1. Introduction
X-ray fluorescence (XRF) spectroscopy is widely employed for the elemental (chemical) analysis of minerals and glasses. In mineral, geological, mining and archaeological analyses, XRF has been used to assess the elemental composition of minerals and other inorganic materials, allowing for the identification of consistency, impurities and contaminants [
1,
2,
3,
4]. Within the glass industry and glass science, XRF spectroscopy is widely used to analyse the chemical composition of commercial and experimental glasses [
5,
6,
7]. XRF has also been widely used to assess the viability and quality of raw materials and minerals for application across multiple manufacturing industries, including glasses, ceramics and refractories [
8,
9,
10,
11].
Due to the high manufacturing temperatures of many commercial glasses (ca. 1450–1500 °C for soda-lime-silica container and float glasses), some components are known to volatilise and escape from the melt into the vapour phase and thence the off-gas system, which can have potentially adverse effects on the final glass product, the furnace refractories and gaseous/particulate emissions. Different species can volatilise at different temperatures, pressures, atmospheres and melt durations. Volatilisation of key glass making components, such as Na and B, has been observed at melting temperatures ranging from 1150 to 1600 °C [
12,
13,
14,
15]. Other alkali components such as Li and K have also been recorded to have volatilised at similar temperatures [
15,
16,
17] although in the case of Li, volatilisation requires longer melt times, higher temperatures, or a reducing melting atmosphere [
17]. As a general rule of thumb, the alkali volatilisation rate increases down the alkali metal series, from Li to Cs. Bingham [
18] summarised literature pertaining to volatilisation from soda-lime-silica and commercial silicate glass types, which includes work by Cable and Chaudhry [
19] that demonstrates the existence of a linear relationship between log (mass loss) and reciprocal temperature for evaporation of volatile species from soda-lime-silica glass melts. Cable and Chaudhry [
19] studied SiO
2-Al
2O
3-Na
2O-CaO glasses and, in addition to the relationship between mass loss and temperature, they also showed that the relation between mass loss, M, and melting time at a given temperature, t, was not necessarily M ∝ t
1/2, as expected for a simple diffusion-based model, and that volatilisation of Na could be described reasonably accurately by a model including both diffusion in the melt and a first-order reaction for surface loss, i.e., a modified diffusion-based model. Cable and Fernandes [
20] explained that volatilisation from glass melts occurs in three separate steps: (i) diffusion of the volatile component/s through the melt to its surface; (ii) evaporation or reaction at the liquid–gas interface; and (iii) transport of the vapour away from the surface in the gas phase. The first stage is diffusion-based and gives rise to the M ∝ t
1/2 relationship but, as noted by Cable and Fernandes [
20], this can fail to describe data accurately in some cases. More recently, Kucuk et al. [
21] studied volatilisation from SiO
2-K
2O and SiO
2-Na
2O-CaO glass melts and found similar results to Cable’s [
19,
20]. However, Matousek and Hlavac [
22] found mass loss from SiO
2-Na
2O-K
2O-PbO glasses, and Zhang et al. [
23] found mass loss from SiO
2-B
2O
3-SrO-BaO and SiO
2-B
2O
3-CaO-SrO-BaO-ZnO-Al
2O
3-TiO
2 glasses, and all obeyed the relationship M ∝ t
1/2. Beerkens [
24] also modelled volatilisation from silicate melts and summarised some of the available literature. The differences in results between these various studies suggest that different mechanisms determine or influence volatilisation from glass melts, with different impacts depending on glass composition, the component/s in question (e.g., a particular component such as Na, or total mass loss) and experimental conditions (e.g., static or flowing melt/atmosphere, different vessel geometries).
