1. Introduction
Adsorption processes of solute species from fluids onto mineral surfaces play a central role in many natural and industrial applications such as surface catalysis, wastewater treatment, and long-term safety of nuclear waste repositories. For nuclear waste repositories, the potential to adsorb radionuclides from solution is one of the key parameters in describing the quality of host rock formations. Here, adsorption can lead to the retention of hazardous elements such as trivalent actinides causing long-term radiotoxicity of the nuclear waste [
1]. Clay rock formations such as the Opalinus clay in (Southern) Germany and Switzerland are of high interest for potential nuclear waste storage due to the high adsorption potential of clay minerals, e.g., [
2,
3].
In such natural systems, mineral surfaces may dissolve during large periods of disequilibrium conditions (e.g., over 100,000 years) due to pore-fluid flow. The dissolution processes create nanotopography and, consequently, modify the reactivity distribution on the surface. The distribution of reactive sites due to the nanotopography has been reported to be a driving factor for dissolution, e.g., [
4,
5]. However, the influence of nanotopography on the adsorption of species to the mineral surface has not been investigated in sufficient detail, so far.
Commonly studied radionuclide adsorbents are the trivalent actinides plutonium, curium, and americium due to their long-term radiotoxicity, e.g., [
6,
7]. A number of experimental studies use the trivalent lanthanide europium as a chemical homolog for the trivalent actinides, e.g., [
6,
8,
9,
10,
11,
12,
13]. Illite is one of the main clay mineral components of the Opalinus clay formation [
2,
3,
14]. However, the phyllosilicate muscovite is often selected as a mineral analog for such studies due to its structural similarity with illite. In contrast to illite, however, it offers large, often perfect cleavage surfaces [
7,
13].
The interaction of radionuclides with phyllosilicates has been the subject of several studies in the past. A pH dependence of europium adsorption has been reported repeatedly. Adsorption starts at a pH value of 3 and increases until pH 8, where up to 100% of the available europium is adsorbed to the phyllosilicate surface [
6,
8,
9,
11,
13]. At low pH values, europium predominantly forms complexes on the surface without loss of coordinated water molecules from its hydration shell, so-called outer-sphere complexes [
8,
11,
13]. With increasing pH, the formation of one or more inner-sphere complexes directly bound to surface sites is indicated by the measured fluorescence spectra [
6,
8,
11]. For muscovite, no exchange reactions with interlayer atoms have been reported [
12]. Inner-sphere complexes on kaolinite preferably bind to sites located in the octahedral layer of the crystal structure [
11], while for montmorillonite, no preference in adsorption position is reported [
10]. One study suggested that on the tetrahedral layer, creating the basal plane of phyllosilicates, adsorption to aluminum substitution sites is preferred over silicon sites due to electrostatic attraction [
15]. The coordination of specific adsorption sites is not discussed in these studies. The binding of derived inner-sphere complexes differs between monodentate [
6,
13,
16] and multidentate complexes [
13,
15]. In conclusion, it is currently unclear where inner-sphere adsorbed complexes are located on rough mineral surfaces and how the nanotopography may influence the binding coordination of the adsorbed species. Therefore, in this study, we investigated how adsorption behavior and efficiency are influenced by complex formation at specific surface sites and how the nanotopography on mineral surfaces may impact adsorption site availability and distribution.
Here, we apply kinetic Monte Carlo (KMC) simulations to the system of a model radionuclide interacting with the muscovite (001) basal plane. KMC is a modeling tool able to simulate surface processes at the atomic scale with system size in the range of hundreds of nanometers. It is often applied to study mineral dissolution [
5,
17,
18,
19] and other processes at a similar time and length scale, such as diffusion, crystal growth, or catalysis [
20]. Dissolution KMC models can simulate the influence of lattice defects and highlight the importance of surface reactivity distribution and atomic site reactivity. The KMC model in this study consisted of two main parts: (1) simulation of muscovite (001) face dissolution using a modified version of an existing model [
5] and (2) simulation of model radionuclide adsorption to the previously dissolved muscovite surface nanotopography. By combining the simulation of dissolution and subsequent adsorption, we aimed to answer what surface sites are preferred for adsorption, where these sites are located on the nanotopographical surface, and how they influence adsorption efficiency and distribution.
