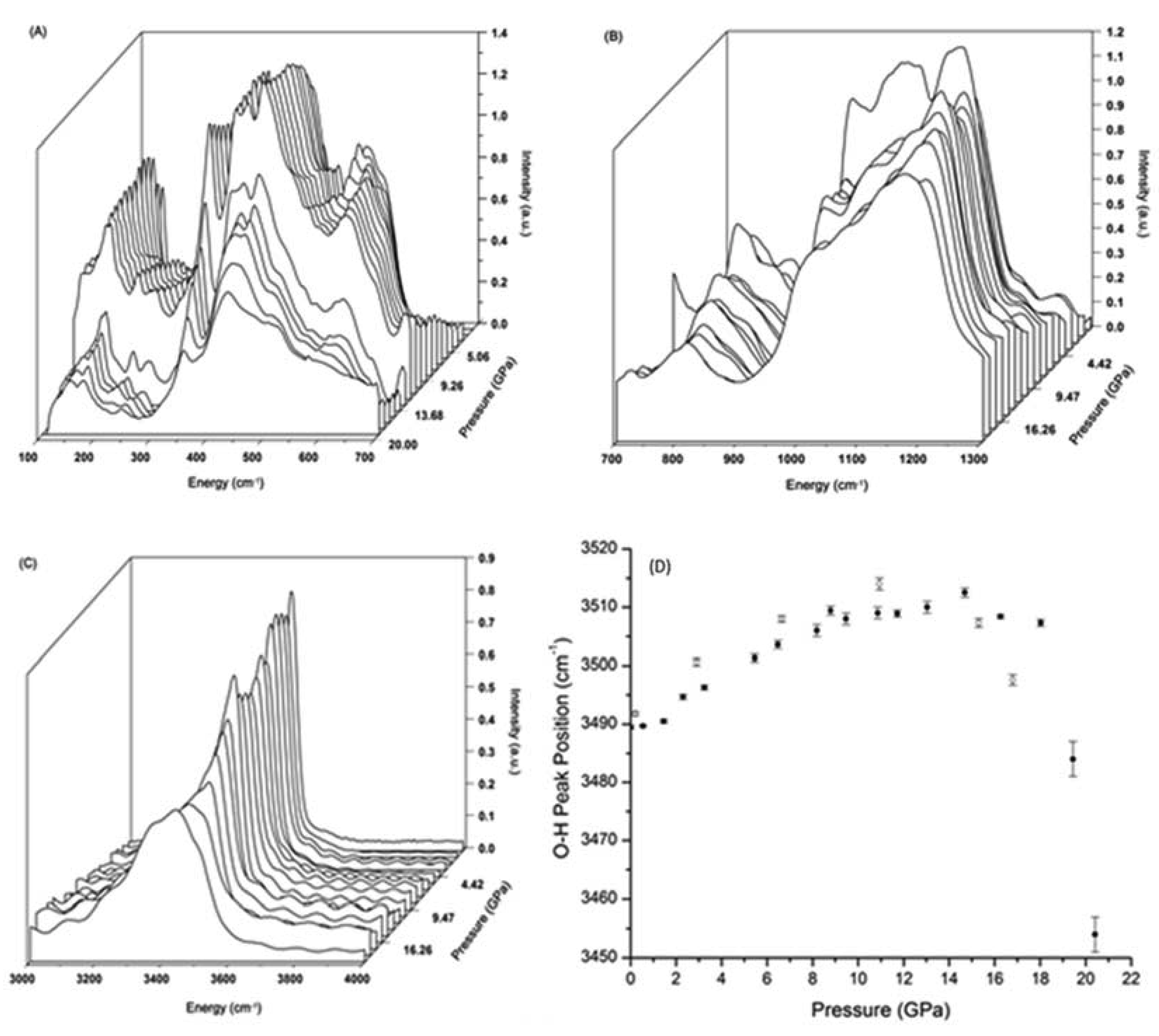
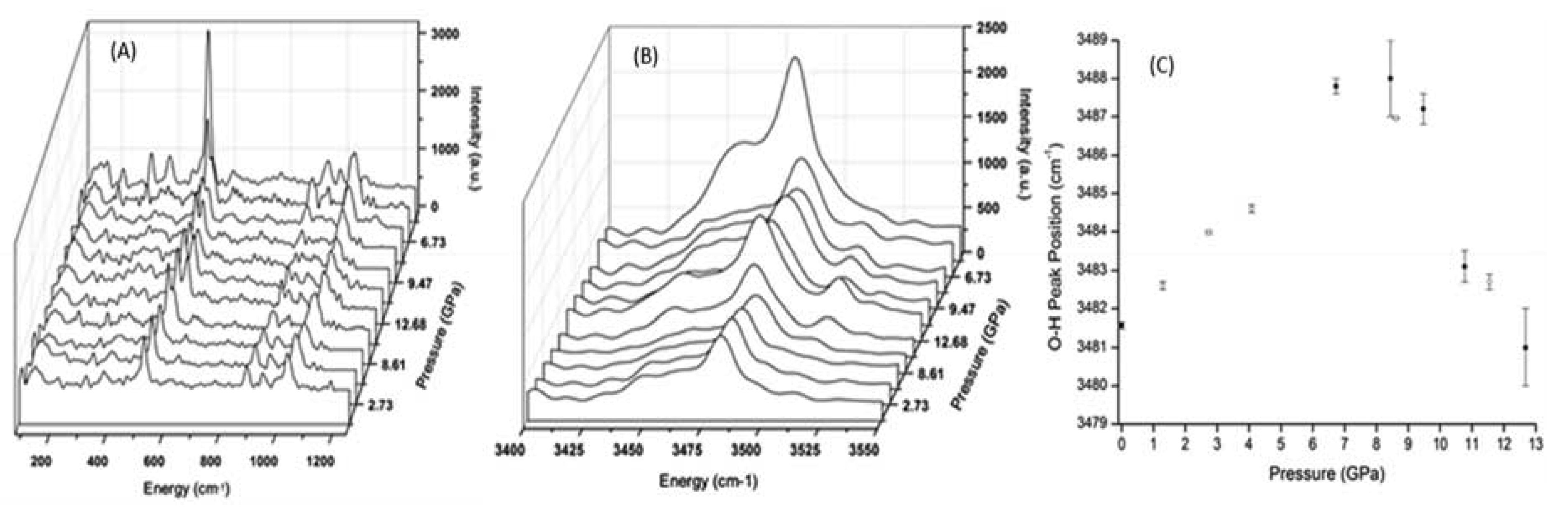
3.2. High Pressure Infrared Spectra of Prehnite
3.3. High Pressure Raman Spectra of Prehnite
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Artioli, G.; Quartieri, S.; Deriu, A. Spectroscopic data on coexisting prehnite-pumpellyite and epidote-pumpellyite. Can. Mineral. 1995, 33, 67–75. [Google Scholar]
- Liou, J.G. Synthesis and stability relations of prehnite, Ca2Al2Si3O10(OH)2. Amer. Mineral. 1971, 56, 507–531. [Google Scholar]
- Perkin, D., III; Westrum, E.F., Jr.; Essene, E.J. thermodynamic properties and phase relations of some minerals in the system CaO–Al2O3–SiO2–H2O. Geochim. Cosmochim. Acta 1980, 44, 61–84. [Google Scholar] [CrossRef]
- Gottschalk, M. Internally consistent thermodynamic data for rock-forming minerals in the system SiO2–TiO2–Al2O3–Fe2O3–CaO–MgO–FeO–K2O–Na2O–H2O–CO2. Eur. J. Miner. 1997, 9, 175–223. [Google Scholar] [CrossRef]
- Peng, S.-T.; Chou, K.-D.; Tang, Y.-C. The structure of prehnite. Acta Chem. Sin. 1959, 25, 56–63. [Google Scholar]
- Papike, J.J.; Zoltai, T. Ordering of tetrahedral aluminium in prehnite. Am. Miner. 1967, 52, 974–984. [Google Scholar]
- Akizuki, M. Al, Si order and the internal texture of prehnite. Can. Miner. 1987, 25, 707–716. [Google Scholar]
- Balić-Žunić, T.; Šćavničar, S.; Molin, G. Crystal structure of prehnite from Komiža. Eur. J. Miner. 1990, 2, 731–734. [Google Scholar] [CrossRef]
- Detrie, T.A.; Ross, N.L.; Angel, R.J.; Welch, M.D. Crystal chemistry and location of hydrogen atoms in prehnite. Miner. Mag. 2008, 72, 1163–1179. [Google Scholar] [CrossRef]