In the nuclear industry, the globally preferred method of long-term radionuclide immobilisation from high-level radioactive wastes (HLW) is vitrification (the process of turning the waste into a chemically and thermally stable glass waste form) [
25]. Retention of radioisotopes such as
137Cs and
99Tc in the melt has proven to be a challenge in HLW vitrification [
26,
27,
28], with experiments showing that over 95% of
99Tc will volatise at temperatures as low as 900 °C, with a melting time of 30 min [
26]. As illustrated by Ojovan and Lee [
29] for Na, K, Rb, Cs, Te and Mo in HLW vitrification, loss of these elements by volatilisation obeys a linear relationship between log (mass loss) and reciprocal temperature. This is consistent with the literature for commercial glass manufacture summarised above, and highlights the need to maintain radioactive waste vitrification processes at temperatures capable of melting the products whilst minimising volatilisation. However, despite this knowledge of the effects of melting temperature, relatively little research has been published on volatilisation of components from radioactive waste-type glasses as a function of melting time. Banerjee et al. [
30] studied Cs volatilisation from SiO
2-B
2O
3-Na
2O-Cs
2O-(CaO, BaO, ZnO) glasses relevant to radioactive waste immobilisation, and confirmed that for most of the glasses and temperatures studied, the volatilisation (measured as Cs loss) could be accurately described by the simple diffusion-based model, M ∝ t
1/2 described above. The three mechanisms described by Cable and Fernandes [
20] were used by Banerjee et al. [
30] to describe this process, namely diffusion of Cs through the glass melt to its surface, followed by transport of Cs through the melt/vapour interface, and then transport into the atmosphere. However, some of their glasses [
30] displayed deviation from this behaviour, suggesting that other processes were involved: the results were thus broadly consistent with Cable’s observations [
19,
20] described above. It is clear that the mechanisms governing volatilisation from glass melts are highly complex, with multiple contributing factors, and with compositional, temperature and experimental conditions dependencies that are not yet fully understood.
Volatilisation from B
2O
3-Li
2O glass melts during XRF fused bead preparation has also been documented [
31], and qualitatively shown to obey similar relationships to those above. Fused beads for oxidised samples such as minerals and oxide glasses often contain added lithium, sodium or ammonium halides (I, Br) as mould release agents [
31]. These are added to facilitate release of the cooled fused beads from the mould and to prevent cracking. Claisse and Blanchette [
31] discussed this in detail, noting in particular the evaporation of Li, B, halides, sulphates and Na. They also noted the need to limit fusion temperatures in order to maintain volatilisation at acceptable levels. They also stated that volatility of the release agents varies with sample and flux composition, and that “it is impossible to determine the exact amount that remains in a fusion bead to facilitate its separation” [
31]. Finally, they noted that a part of the release agent is distributed uniformly in the fused bead, while part is present as a film on the surface of the bead.
The majority of previous studies, including those summarised above, have considered volatilisation of major species (e.g., B, alkalis) or total mass loss from glass melts. Few have studied, at least from a fundamental perspective, losses of particularly volatile species such as halides or sulphates, as a function of melting time or melting temperature. One such study by Tatevosyan et al. [
32] considered mass loss (principally F) from SiO
2-CaO-Na
2O-K
2O-Al
2O
3-F glasses, and showed a deviation from linearity when mass loss was plotted as a function of (time)
1/2, i.e., a deviation from M ∝ t
1/2. Another study by Parker et al. [
33] on SiO
2-Al
2O
3-Na
2O-CaO-F glasses showed that, at relatively low temperatures below 950 °C, volatilisation (principally of F but also other constituents) was a diffusion-controlled process (as demonstrated by a (time)
1/2 dependency), but at higher temperatures more complex behaviour was observed, involving cellular convection currents driven by surface energy and density gradients. Fluorine, as a halogen, may behave differently in structural terms and in volatilisation rates from other halogens in oxide glasses. However, fluorine can reasonably be expected to be governed by the same relationships as other halides, in terms of volatilisation from oxide glass melts. The fundamental volatilisation behaviour (e.g., in terms of time, temperature) of other halides in oxide glass melts, and particularly those halides relevant to radioactive waste vitrification (Cl and I) has thus, to date, received little attention. However, it has been demonstrated that in radioactive waste borosilicate glass melting, halogen volatilisation increases in the order F < Cl < I [
34], following the same trend as the alkalis (see above).