2. Materials and Methods
The mineral muscovite (KAl
2(AlSi
3O
10)(OH)
2) belongs to the group of 2:1 dioctahedral phyllosilicates with a crystal structure of the monoclinic polytype 2M
1. It is a non-swelling phyllosilicate. Thus, we do not consider the interlayer composed of potassium in the simulation. The simulation focuses on the basal plane (001) face of the muscovite crystal. In the tetrahedral layer, 25 % of the positions are substituted by aluminum for silicon, with no ordering in the substitution. The lattice parameters used in this study are a = 5.1918 Å, b = 9.0153 Å, c = 20.0457 Å, β = 95.735° [
21]. The selected structural data are representative for muscovite and sufficient for simulating its general behavior. They have been used for simulations previously, e.g., [
5]. The model presented in this study is based on literature data for both the crystal structure data (above) and the KMC parameterization (below).
Kinetic Monte Carlo models simulate a sequence of individual events occurring in a predefined system. This method has been applied in several studies for the investigation of crystal growth [
22], dissolution [
4], and surface adsorption [
23]. The probability of any event can be described by a Boltzmann distribution:
where
P is the event probability, Δ
E is the event activation energy,
k is the Boltzmann constant, and
T is the temperature. All successful event attempts over the sum of all trials describe the event probability. When multiple activation steps are required for an event to occur, for example, the dissolution of an atom with n neighbors, Equation (1) can be modified to:
A heterogeneous system has multiple possible combinations of coordinated neighbors resulting in a set of events, where all probabilities can be calculated by Equation (2). When all probabilities are available, a series of discrete individual events in the form of multiple iterations is simulated. The algorithm used in the present KMC model is the “Divide-and-Conquer” [
24] or “N-fold” [
25] algorithm. Here, the probability of a given event is integrated over the whole system:
where
Ni is the number of positions in state
i with the probability
Pi in the system. For all possible states, the integrated probability is calculated during each iteration because the number of positions changes with proceeding dissolution. Then, a random number
x between zero and one is generated. The complete range is equivalent to the sum of all probabilities, where each individual event probability covers an interval based on its ratio to the sum. Random number
x falls into one of the probability intervals, and the respective state
i is selected:
Following, a second random number is used to select a position in the system belonging to state i and carry out the associated event, dissolution, or adsorption. Then a new iteration begins.
The muscovite KMC dissolution simulation is a modified version closely based on the model published in [
5], where a more detailed description is available. It simulates the detachment of cations in the crystal structure that are connected by oxygen bonds. Three different atom types, tetrahedral silicon, aluminum, and octahedral aluminum, are considered. Bonds exist internally as well as between the differently coordinated layers. According to aluminum-aluminum bond avoidance, no tetrahedral aluminum may be in a position as first neighbor to another tetrahedral aluminum [
26]. For the tetrahedral positions, the dissolution probability is calculated based on Equation (2) by:
where the maximum for intralayer bonds is three (
n +
m ≤ 3) and the maximum for interlayer bonds is two (
l ≤ 2). For octahedral aluminum sites:
the intralayer maximum is three (l ≤ 3) and the interlayer maximum is five (n + m ≤ 5), based on the crystal structure. Correction factors are applied to Equations (5)–(7) to compensate for the long-range influence of the crystal lattice and the influence of the second coordination sphere. Before the start of the dissolution by iterations, pits are placed into the (001) face with depths between 1 and 8 unit cells in c direction. The system is simulated with periodic boundary conditions, and thus no (hk0) faces are simulated.
The KMC simulation of radionuclide adsorption is added to the existing dissolution KMC model. A conceptual overview is shown in
Figure 1. The surface generated after the last dissolution iteration is used as a starting point for adsorption simulation. Both simulation parts are explicitly separated and do not run in parallel. This is due to the large time scale difference on which dissolution [
27] and adsorption [
7] occur. A general equation of radionuclide adsorption probability is based on Equation (2):
where the sum of
n,
m, and
l depends on the maximum denticity of the complexes allowed in the system. In the current study, the focus is on monodentate adsorption complexes, i.e., only a single bond to the surface may exist (
n or
m or
l = 1). Adsorption positions on the muscovite surface are defined based on the topography after dissolution. For monodentate complexes, each surface cation provides one adsorption site and only a monolayer of adsorbed atoms is allowed. During the adsorption iteration, one of the three possible adsorption events is selected: (1) adsorption to silicon, (2) adsorption to tetrahedral aluminum, and (3) adsorption to octahedral aluminum. Then a respective random adsorption site is chosen, and a radionuclide atom is placed there, blocking any further adsorption at this site. In the adsorption part of the KMC, no real-time is currently calculated. An internally comparable simulation time is computed by counting the required iterations to place the selected amount of radionuclide atoms on the surface. If a selected position is already occupied, the iteration is restarted and the timer increased. Thus, with the increasing occupation of surface sites and lower availability of high probability adsorption sites, the simulation timer increases in larger increments per successful event.
