
X-ray Total Scattering Study of Phases Formed from Cement Phases Carbonation



Abstract
:1. Introduction
2. Materials and Methods
2.1. Total Scattering Synchrotron X-ray Powder Diffraction
2.1.1. Rietveld Analysis
2.1.2. PDF Analysis
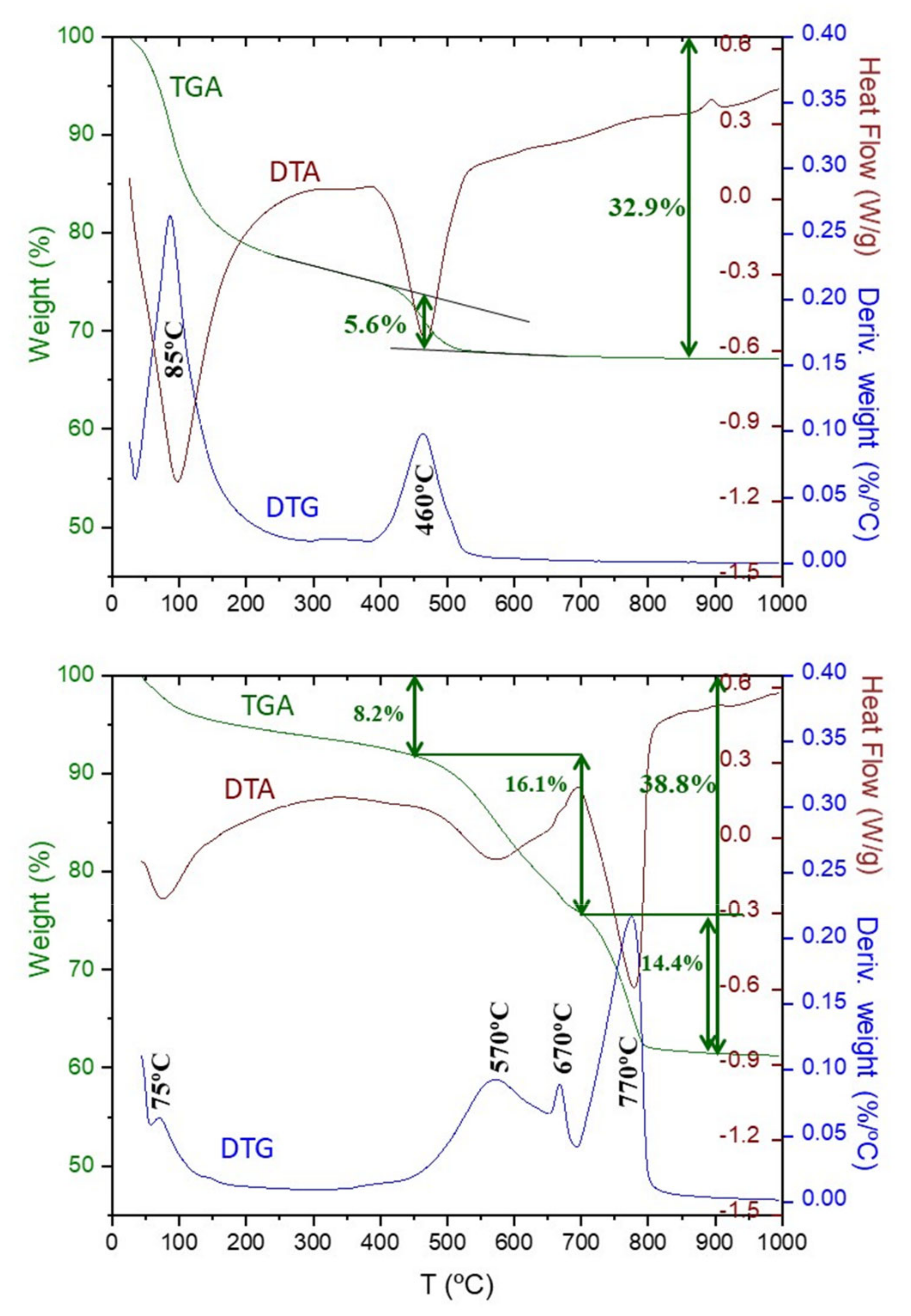
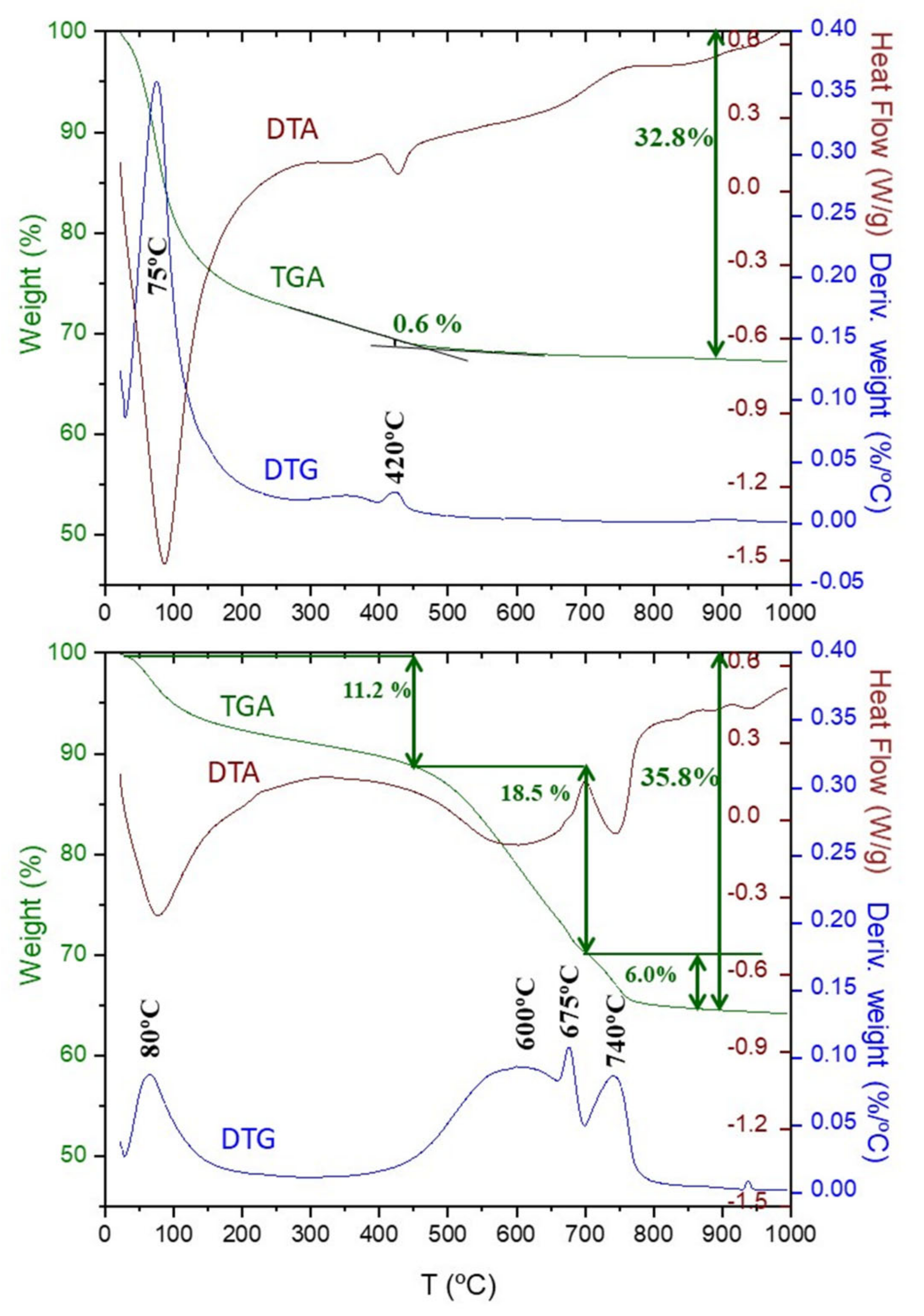
2.2. Thermal Analysis (TA)
2.3. Particle Size Distribution (PSD) Analysis
2.4. BET Surface Area Determination
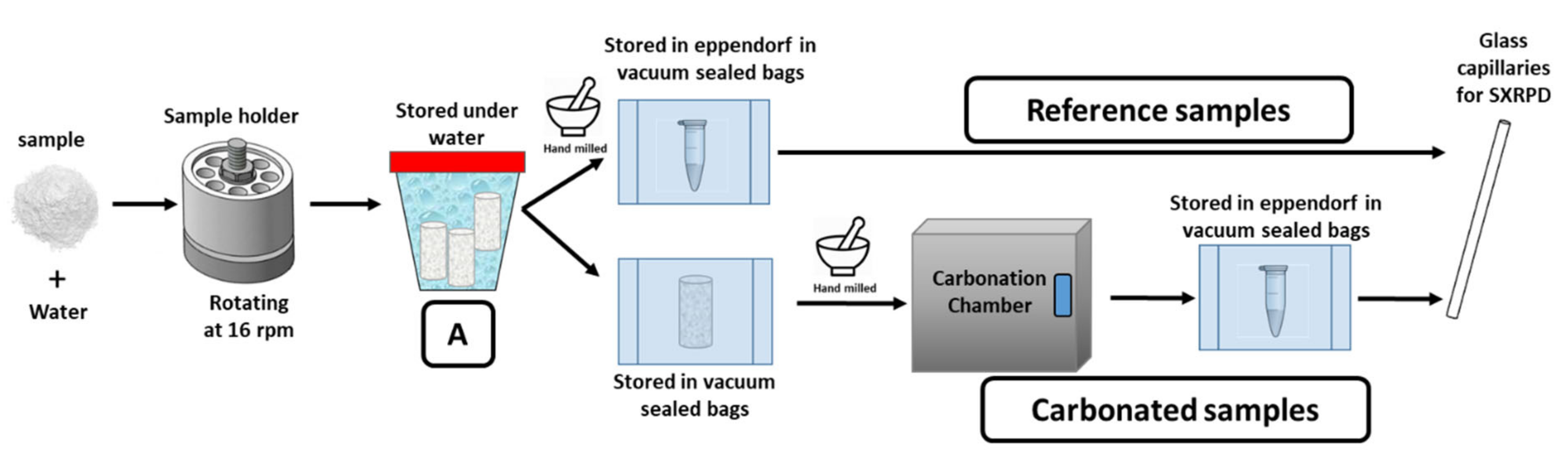
2.5. Samples Description