The production of fused beads is a common method of sample preparation for XRF spectroscopy. The fused bead method is often preferred to other techniques [
35,
36,
37,
38,
39] due to the ability to form a sample of the correct size, shape and finish for analysis without time-consuming sectioning and polishing, whilst reducing the environment-specific effect on the intensity of characteristic element lines [
31,
35,
39]. With the fused bead method, the sample is mixed with a flux (e.g., lithium tetraborate, Li
2B
4O
7), melted at high temperature, and either poured into a disc-shaped mould, or otherwise formed in the required dimensions, to form the bead. The flux is used to reduce the melting point of the sample so that the sample can be entirely and homogeneously dissolved in the matrix, and so that a bead can be produced at temperatures as low as 900–1050 °C [
31,
35,
40]. The flux generally contains low-Z elements, which are not readily detected using laboratory-scale XRF spectrometers, e.g., Li, B and O as in lithium tetraborate, Li
2B
4O
7. However, the presence of mould-release agents consisting of halides (I, Br) can give rise to “an efficient albeit undesirable filter of X-rays” [
31] so using the smallest possible quantity of release agent is advisable [
31].
The volatile nature of certain components including, but not limited to, the alkalis and boron, as described above, raises the question as to whether the very application of the fused bead process leads to analytical inaccuracies due to volatilisation during XRF fused bead sample preparation, and the extent to which this affects the results. The research presented here sought to address this question by performing XRF analysis on fused beads of standard materials of known composition. Four standard materials were chosen, all of which are British Chemical Standard Certified Reference Materials (BCS-CRMs): BCS-CRM-529 (Anorthic Feldspar) [
41], BCS-CRM-532 (Swedish Feldspar) [
42], BCS-CRM-375 (Soda Feldspar) [
43] and BCS-CRM-525 (Low-Iron Float Glass) [
44]. In order to investigate the effects of fused bead melting time on volatilisation and analytical stability and reproducibility, a number of samples were produced for each reference material; melting times ranged from 11 min to 106 min, corresponding to total process times (melting + cooling) of approximately 16 and 111 min. The volatilisation and reproducibility behaviour of each element was then studied from the XRF spectra for each reference sample and each total process time. The melting temperature, as discussed above, affects volatilisation losses but, as will be shown, it can also affect fused bead quality and analytical reproducibility. In order to focus on studying the effects of melting time in the present work, all fused beads were melted at a fixed temperature of 1065 °C. By comparison, a key relevant ISO standard method [
11] stipulates the melting of fused beads at 1200 ± 50 °C for 5 min. As shown by several authors including, for example, Ojovan and Lee [
29], volatilisation of alkalis from glass melts (e.g., in mg/cm
2) can be expected to be one order of magnitude greater using the ISO standard [
11] compared to the method we have used here, owing to the substantially higher temperatures used. However, the 5 min melting time for the ISO standard method [
11] is considerably shorter than the times used in this study. Consequently, we have focussed here on determining the effects of fused bead melting time, not only in terms of the relationships governing volatilisation losses, but also on the ability to reproducibly manufacture crack-free fused beads that can be readily retrieved from the mould and which provide stable and reproducible analytical results. In planned future work we will seek to investigate the effects of fused bead melting temperature in order to provide a deeper assessment.
2. Materials and Methods
The materials used in this research were British Chemical Standard-Certified Reference Materials (BCS-CRMs) provided by the Bureau of Analyzed Samples Ltd. (BAS). The samples were Anorthic Feldspar (CRM 529 [
41]), Swedish Feldspar (CRM 532 [
42]), Soda Feldspar (CRM 375 [
43]), and Low-Iron Float Glass (CRM 525 [
44]). The certified reference analytical compositions for each CRM sample are reproduced in
Table A1,
Table A2,
Table A3 and
Table A4. The CRMs were prepared for fused bead generation by mixing approximately 1 g of sample, with approximately 10 g of lithium tetraborate (Li
2B
4O
7) flux. The precision of weighing ensured that all mixtures were at a flux: sample ratio of 9.99 to 10.04. The flux was doped with 0.5 wt% of lithium iodide (LiI) as a non-wetting (release) and anti-cracking agent [
11,
31]. Attempts to make fused beads with approximately 1 g of CRM 532 resulted in fused beads cracking on cooling so the samples with this CRM were made with approximately 0.76 g of sample, to 10 g of lithium tetraborate and 0.5 wt% of lithium iodide.