KMC models require a parameterization for the activation energies of the events in the system. The parameterization for muscovite dissolution is described in detail in [
5] and will not be discussed here. Calculation of activation energies for the involved processes is complex and time-consuming. No activation energies for the adsorption of trivalent actinides or europium to muscovite surface sites or to the individual building blocks of the muscovite structure can currently be found in the literature. For a first approximated model, we used purely qualitative, estimated values based on considerations or findings from the literature for a model radionuclide. Three different adsorption activation energies were required: Si-RN, Al(Tet)-RN, and Al(Oct)-RN. For these three parameters, the values were chosen in relation to each other due to the following assumptions. The adsorption to tetrahedral layer sites prefers aluminum substitution over silicon sites. This follows the assumption made by [
15]: adsorption to the tetrahedral layer is driven by its negative surface charge and electrostatic attraction to the positively charged radionuclide. Negative charges are located at aluminum substitutions due to the charge differences between Si
4+ and Al
3+. In comparison to the tetrahedral sites, the activation energy of adsorption to octahedral aluminum was selected to be smaller. Octahedral sites are preferable adsorption sites in kaolinite [
11], and the (
hk0) faces have been shown to offer important adsorption sites due to an accessible octahedral layer [
28]. Following these assumptions, the order of adsorption activation energies in the system was Si-RN > Al(Tet)-RN > Al(Oct)-RN. The value for adsorption to silicon was arbitrarily set to 50 (
Table 1). To create a large contrast in adsorption probability, the values for both aluminum sites were set to be around three-fifths of the silicon value or 30. To highlight the preferable adsorption to the octahedral layer sites, a smaller difference of 5 was set between Al(Tet) and Al(Oct) radionuclide adsorption, resulting in values of 33 and 28, respectively.
The number of radionuclide atoms to be adsorbed on the muscovite surface was calculated by using the surface area and the alpha-spectrometry measurement of 1.4 Pu
3+ atoms per area of one unit cell [
7]. For the selected surface size, this resulted in ~300,000 adsorbed radionuclide atoms. All KMC simulation parameters are listed in
Table 1.
The influence of nanotopography on the adsorption behavior was studied using two different cases: in case (I), one single etch pit with a depth of six unit cells (12 TOT layers or 12 nm) was placed on the surface (
Figure 2IA). For case (II), six etch pits were randomly distributed over the surface, each with a depth of one unit cell (2 TOT layers or 2 nm) (
Figure 2IIA). Both cases initially had twelve atomic steps that consisted of one TOT layer each, and the same number of atoms was dissolved from the surface. By comparing the two cases, the impact of the two different nanotopographies with the same number of steps was observable.
3. Results
During the dissolution of the basal plane, the etch pits in both cases follow preferred growth in [110] direction [
5]. This direction alternates between underlying TOT layers by 120° due to the layer rotation in 2M
1 phyllosilicate polytypes. In positions where the growth of a lower rotated layer is inhibited by an overlying layer, pit walls consist of two TOT layer units as opposed to the regular step height of one TOT layer (
Figure 3). In case (II), the availability of multiple initial etch pits can lead to the coalescence of steps originating from different pits. Coalescence occurs when the initial pit positions are in close vicinity or if they are located along the [110] direction with respect to each other. This process causes the loss of individual steps in comparison to case (I) where no coalescence is possible.
The applied parameterization for radionuclide adsorption leads to three different possible adsorption positions distributed over the muscovite surface: (1) silicon, (2) tetrahedral aluminum, and (3) octahedral aluminum. With these considerations, the distribution of adsorbed radionuclide atoms with an average distribution of 1.4 atoms per area of a unit cell does not seem to be influenced by the nanotopography (
Figure 2C and
Figure 3C). In the early stages of adsorption, a slightly higher concentration of occupied positions along steps can be found (
Figure 3B). With proceeding adsorption, the distribution of occupied positions does not show areas of higher concentration and any difference in the distribution to the final state. The overall observed adsorption pattern follows a random distribution of occupied positions over the muscovite (001) surface. No difference in adsorption concentration between atomically flat surface areas and the steps around etch pits is observable. Both cases show the same distribution of adsorbed radionuclides, and no influence can be attributed to the different nanotopographic surface portions between the two cases. In total, around 35% of the available muscovite basal plane adsorption sites are occupied at the simulation end.