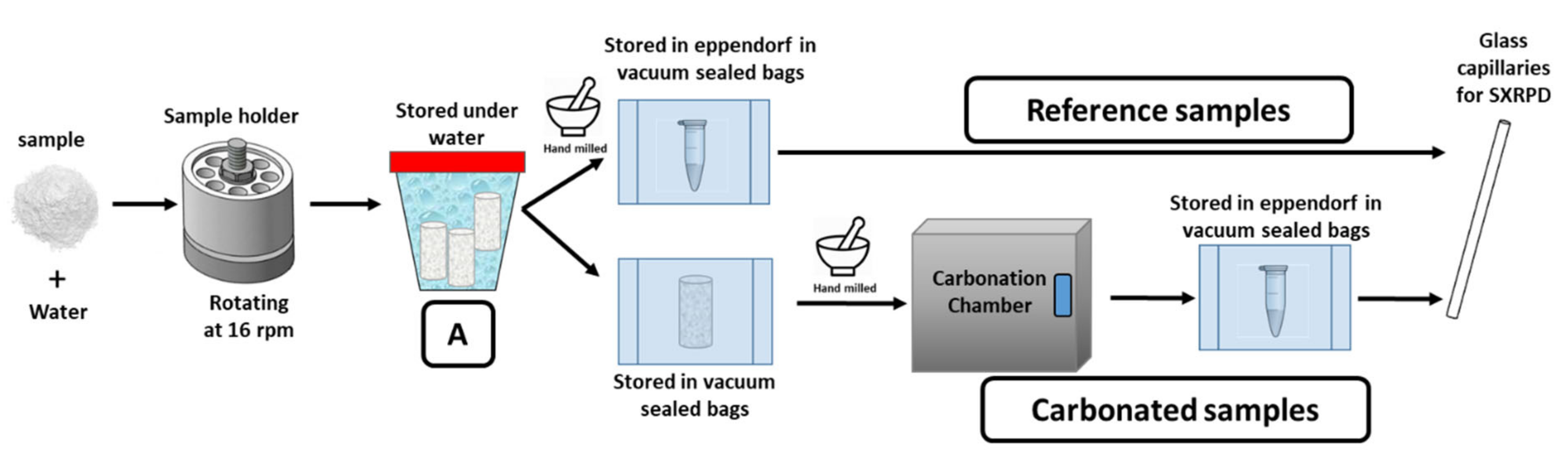
2.6. Hydration and Accelerated Carbonation
3. Results and Discussion
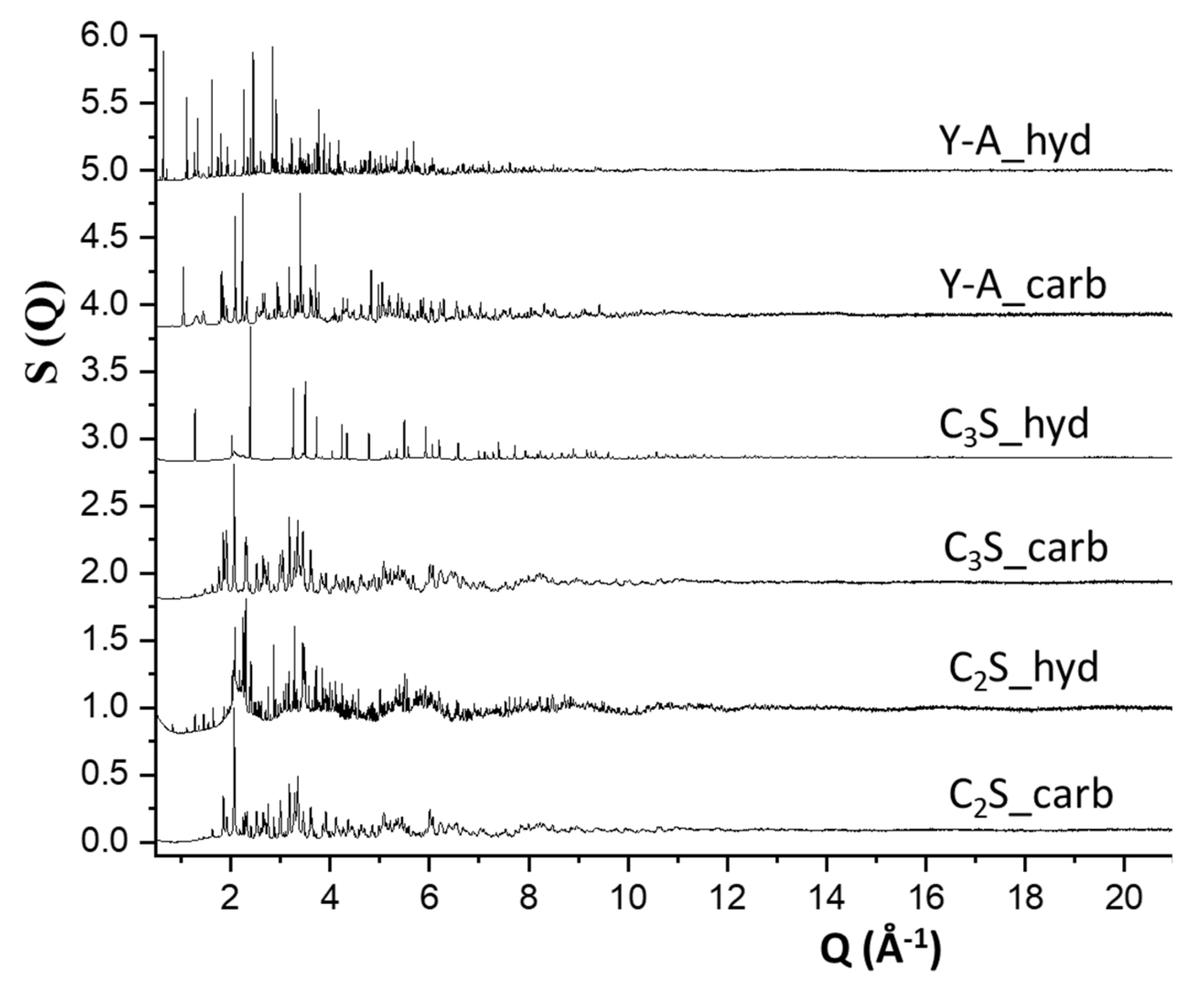
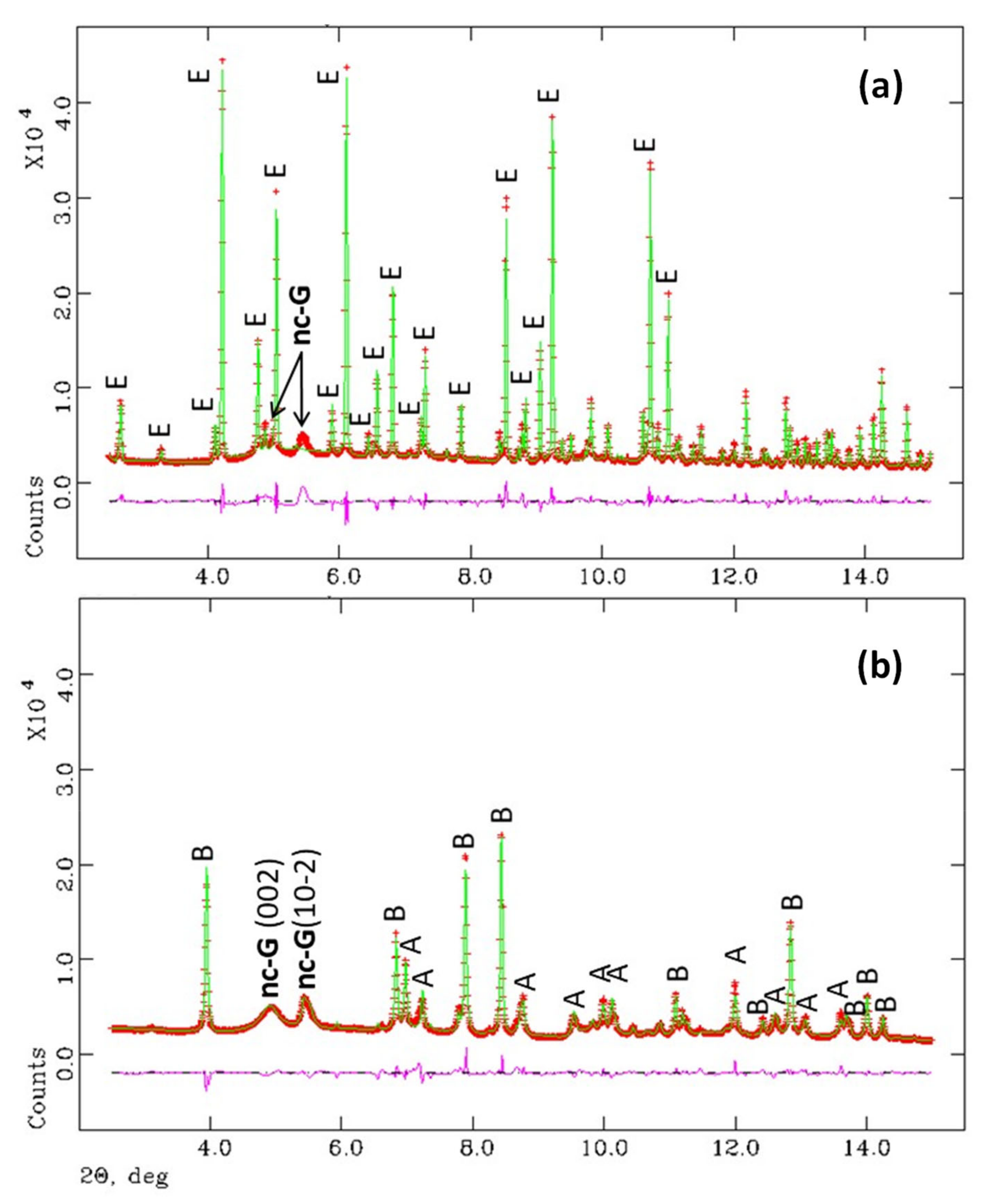
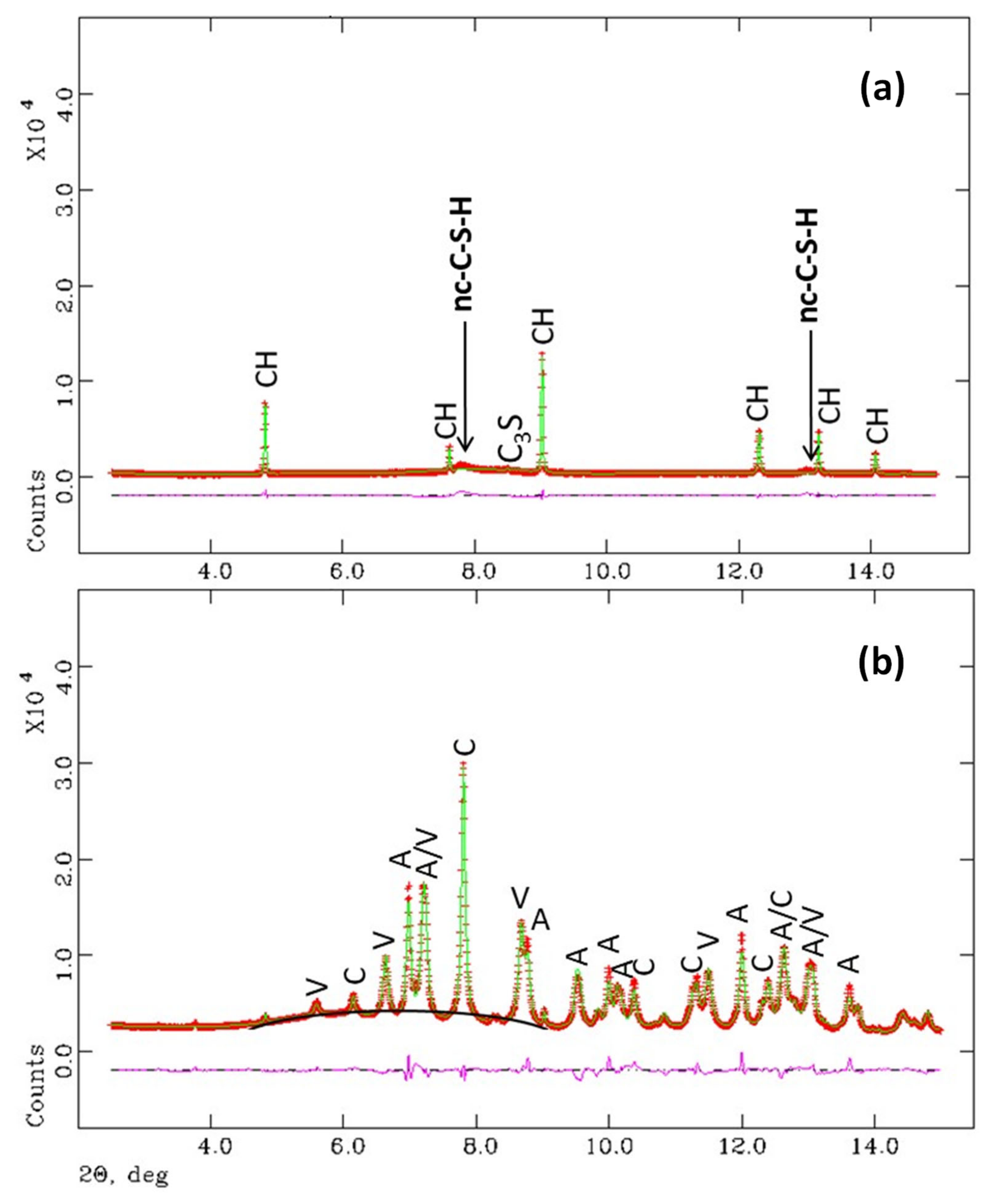
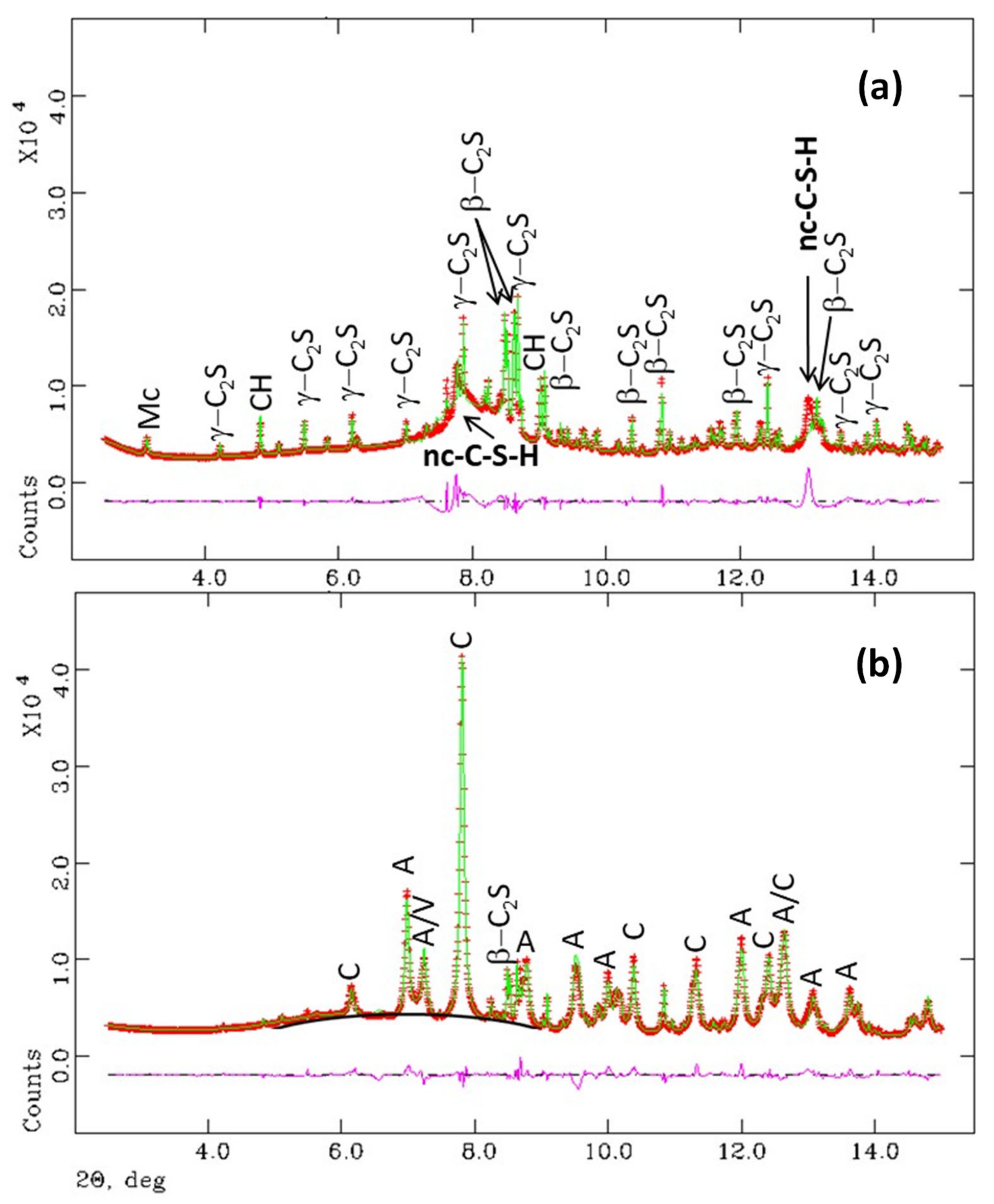