The fused beads were prepared using a Claisse LeNeo Fused Bead maker [
45], by melting in 5% Au–95% Pt casting bowls at 1065 °C, with fixed, programmed rocking motions during melting and fixed, programmed blown air during cooling. The total process has two elements: the melting time and the cooling time, during which the melt will be automatically poured into a 5% Au-95% Pt mould and air cooled under fixed and highly reproducible conditions [
45]. The melting and total process times are detailed in
Table 1. Mass loss from fusion was recorded for each fused bead. The shortest total process times used in this study, 16 min and 15 s, failed to produce fused beads with sufficient consistency, and over 50% of the beads made were subject to catastrophic cracking, either during cooling or when removed from the 5% Au-95% Pt mould. This cracking is attributed to the CRM sample materials not fully and homogeneously dissolving in the Li
2B
4O
7 flux and thereby causing local thermal expansion variations, leading to crack formation upon cooling. Consequently, the 16 min 15 s data were not reproduced here and can be considered too brief to produce consistent and useable fused beads for feldspar and glass sample materials under the preparation conditions studied. It is possible that with higher melting temperatures, as in [
11], such problems might be avoided. However, in doing so these problems may potentially be replaced by other problems associated with higher volatilisation losses [
18,
19,
20,
21,
22,
23,
24,
25,
26,
27,
28,
29,
30,
31,
32,
33].
All XRF data were collected with a PANalytical MagiX PRO XRF spectrometer (Malvern Panalytical Ltd., Malvern, UK). using a Rh anode X-ray tube. The XRF data were analysed using a modified version of the OXI program [
46], a Wide Range Oxide XRF program developed and verified in-house. In this case our calibration curve for the oxide standards for the sixteen elements used in this version of OXI had multiple data points to increase accuracy. Details of these elements, numbers of data points and calibration ranges are given in
Table 2 and details of the operational conditions for each of the 16 elements in the OXI program are given in
Table 3. In addition to collection of XRF data on every fused bead, further XRF data for CRM-525 (Low-Iron Float Glass) were collected 4 separate times for each fused bead to enable quantification of any measurement-to-measurement variabilities which could thence be eliminated (if sufficiently small) from consideration as potential origins of any differences observed between XRF analyses for different fused bead melting times. Mass loss on fusion was established (
Table A1,
Table A2,
Table A3 and
Table A4) by mass balance before fusion and after fusion, and is reproduced here as weight (%).
Further XRF data were collected using the PANalytical standardless IQ+ software (Malvern Panalytical Ltd., Malvern, UK). This software can analyse for elements from F to Am. IQ+ results are semi-quantitative so they were not used for analyses of these samples. However, these IQ+ results were particularly useful in measuring how the intensities of the iodine Kα
1 line, and hence the abundance of I in the fused beads, decreased with increasing melting time.
Table 4 shows details of the operational conditions for each of the IQ+ measurements, which were carried out over 10 different energy ranges.