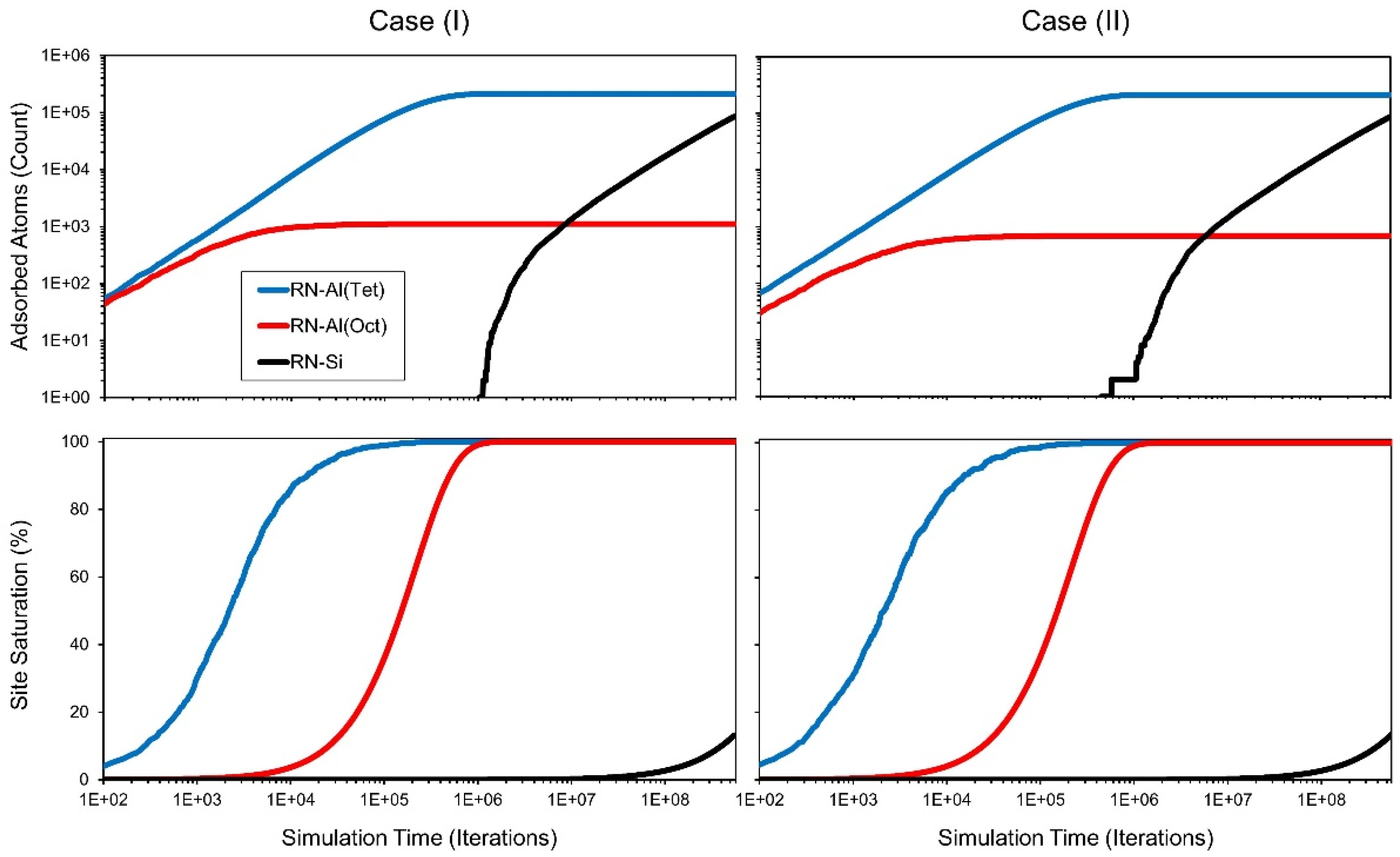
More details of the adsorption behavior become observable by analyzing the number of occupied adsorption sites over simulation time (
Figure 4). The distribution of adsorption sites with proceeding simulation follows two main phases. Phase one shows radionuclide adsorption to both octahedral and tetrahedral aluminum sites and no adsorption to silicon. Case (II) has on average over ten model runs around 60% of octahedral adsorption sites compared to the octahedral sites found on the muscovite surface in case (II). The coalescence of the steps on the case (II) surface leads to a reduction of the total step length in the system and therefore a loss of access to the octahedral layer via these steps. This difference has only a minor impact on the total adsorption behavior because less than one percent of all available surface adsorption sites are coordinated to octahedral aluminum. Octahedral aluminum sites have a high probability to dissolve when exposed to the surface during the dissolution simulation due to low activation energies for bond hydrolysis to their coordinated neighbors. Because of their low availability and high adsorption affinity octahedral aluminum adsorption sites are the first site to become fully saturated. Following this saturation, adsorption exclusively occurs at tetrahedral aluminum sites. These sites reach full saturation with proceeding simulation. Phase two starts when the first radionuclide atom adsorbs to silicon surface atoms. Here, due to the high activation energy selected for this reaction, the speed of adsorption decreases. A high number of adsorption attempts are aimed at now occupied aluminum sites increasing the number of timer counts per successful adsorption attempt. The simulation ends when the adsorption of 300.000 radionuclide atoms is completed. At this point, around 13% of all silicon sites are occupied. In both cases, phase two starts at the same time due to the same amount of tetrahedral aluminum adsorption sites. In the current model, the adsorption is dominated by the tetrahedral layer. No influence of the coordination environment of adsorption sites is considered in the current KMC model, and therefore no influence of topography is created. Except for an increase in adsorption speed for the first few hundred atoms due to the octahedral sites, there is no difference to a simulation performed on an atomically flat muscovite basal plane.