- Detrie, T.A.; Ross, N.L.; Angel, R.J.; Diego Gatta, G. Equation of state and structure of prehnite to 9.8 GPa. Eur. J. Miner. 2009, 21, 561–570. [Google Scholar] [CrossRef]
- Angel, R.J.; Bujak, M.; Zhao, J.; Gatta, D.; Jacobsen, S.D. Effective hydrostatic limits of pressure media for high-pressure crystallographic studies. J. Appl. Cryst. 2007, 40, 26–32. [Google Scholar] [CrossRef]
- Mao, H.; Xu, J.; Bell, P.M. Calibration of the ruby pressure gauge to 800 kbar under quasi-hydrostatic conditions. J. Geophys. Res. 1986, 91, 4673–4676. [Google Scholar] [CrossRef]
- SeaSolve Software Inc. PeakFit v4.12 (1999–2003); SeaSolve Software Inc.: San Jose, CA, USA, 2000. [Google Scholar]
- Šontevska, V.; Jovanovski, G.; Makreski, P. Minerals from Macedonia. Part XIX. Vibrational spectroscopy as identificational tool for some silicate minerals. J. Mol. Struct. 2007, 834–836, 318–327. [Google Scholar] [CrossRef]
- Boev, B.; Jovanovski, G.; Makreski, P. Minerals from Macedonia. Part XX. Geological Setting, Lithologies, and Identification of the Minerals from Ržanovo Fe Ni deposit. Turkish J. Earth Sci. 2009, 18, 631–652. [Google Scholar]
- McKeown, D.A.; Bell, M.I.; Etz, E.S. Raman spectra and vibrational analysis of the trioctahedral mica phlogopite. Am. Miner. 1999, 84, 970–976. [Google Scholar] [CrossRef]
- Saniger, J.M. Al–O infrared vibrational frequencies of γ alumina. Mater. Lett. 1995, 22, 109–113. [Google Scholar] [CrossRef]
- White, A.J.R.; Laukamp, C.; Stokes, M.A.; Legras, M.; Pejcic, B. Vibrational spectroscopy of epidote, pumpellyite and prehnite applied to low-grade regional metabasites. Geochem. Expl. Env. Analy. (GEEA) 2017, 17, 315–333. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Infrared Peaks (cm−1) | Peak FHWM (cm−1) | Raman Peaks (cm−1) | Peak FHWM (cm−1) |