4. Discussion
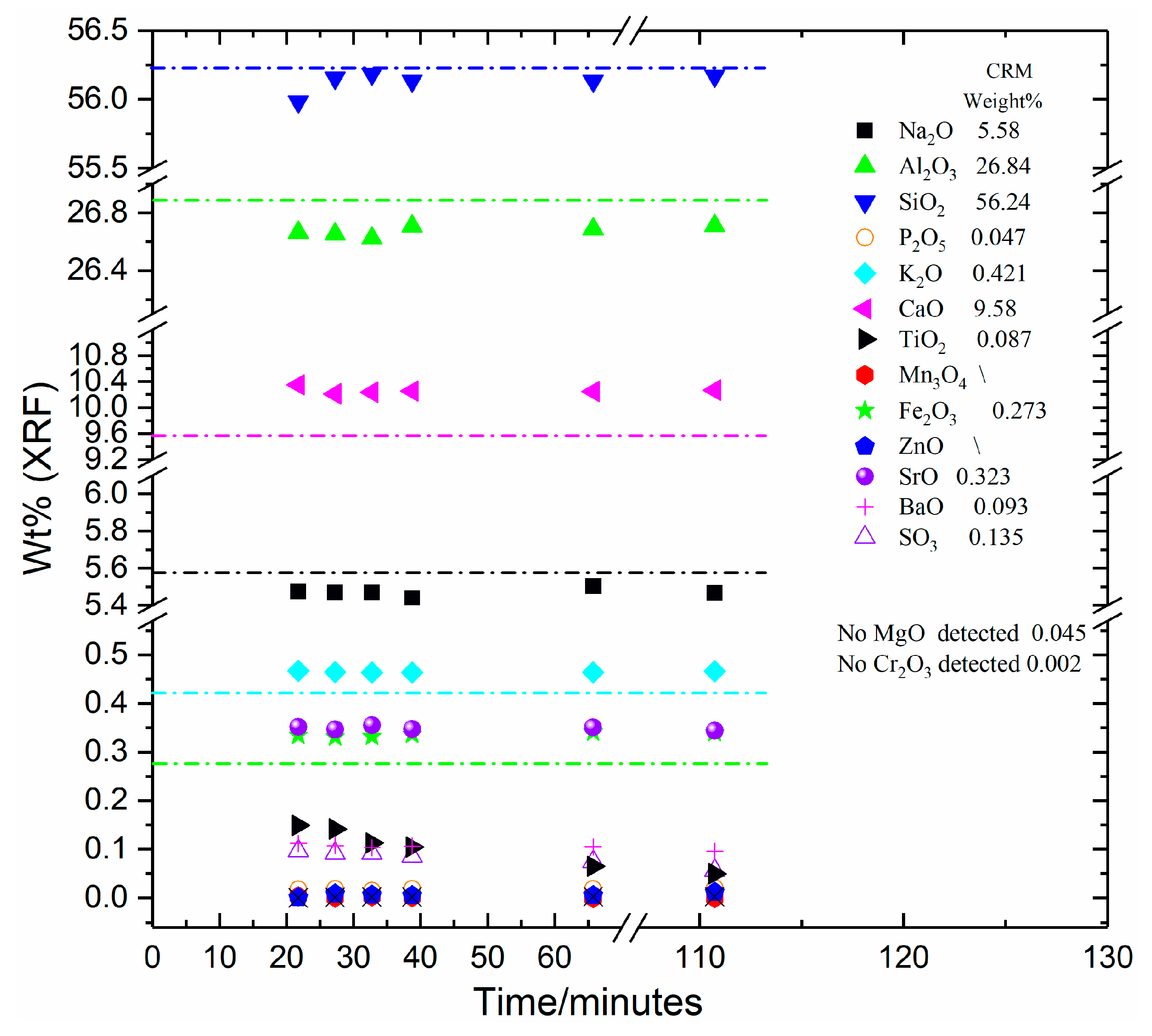
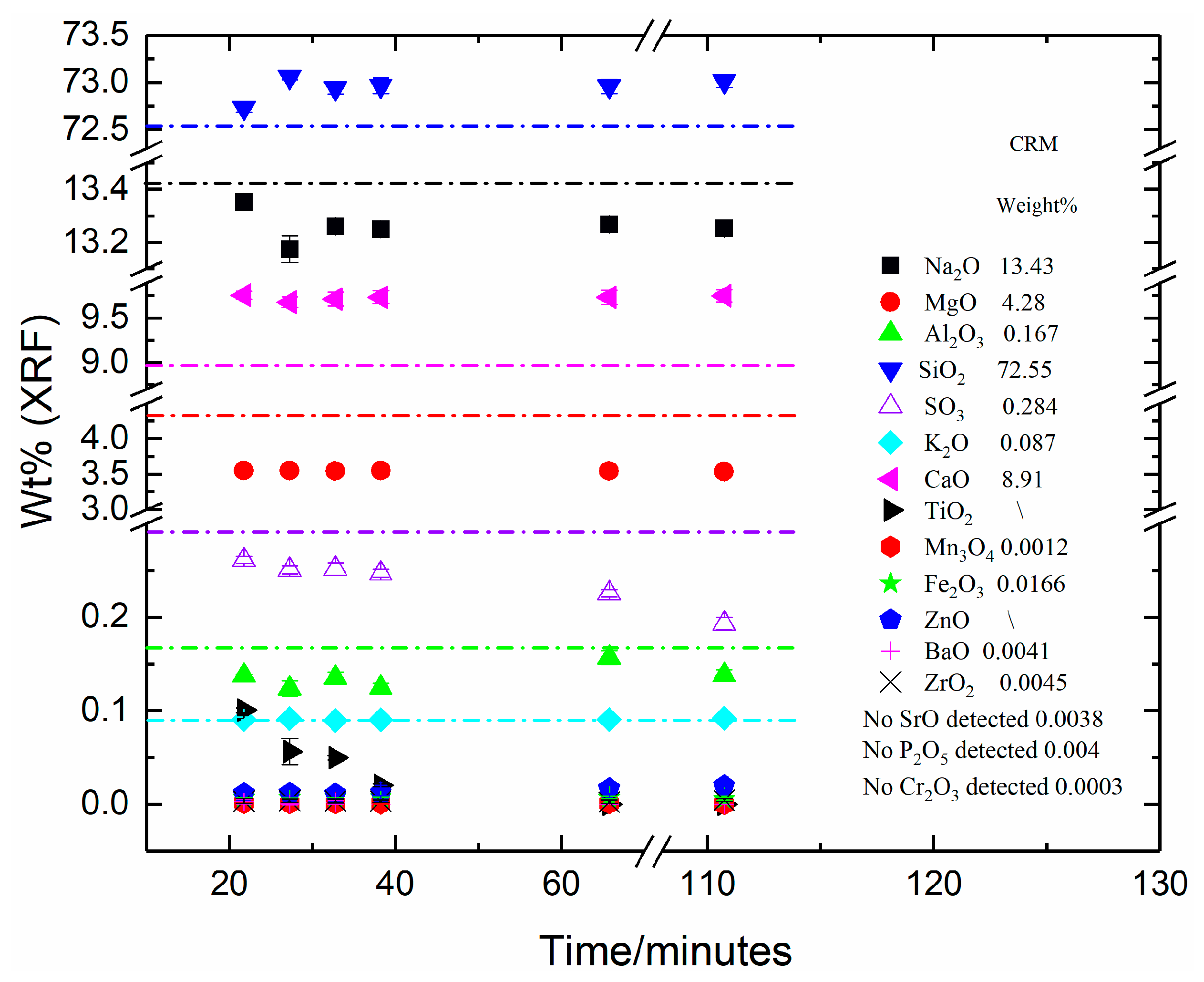
As shown in
Figure 1,
Figure 2,
Figure 3 and
Figure 4 and
Table A1,
Table A2,
Table A3 and
Table A4, all four CRM materials exhibited sample-to-sample fluctuations in XRF-analysed composition and differences from the certified compositions (from [
41,
42,
43,
44], reproduced in
Appendix A,
Table A1,
Table A2,
Table A3 and
Table A4). Sample-to-sample fluctuations were notable with fused beads with shorter process times (i.e., the samples with total process times of 27 min 45 s and 21 min 45 s; the 16 min 15 s samples were too variable and often cracked during removal from the mould, so mostly these were not analysed). These differences and variabilities in XRF analysis results can be attributed to some combination of: (i) inaccuracy of the XRF OXI program; (ii) imprecision or instability of the XRF spectrometer; and/or (iii) fused bead (sample) related issues.