4. Discussion
The parameterization of activation energies of specific sites is the controlling factor of the simulations. For dissolution, the first and second coordination shells of a site determine the total activation energy barrier, and thus a large number of possible combinations exist [
5]. This coordination environment influences the reactivity of any given site on the surface. The site reactivity based on the site coordination can affect the activation energy of adsorption as has been shown for other systems [
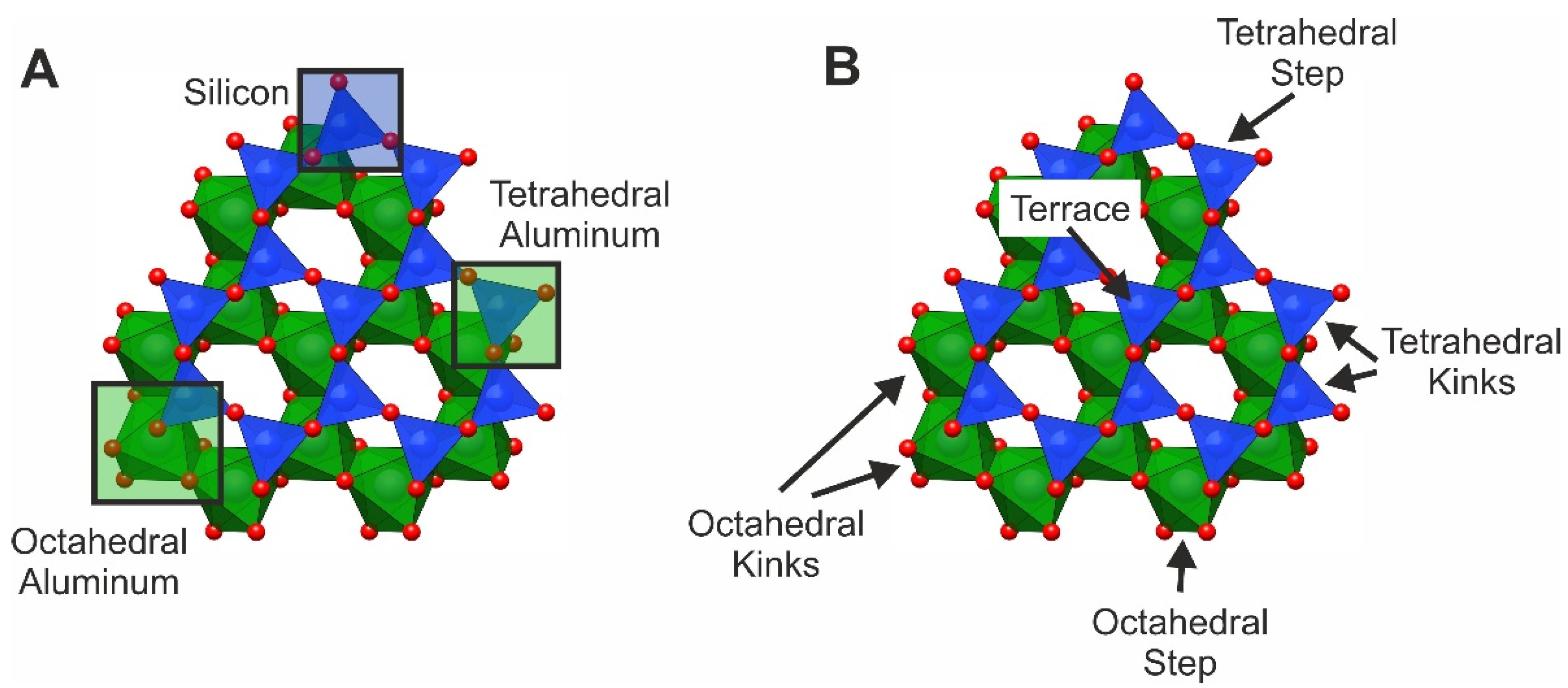
18]. Considering the coordination of the surface site and the effect on the reactivity toward adsorption is required for a more sensitive description of surface adsorption. For the muscovite (001) surface, differentiating between the basic sites of kink, step, and terrace positions would result in eleven adsorption sites: two kink positions for Si, Al(Tet), and Al(Oct), one step position for Si, Al(Tet), and Al(Oct), and one terrace position for Si and Al(Tet). This complexity can be further increased by considering the difference between the upper and lower tetrahedral layer and the orientation of the kink and step sites. When activation energies of adsorption to these sites differ in relation to each other, a direct influence on the distribution of adsorbed complexes can be observed. Currently, a parameterization of the described sites is not possible due to missing literature data. In the present simulation, three different sites are available with activation energies based on literature assumptions (
Figure 5A) [
11,
15]. The variability of the adsorption energies is based on the chemistry (Si, Al) and the coordination environment (tetrahedral, octahedral) of the respective surface site.