|---|---|---|---|
| 128 (3) | 27 | 112 (1) | 21 |
| 146 (2) | 21 | 138 (1) | 27 |
| 213 (1) | 26 | 161 (1) | 16 |
| 243 (1) | 16 | 216 (1) | 17 |
| 296 (1) | 27 | 317 (1) | 18 |
| 343 (1) | 33 | 350 (1) | 27 |
| 377 (1) | 21 | 381 (1) | 25 |
| 423 (2) | 55 | 464 (1) | 21 |
| 475 (1) | 35 | 492 (1) | 22 |
| 505 (1) | 36 | 518 (1) | 20 |
| 542 (1) | 38 | 541 (1) | 14 |
| 574 (5) | 44 | 606 (1) | 26 |
| 755 (2) | 47 | ||
| 812 (1) | 36 | ||
| 876 (1) | 33 | ||
| 931 (9) | 44 | ||
| 976 (9) | 40 | 940 (1) | 30 |
| 998 (25) | 62 | 985 (1) | 16 |
| 1033 (19) | 33 | ||
| 1080 (13) | 52 | 1074 (1) | 28 |
| 1118 (16) | 48 | ||
| 1152 (24) | 47 | ||
| 3453 (1) | 41 | 3456 (1) | 26 |
| 3475 (6) | 18 | ||
| 3490 (1) | 35 | 3419 (1) | 13 |
| 3494 (1) | 13 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ross, N.L.; Detrie, T.A.; Liu, Z. High-Pressure Raman and Infrared Spectroscopic Study of Prehnite. Minerals 2020, 10, 312. https://doi.org/10.3390/min10040312
Ross NL, Detrie TA, Liu Z. High-Pressure Raman and Infrared Spectroscopic Study of Prehnite. Minerals. 2020; 10(4):312. https://doi.org/10.3390/min10040312
Chicago/Turabian StyleRoss, Nancy L., Theresa A. Detrie, and Zhenxian Liu. 2020. "High-Pressure Raman and Infrared Spectroscopic Study of Prehnite" Minerals 10, no. 4: 312. https://doi.org/10.3390/min10040312
APA StyleRoss, N. L., Detrie, T. A., & Liu, Z. (2020). High-Pressure Raman and Infrared Spectroscopic Study of Prehnite. Minerals, 10(4), 312. https://doi.org/10.3390/min10040312