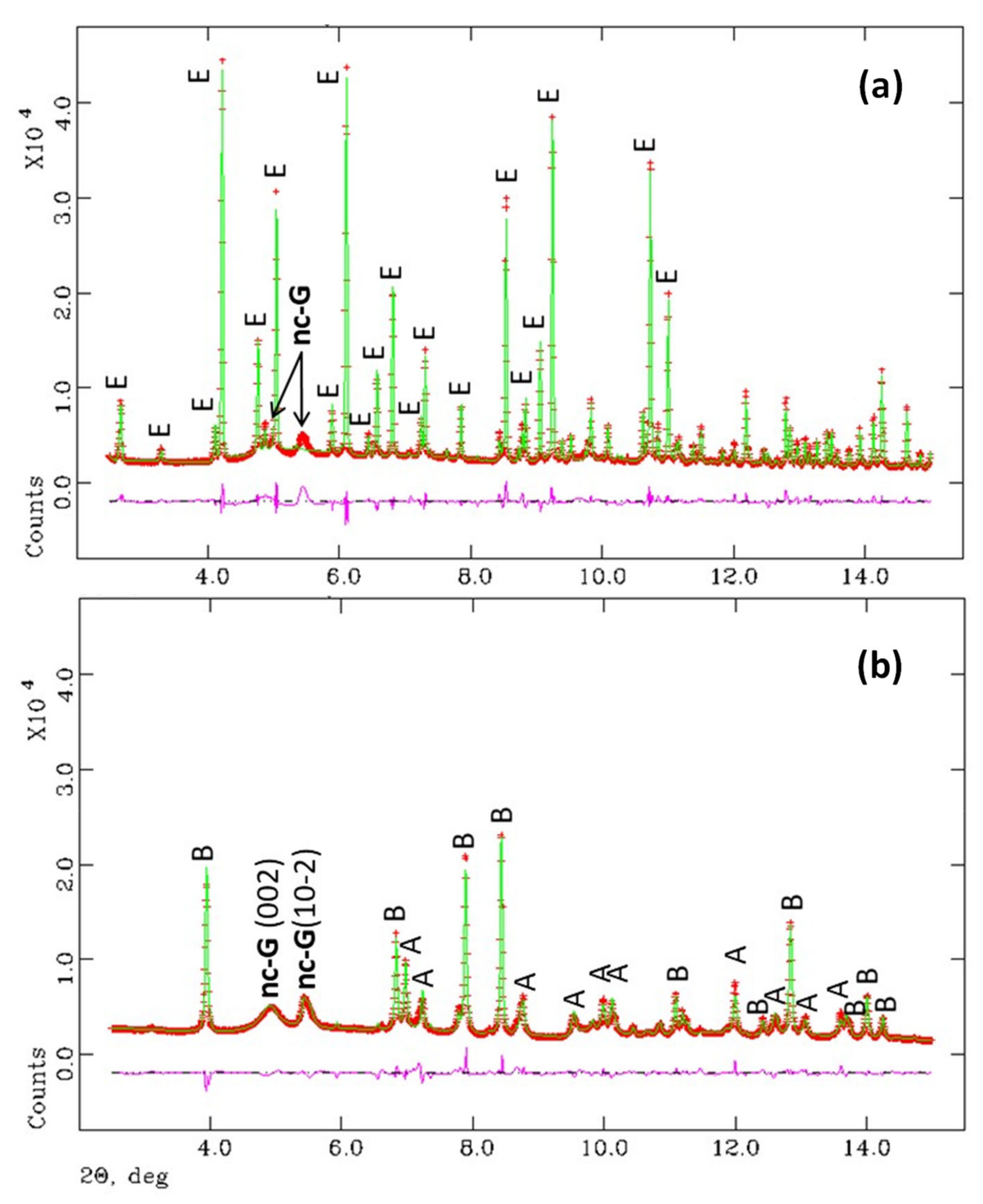
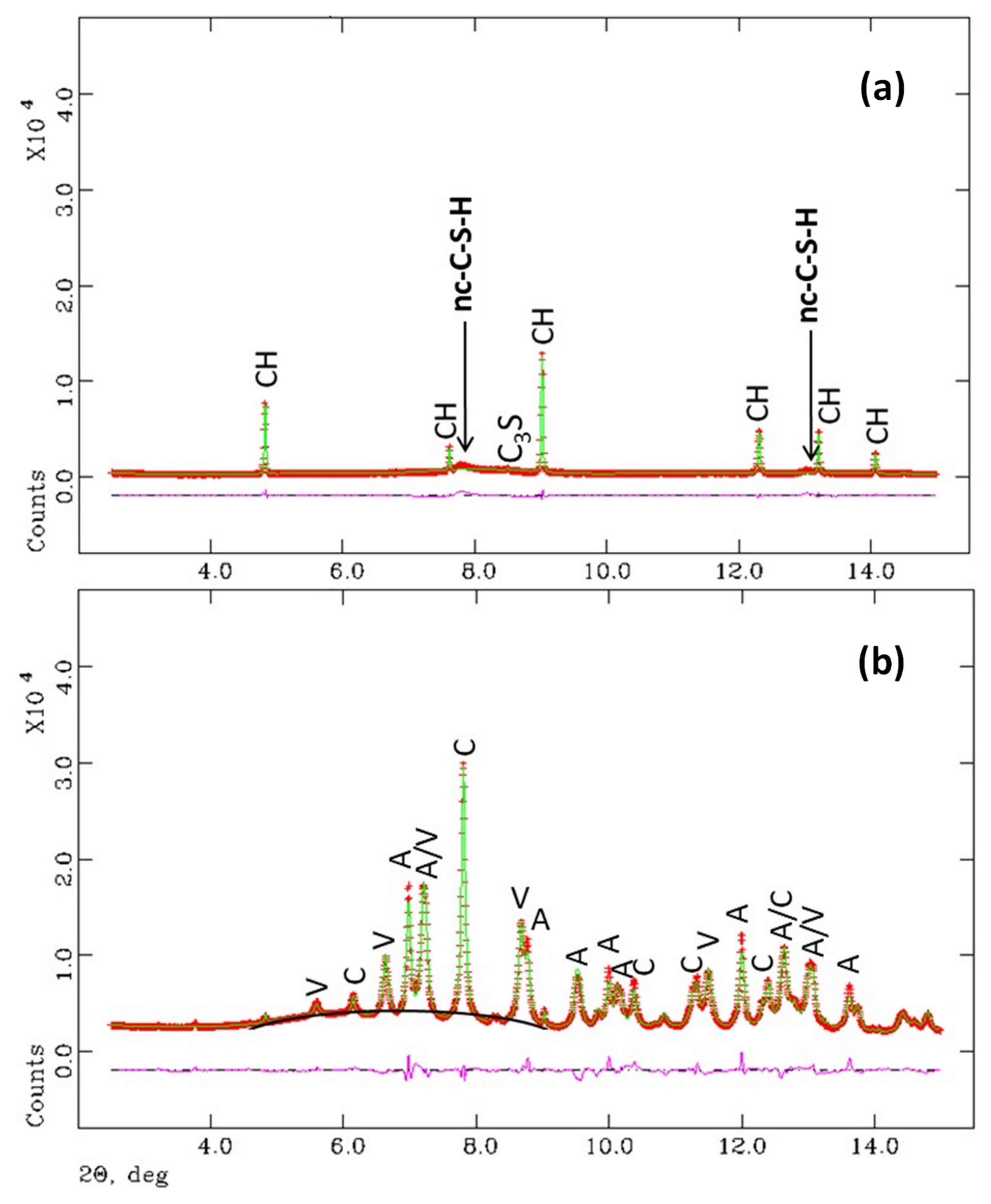
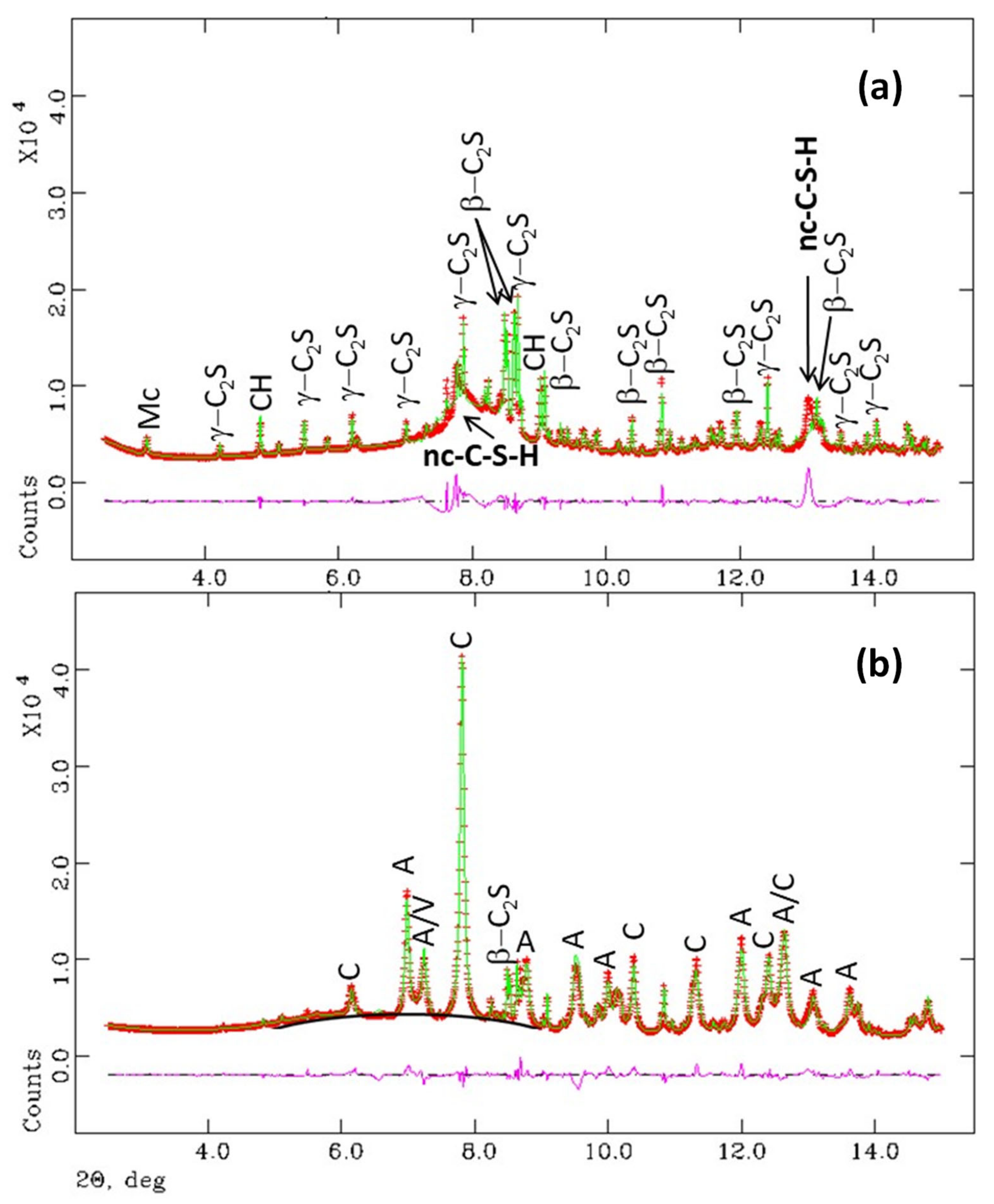
3.1. Synchrotron Rietveld Quantitative Phase Analysis
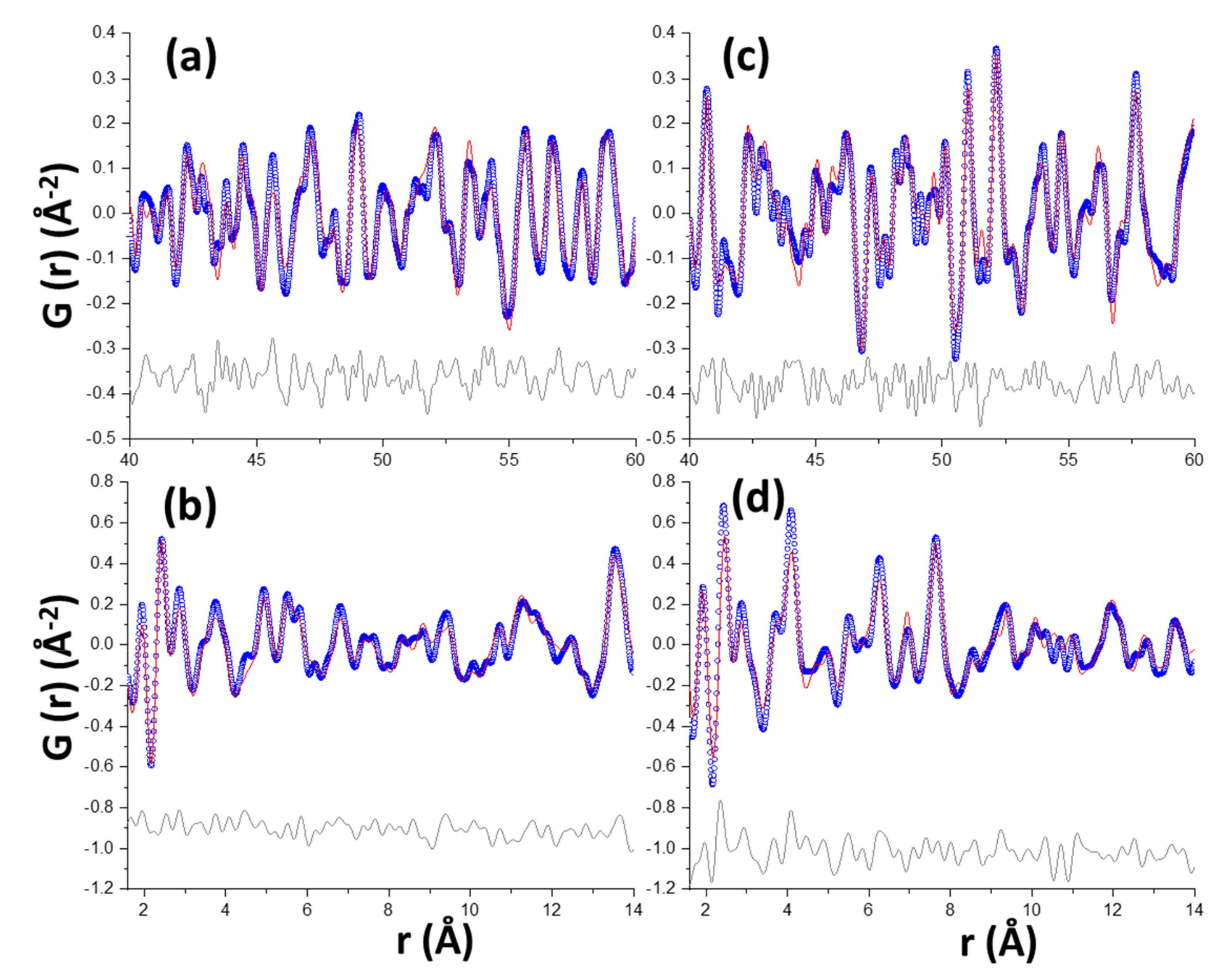
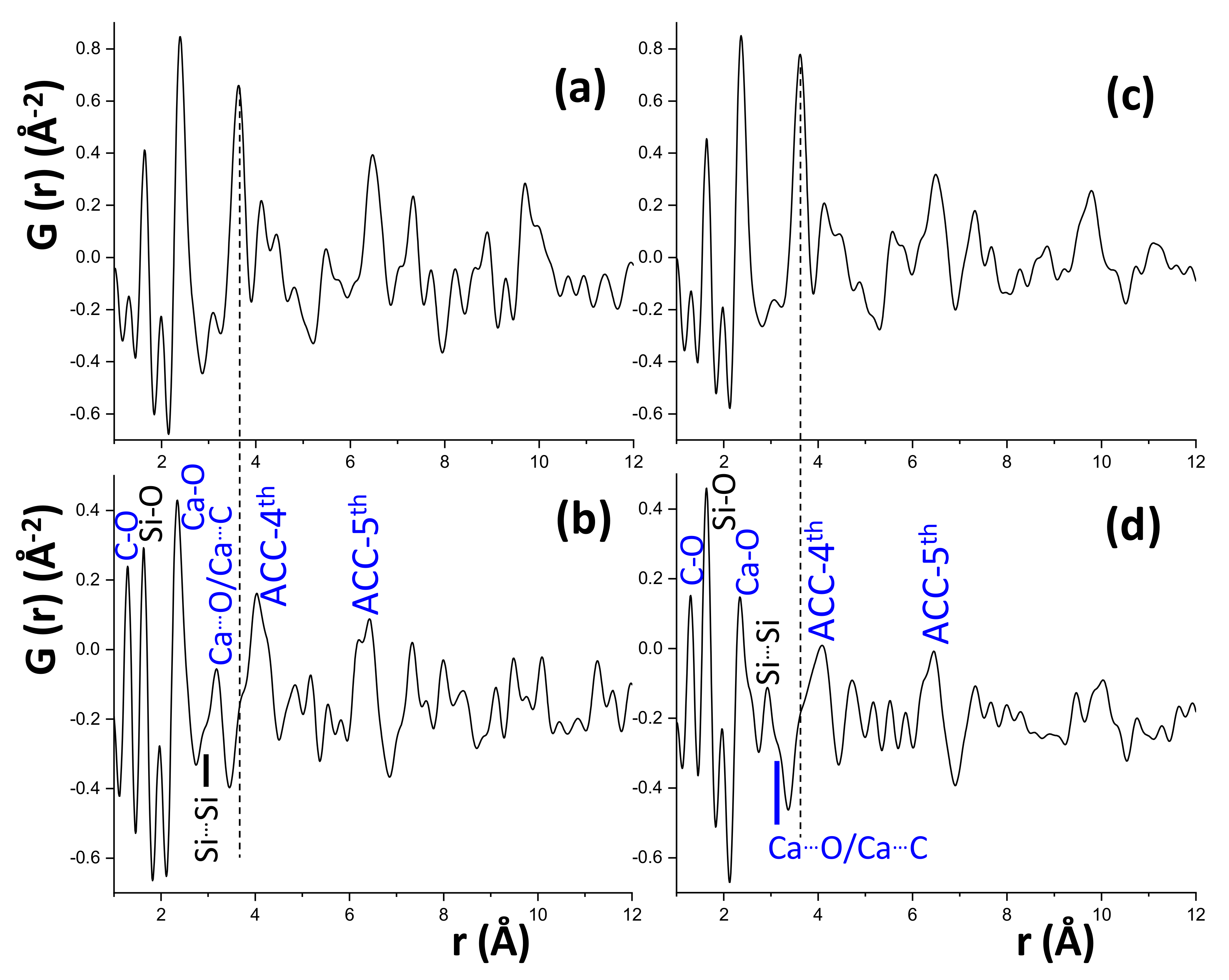