The order of adsorption site saturation directly follows the implemented parameterization of adsorption activation energies from low to high (Al(Oct) > Al(Tet) > Si). Due to the reduced sensitivity and variability of the parameterization, only three different events are possible in the KMC simulation, resulting in a predictable outcome. The model is able to simulate the adsorption of radionuclides on the muscovite basal plane and consider the importance of variable atomic adsorption sites and their reactivity that can be found on surfaces with nanotopography. However, the influence of the nanotopography needs to be addressed in the parameterization leading to a more precise description of site-specific adsorption (
Figure 5B). In the current model, the sole nanotopographic impact is the accessibility of the octahedral layer, controlling adsorption behavior in the very early stages of the process only. The accessibility of the octahedral sites also highlights the importance of the (
hk0) muscovite faces for the adsorption of radionuclides [
28]. Coalescence of steps has been observed and causes a reduced number of octahedral surface sites, and thus it can be an important factor for the adsorption efficiency of a crystal surface.
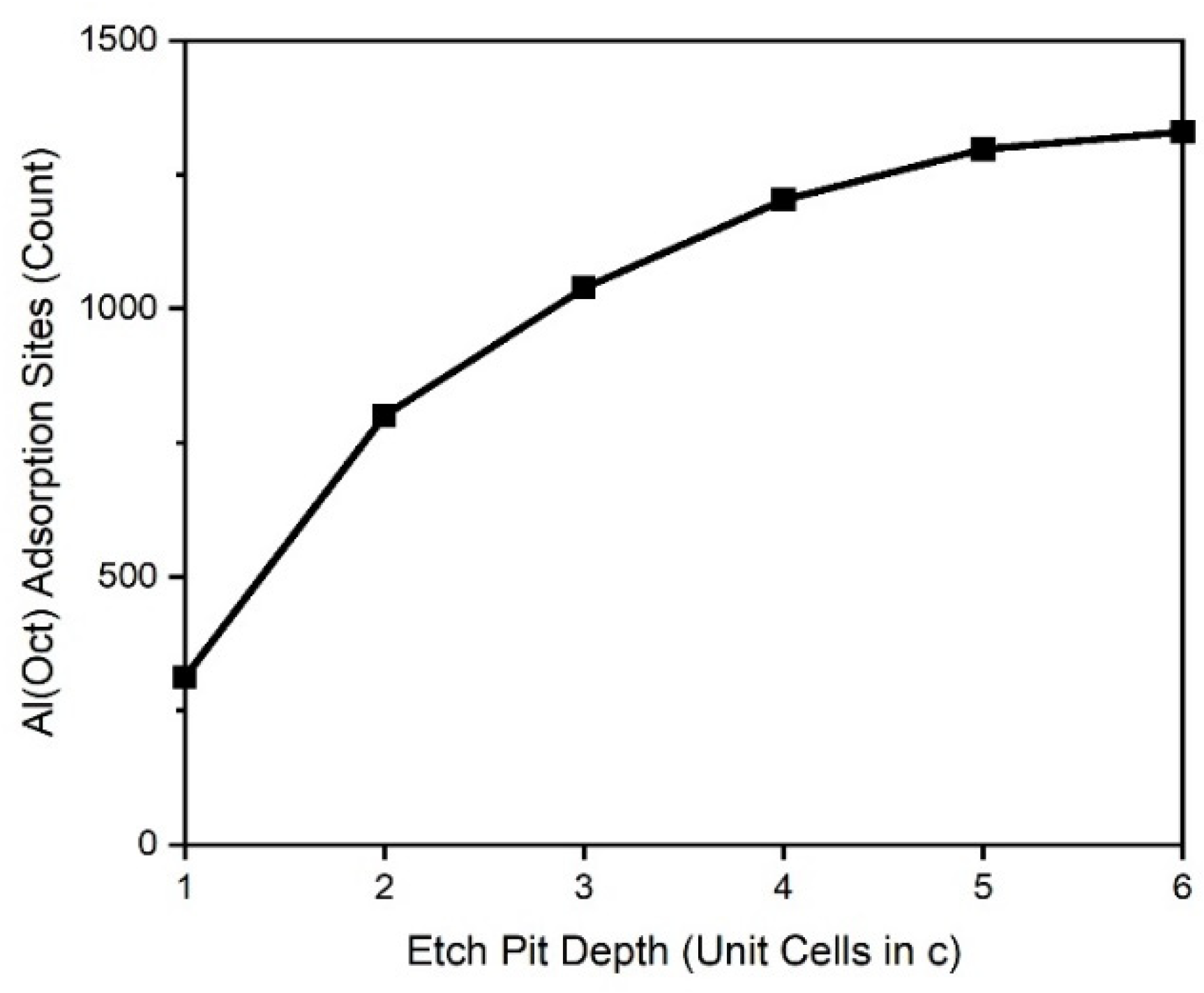
Despite the incomplete adsorption parameterization, the model can show the surface sites that are available as bonding partners on the muscovite surface. The KMC model can show how the change in nanotopography affects the availability of these specific adsorption sites and the changes with proceeding surface dissolution. As we identified, the octahedral aluminum surface sites appear to play an important role in radionuclide adsorption on sheet silicates. Such sites are not available on atomically flat muscovite or similar sheet silicate (001) surfaces. Octahedral aluminum becomes exposed to the surface and is available for adsorption reactions after steps and pits developed on the (001) surface.
Figure 6 shows the availability of octahedral aluminum sites for adsorption depending on the etch pit depth. An increase in etch pit depth leads to an increase of exposed octahedral sites. At higher pit depths, the number of octahedral sites seems to become constant. This effect highlights the importance of surface nanotopography on the availability of surface sites and the efficiency to adsorb species from solution. Such an analysis can be performed for any surface site depending on atom type, crystal structure, and coordination.