In order to identify the source/s of the observed differences in results, and to eliminate candidate causes along the way, we will first consider (i) above: inaccuracy (and thus imperfect calibration) of the XRF Wide Range Oxide (OXI) program we have used. Whilst, as shown in
Table 2, the calibration curve contains many independent data points for most elements and is thereby considered robust, it is seen, for example, that our Wide Range Oxide (OXI) program consistently overestimated the SiO
2 content of CRM-532 (Swedish Feldspar) (
Figure 2) by of the order of 0.7 wt%; and underestimated the SiO
2 content of CRM-375 (Soda Feldspar) (
Figure 3) by of the order of 1.5 wt%. Whilst such inaccuracies are not desirable, they are particularly challenging to completely eliminate from Wide Range Oxide programs that are applied over a large range of sample compositions, as we have done here. However, inaccuracies such as these are systemic and apply to all samples measured, provided they are analysed under the same conditions, as we have carried out in this study. Hence the prospect of point (i) above being responsible for all of the variabilities observed in analysed compositions (
Figure 1,
Figure 2,
Figure 3 and
Figure 4), as a function of fused bead total process time, cannot be completely eliminated as a contributing factor, but it was demonstrably not the primary cause.
Point (ii) above, imprecision or instability of the XRF spectrometer, was directly assessed through the repeated measurements (four each) for fused beads representing each time-step for CRM-525, Low-Iron Float Glass (
Figure 4,
Table A4). The obtained Standard Deviations from these measurements were predominantly very low and, as shown in
Figure 4 were, in most cases, smaller than the size of the data markers. Consequently, the observed variations in analysed composition were, for the most part, larger than the Standard Deviations associated with the measurements. This confirms that point (ii) can also be discounted as the main cause of the observed variations in the analysed composition as a function of fused bead total processing times–again, whilst it cannot be completely eliminated as a contributing factor, it was demonstrably not the primary cause.
Points (i) and (ii) above have been largely eliminated as the primary sources of the observed differences and variabilities in XRF analysis results shown in
Figure 1,
Figure 2,
Figure 3 and
Figure 4 and in
Table A1. This leaves point (iii), fused bead (sample) related issues. An inexhaustive list of potential causes of these issues includes poorly soluble or highly refractory sample material leading to incomplete dissolution in the fused bead; chemically and/or physically inhomogeneous fused beads; contamination of the fused bead from the mould, raw materials or other sources during melting or preparation for analysis; and incorrect or imperfect placement of the fused bead inside the XRF spectrometer. The variability of analysed composition discussed above is observed for the shorter melting times for all four CRM materials, although for the CRM-525 Low-Iron Float Glass sample, the shortest melting time gave the closest analysed composition to the certified composition. This makes it unlikely that contamination or incorrect placement of the XRF samples in the spectrometer could be responsible for these changes as they are consistent and systematic. Consequently, some combination of incomplete fusion and/or inhomogeneity of the fused beads are the most likely explanations for some of the feldspar samples. As noted earlier, CRM-375 Soda Feldspar has larger particle sizes and may need longer fusion times as a result.
Volatilisation during fused bead preparation affects not only the Li
2B
4O
7 flux and LiI mould-release/anti-cracking agent, but also certain components within the samples studied, in particular halides and sulphates but also alkalis, boron and other relatively volatile constituents [
12,
13,
14,
15,
16,
17,
18,
19,
20,
21,
22,
23,
24,
26,
28,
29,
30,
31,
32,
33,
34]. In the case of the samples studied here, losses of iodine and sulphur (as SO
3) were notable. The intensity of the iodine Kα
1 line, and total analysed content of SO
3, in various fused beads of Anorthic Feldspar (CRM-529) [
41] were obtained at various fused bead melting times, and both sets of data are shown in
Figure 5. Since iodine was not included in our OXI Wide Range Oxide XRF program, the extent of volatilisation of iodine in fused beads was established from the decrease in the intensity of the IKα
1 line obtained using the IQ+ standardless program as the melting time increased. Iodine concentration in fused beads is shown to decrease roughly linearly (R
2 = 0.861) with decreasing (time)
1/2, and iodine essentially completely volatilised when (time)
1/2 reached approximately 10 min
1/2 (data) or 9 to 10 min
1/2 (linear fit), i.e., when melting time reached approximately 80 to 100 min. This behaviour is also reflected in the analysed “apparent content” of TiO
2 in all samples. As noted in
Section 3, the I Lβ
1,2,3 lines overlap with the Ti Kα
1,2 lines, so the apparent volatilisation of Ti, as implied by the decreasing analysed TiO
2 contents shown in
Figure 1,
Figure 2,
Figure 3 and
Figure 4, is readily explained by the loss of I (
Figure 5) and not by the volatilisation of Ti, which is highly unlikely under these melting conditions.