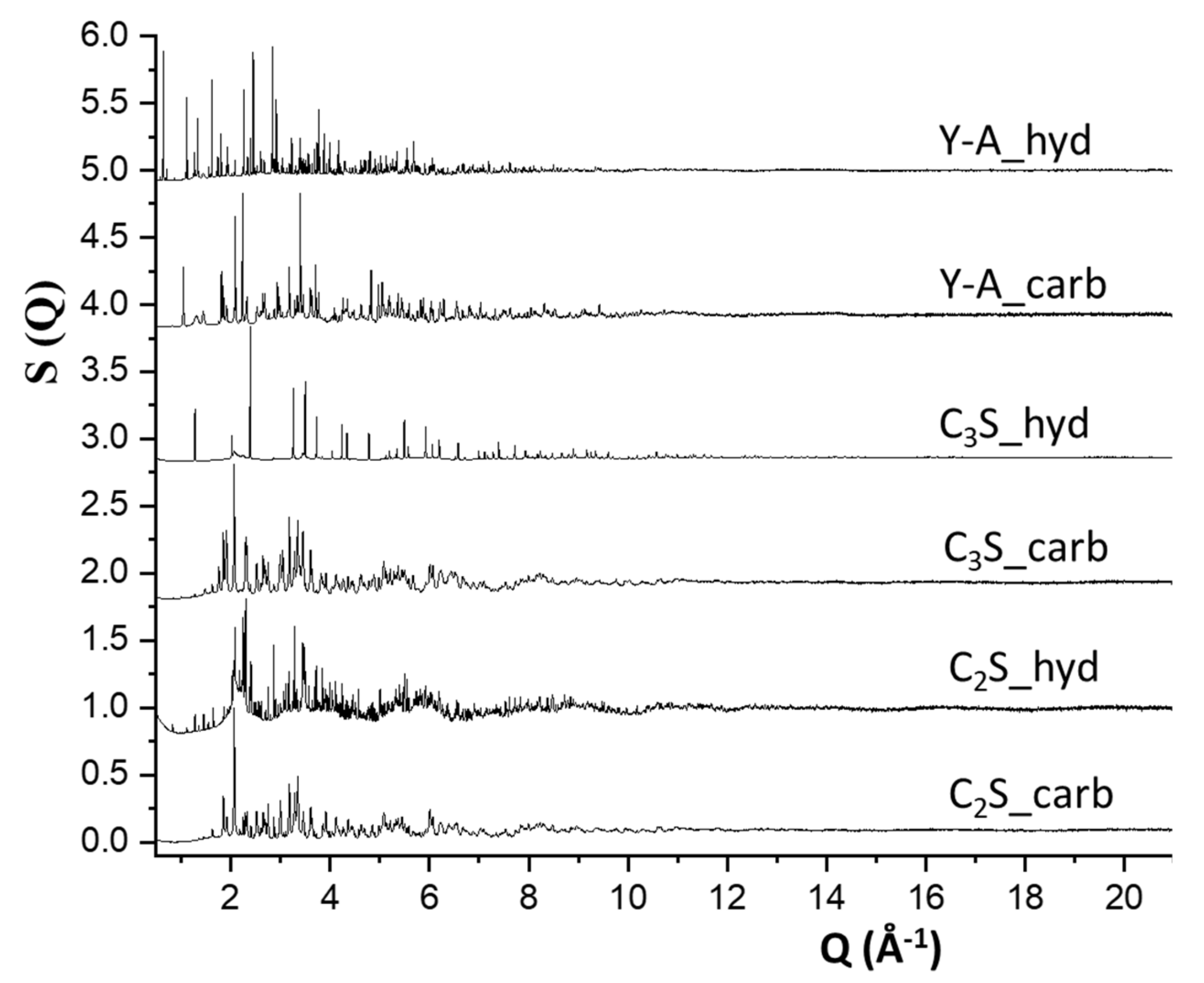
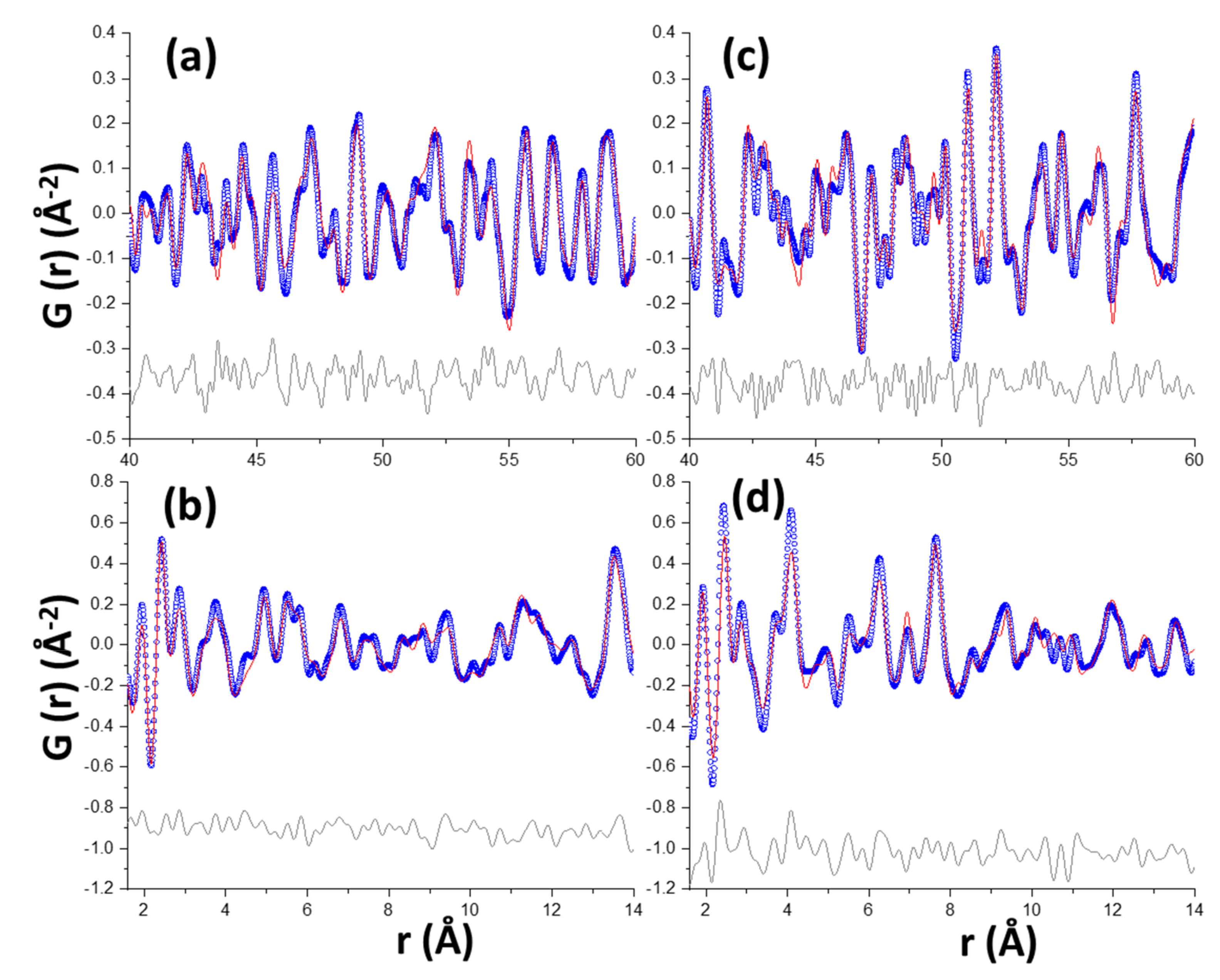
3.2. Synchrotron Pair Distribution Function Analysis
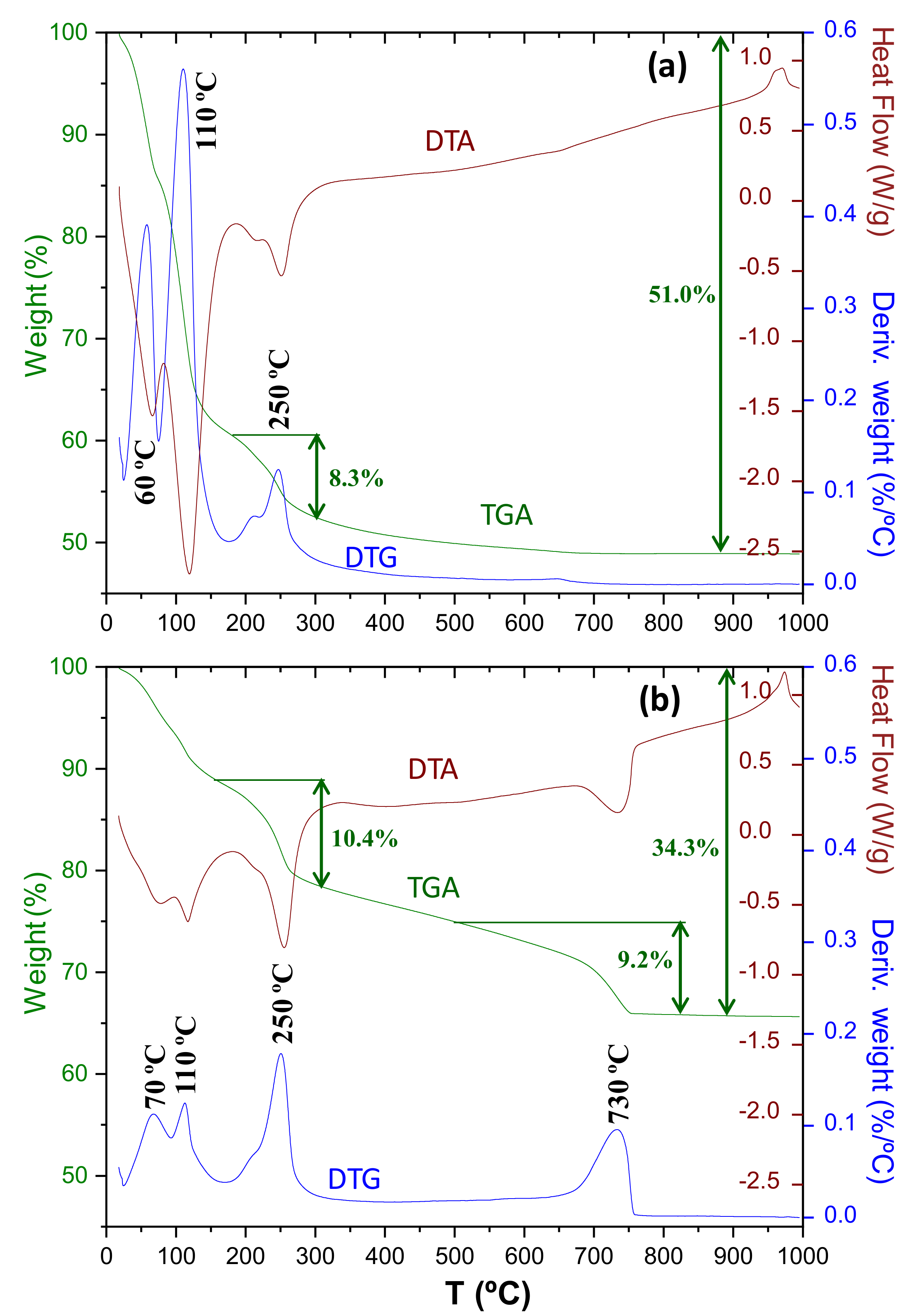
3.3. Thermal Analysis
4. Conclusions
- The reaction of ye’elimite with the stoichiometric amount of anhydrite yields crystalline ettringite and gibbsite with nanocrystalline nature (average particle size ~3 nm). After accelerated carbonation (3%CO2 and RH = 65%), bassanite, aragonite and gibbsite are formed and quantified. The particle size of gibbsite under these conditions was larger, ~5 nm. Chiefly, PDF analysis indicated that any amorphous content should be low and no appreciable amounts of amorphous calcium carbonate are formed in these experimental conditions.