Most radionuclide atoms adsorb to tetrahedral aluminum in the KMC simulation. Therefore, any distribution of tetrahedral aluminum directly leads to a distribution of the adsorbed radionuclide atoms. In the present muscovite KMC model, 25% of all tetrahedral sites are occupied by aluminum, which is randomly distributed. However, a short-range ordering in two dimensions has been observed in the muscovite tetrahedral layer, which would directly influence the adsorption distribution [
29].
The key for a precise description of adsorption on crystal surfaces controlled by nanotopography is a site-specific parameterization of all possible adsorption sites. As we showed in this study, the surface atom type (element, oxygen coordination) of the adsorption site is crucial for the parameterization. However, the influence of the neighboring cations coordinated to the adsorption sites needs to be considered. This is a key factor for the dissolution mechanism and influences the reactivity of the surface site. Such a parameterization requires ab initio methods such as density functional theory (DFT) calculations. With this method, a calculation of the activation energies needed in the KMC system is possible. Comparable DFT calculations have been performed for the europium adsorption complexes on hematite [
30], but no literature data are currently available for muscovite. The consideration of different surface sites, such as kink, step, or terrace positions, leads to a large number of possible adsorption positions and a more heterogeneous distribution that cannot be predicted. The nanotopography is directly responsible for the distribution of these sites, and its influence on adsorption cannot be estimated without site-specific adsorption energies. By using a Monte Carlo modeling approach, one study has shown that Cl
- anions for solution preferentially adsorb at kink followed by step and terrace sites on a calcite surface [
18]. This system is not transferable to the interaction of muscovite with positively charged radionuclides but highlights the impact of surface atom coordination on the adsorption reactions.