As shown in
Figure 5, a linear fit to the iodine loss data is imperfect, and the relationship is clearly non-linear. This has important implications for XRF fused bead preparation and also wider implications, for example for vitrification of iodine-bearing radioactive wastes. The simple diffusion-based model discussed in
Section 1 does not fully describe the loss of iodine as a function of melting time, and implies that the additional mechanisms described earlier [
19,
20,
21,
30,
33], including those observed for fluorine, are in evidence. Claisse and Blanchette [
31] noted the presence of some of the halide (iodine here) within the fused bead and some on the surface (to promote mould release), and this may help to explain the observed deviations from linearity for iodine in
Figure 5. With a film on the bead surface when molten, direct mass transfer across the liquid/gas interface will augment diffusion of iodine from within the melt to the surface. It also underlines the need to minimise melting temperatures in addition to melting times—as shown by Banerjee et al. [
30] and Parker et al. [
33], above certain temperatures (depending on glass composition among other factors), volatilisation rates can become non-linear and thus it may be that using a lower fusion temperature would reduce or remove the non-linear behaviour observed here (
Figure 5). It is known that iodine is particularly volatile from oxide glass melts [
26,
34]. Conversely to the volatilisation behaviour of iodine, there is also clear evidence for the volatilisation of sulphate, SO
3. However, SO
3 loss displays a strongly linear relationship with (time)
1/2 for the Anorthic Feldspar fused beads (CRM-529,
Figure 1,
Figure 5 and
Table A1) and for the Low-Iron Float Glass (CRM 525 [
44],
Figure 4 and
Table A4). This confirms that sulphate volatilisation obeys M ∝ t
1/2, i.e., volatilisation of SO
3 can be accurately described as a diffusion-based process. Sulphate volatilisation from silicate glass melts is well known in glass manufacture [
18] and has also been documented in XRF fused bead preparation [
31]. Here we have thus demonstrated that the mechanism of sulphate volatilisation is solely or predominantly diffusion based.
The loss of iodine and sulphate during fused bead melting are both governed (SO3 solely or predominantly; and I only partially), by diffusion-based mechanisms, as demonstrated by a linear relationship with (time)1/2. For iodine, additional transport mechanisms accelerate mass loss, particularly in the early stages of melting. This underlines the necessity to keep both melting times and melting temperatures to the minimum necessary to achieve good quality fused beads that provide reproducible results. It also illustrates how “over-melting” (i.e., melting for longer than necessary and/or melting hotter than necessary) of samples containing particularly volatile species such as halides and sulphates, can lead to under-estimation of the amounts originally present due to volatilisation losses which can be non-negligible, even at short melting times.
There are many variable parameters impacting upon XRF fused bead preparation quality, consistency and the accuracy, precision and repeatability of analysis [
31], and here it has only been possible to study a fraction of these. As noted in
Section 1, these parameters include both time and temperature of fused bead preparation, but added to this must be preparation method (e.g., manual or automatic; static or agitated; melt vessel dimensions and surface area to volume ratio) as discussed elsewhere [
11,
31]. Further parameters requiring attention include fused bead compositions, mixtures and media [
11,
31], effects of particle size of the sample material and its thermal behaviour (e.g., melting point, viscosity-temperature profile, refractoriness and/or acid-base nature [
31]). Multiple analyses involving a number of different fused beads produced under the same conditions, e.g., several independently-produced CRM-525 fused beads melted for the same time under the same conditions, are also needed to further establish consistency and reproducibility of results. All of these factors are recommended for further study, and the authors aim to address the effects of more of these variable parameters in future publications.

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}