Another influence on the adsorption of radionuclides is the protonation of surface sites. In aqueous systems, the muscovite surface is in permanent contact with water molecules. Depending on the pH conditions, surface oxygen atoms can occur as protonated or deprotonated sites. The muscovite basal plane has a permanent negative charge due to the aluminum-silicon substitution in the tetrahedral layer. Basal plane surface oxygen atoms are bound by two tetrahedral cations (e.g., the siloxane group –Si-O-Si-; see
Figure 5). These surface groups are difficult to hydrolyze, and they are also less affected by protonation because all bonds are saturated and charge-neutral [
31]. The permanent negative charge of the basal plane attracts the positively charged radionuclide cations. It is currently unclear at which sites inner-sphere adsorption on the basal plane can occur. Meleshyn [
15] simulated a favorable inner-sphere adsorption position directly above tetrahedral aluminum substitution sites where the radionuclide is then coordinated to the three oxygen atoms bound to aluminum. Another possible adsorption site is the hexagonal cavity on the muscovite basal plane, although it is unknown if inner-sphere adsorption is possible at this site. Muscovite edge surfaces or steps and pits on the basal plane have dangling oxygen atoms that are bound to one surface cation only. These oxygen atoms can be protonated to form surface OH groups. This study focused on alkaline pH (>8) conditions due to the high adsorption affinity toward radionuclides. At these pH values, deprotonation of OH groups on the edge surface sites has been reported [
31,
32]. On kaolinite edge sites, preferred deprotonation of the aluminum site (Al-OH) has been observed [
32]. For muscovite edge sites, the deprotonation is reported to occur on the silicon sites (Si-OH), while aluminum sites can be further protonated to Al-OH
2 [
31]. Deprotonation of any edge sites creates a negative surface charge that attracts cations from the solution and leads to an increased inner-sphere adsorption at deprotonated oxygen atoms [
32]. Another adsorption mechanism is a proton exchange during the inner-sphere adsorption reaction, e.g., leading to the formation of an inner-sphere tridentate complex and three hydronium (H
3O
+) ions or water molecules if radionuclide hydroxide complexes are involved [
30,
31,
33]. Due to the different coordination of surface oxygen atoms, it can be expected that the adsorption mechanism differs between basal and edge surfaces on muscovite. However, there is no precise description of the involved adsorption mechanisms and adsorption complex structures currently available in the literature. It is possible to address the influence of surface protonation on the KMC adsorption activation energies during the parameterization calculation, e.g., with DFT [
30]. In the DFT simulations, it is possible to directly investigate the interaction of the surface OH group with the dissolved radionuclide. This interaction is then included in the overall activation energy barrier used in the KMC simulation.
To validate simulated results, new experimental insight on the distribution of adsorbed atoms on surfaces with nanotopography is required for a better understanding of the involved mechanisms. Currently, mostly spectroscopic data without spatial resolution are available. This allows us to identify the complexes formed during adsorption of the respective species but cannot show any spatial variability due to nanotopography. A few examples for such experimental analyses are available from the literature. In an experimental study [
34], atomic force microscopy (AFM) was used to visualize the adsorption or Rb
+ ions to an atomically flat muscovite surface. This technique is able to show the distribution of single atoms on the surface and any pattern therein. The investigated surface area is smaller than the KMC surfaces simulated in this study, and therefore a direct comparison between KMC and such an experimental approach is possible. Another recent method is the spatially resolved time-resolved laser fluoresce spectroscopy (µTRLFS). This method was used to investigate the spatial distribution of adsorption complexes on a granite surface [
35]. Combined with a measurement of surface nanotopography, a correlation between the two parameters might be possible leading to an improved mechanistic understanding. Overall, a combination of surface-sensitive analytical techniques and improved parametrization of numerical approaches are required to provide quantitative insight into the effect of surface nanotopography on surface reactivity.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}