- Crystalline CaCO3 polymorph contents (vaterite, aragonite and calcite) were highly variable in the three studied samples and we still cannot rationalise the conditions for their formation.
- C-S-H carbonates give: (i) amorphous silica gel; (ii) amorphous calcium carbonate, and (iii) variable contents of crystalline calcium carbonates. The existence of both coexisting amorphous components has been firmly established from the differential PDF study, being the most relevant and novel result from this work.
- Carbonate decomposition in the 500–700 °C temperature range (modes II and III) is intimately joined to C-S-H carbonation. Larger C-S-H content led to larger contribution of the II and III modes. Modes-II and III contain the weight loss from amorphous calcium carbonate but they should also have a contribution from metastable crystalline calcium carbonates (vaterite and aragonite).
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phase Name | ICSD | Ref. |
|---|---|---|
| t-C3S | 4331 | [64] |
| β-C2S | 81096 | [65] |
| γ-C2S | 81095 | [65] |
| CH | 202220 | [66] |
| AFt | 155395 | [67] |
| Calcite | 80869 | [68] |
| Vaterite | 15879 | [69] |
| Aragonite | 157994 | [70] |
| Bassanite | 79529 | [71] |
| Monocarbonate | 59327 | [72] |
| nano-Gibbsite | 6162 | [73] |
References
- World Business Council for Sustainable Development—IEA. Cement Technology Roadmap 2009: Carbon Emissions Reductions up to 2050; OECD Publishing: Paris, France, 2009. [Google Scholar]
- UN Environment; Scrivener, K.L.; John, V.M.; Gartner, E. Eco-efficient cements: Potential, economically viable solutions for a low-CO2, cement-based materials industry. Cem. Concr. Res. 2018, 114, 2–26. [Google Scholar] [CrossRef]
- Krausmann, F.; Wiedenhofer, D.; Lauk, C.; Haas, W.; Tanikawa, H.; Fishman, T.; Miatto, A.; Schandl, H.; Haberl, H. Global socioeconomic material stocks rise 23-fold over the 20th century and require half of annual resource use. Proc. Natl. Acad. Sci. USA 2017, 114, 1880–1885. [Google Scholar] [CrossRef] [Green Version]
- Akhtar, A.; Sarmah, A.K. Construction and demolition waste generation and properties of recycled aggregate concrete: A global perspective. J. Clean. Prod. 2018, 186, 262–281. [Google Scholar] [CrossRef]
- Wu, H.; Zuo, J.; Zillante, G.; Wang, J.; Yuan, H. Construction and demolition waste research: A bibliometric analysis. Archit. Sci. Rev. 2019, 62, 354–365. [Google Scholar] [CrossRef]
- Torgal, F.P.; Miraldo, S.; Labrincha, J.A.; de Brito, J. An overview on concrete carbonation in the context of eco-efficient construction: Evaluation, use of SCMs and/or RAC. Constr. Build. Mater. 2012, 36, 141–150. [Google Scholar] [CrossRef] [Green Version]
- Šavija, B.; Lukovic, M. Carbonation of cement paste: Understanding, challenges, and opportunities. Constr. Build. Mater. 2016, 117, 285–301. [Google Scholar] [CrossRef] [Green Version]
- Ashraf, W. Carbonation of cement-based materials: Challenges and opportunities. Constr. Build. Mater. 2016, 120, 558–570. [Google Scholar] [CrossRef]
- Black, L.; Breen, C.; Yarwood, J. Structural features of C–S–H(I) and its carbonation in air—A raman spectroscopic study. Part II: Carbonated phases. J. Am. Ceram. Soc. 2007, 917, 908–917. [Google Scholar] [CrossRef]
- Castellote, M.; Andrade, C.; Turrillas, X.; Campo, J.; Cuello, G.J. Accelerated carbonation of cement pastes in situ monitored by neutron diffraction. Cem. Concr. Res. 2008, 38, 1365–1373. [Google Scholar] [CrossRef]
- Castellote, M.; Fernandez, L.; Andrade, C.; Alonso, C. Chemical changes and phase analysis of OPC pastes carbonated at different CO2 concentrations. Cem. Concr. Res. 2009, 42, 515–525. [Google Scholar] [CrossRef]
- Morandeau, A.; Thiéry, M.; Dangla, P. Investigation of the carbonation mechanism of CH and C-S-H in terms of kinetics, microstructure changes and moisture properties. Cem. Concr. Res. 2014, 56, 153–170. [Google Scholar] [CrossRef] [Green Version]
- Morandeau, A.E.; White, C.E. In situ X-ray pair distribution function analysis of accelerated carbonation of a synthetic calcium–silicate–hydrate gel. J. Mater. Chem. A 2015, 3, 8597–8605. [Google Scholar] [CrossRef]
- Saillio, M.; Baroghel-Bouny, V.; Pradelle, S.; Bertin, M.; Vincent, J.; d’Espinose de Lacaillerie, J.-B. Effect of supplementary cementitious materials on carbonation of cement pastes. Cem. Concr. Res. 2021, 142, 106358. [Google Scholar] [CrossRef]
- Fernández-Carrasco, L.; Torréns-Martín, D.; Martínez-Ramírez, S. Carbonation of ternary building cementing materials. Cem. Concr. Compos. 2012, 34, 1180–1186. [Google Scholar] [CrossRef]
- Martínez-Ramírez, S.; Fernández-Carrasco, L. Carbonation of ternary cement systems. Constr. Build. Mater. 2012, 27, 313–318. [Google Scholar] [CrossRef]
- Hargis, C.W.; Lothenbach, B.; Müller, C.J.; Winnefeld, F. Carbonation of calcium sulfoaluminate mortars. Cem. Concr. Compos. 2017, 80, 123–134. [Google Scholar] [CrossRef]
- Chen, B.; Horgnies, M.; Huet, B.; Morin, V.; Johannes, K.; Kuznik, F. Comparative kinetics study on carbonation of ettringite and meta-ettringite based materials. Cem. Concr. Res. 2020, 137, 106209. [Google Scholar] [CrossRef]
- Nishikawa, T.; Suzuki, K.; Ito, S.; Sato, K.; Takebe, T. Decomposition of synthesized ettringite by carbonation. Cem. Concr. Res. 1992, 22, 6–14. [Google Scholar] [CrossRef]
- Zhou, Q.; Glasser, F.P. Kinetics and mechanism of the carbonation of ettringite. Adv. Cem. Res. 2000, 12, 131–136. [Google Scholar] [CrossRef]
- Grounds, T.; Midgley, H.G.; Novell, D.V. Carbonation of ettringite by atmospheric carbon dioxide. Thermochim. Acta 1988, 135, 347–352. [Google Scholar] [CrossRef]
- Jiang, Y.F.; Liu, C.L.; Xue, J.; Li, P.; Yu, J.G. Insights into the polymorphic transformation mechanism of aluminum hydroxide during carbonation of potassium aluminate solution. CrystEngComm 2018, 20, 1431–1442. [Google Scholar] [CrossRef]
- Cuesta, A.; Santacruz, I.; de la Torre, A.G.; Dapiaggi, M.; Zea-Garcia, J.D.; Aranda, M.A.G. Local structure and Ca/Si ratio in C-S-H gels from hydration of blends of tricalcium silicate and silica fume. Cem. Concr. Res. 2021, 143, 106405. [Google Scholar] [CrossRef]
- Taylor, H.F.W. Cement Chemistry, 2nd ed.; Thomas Telford Pub: London, UK, 1997; ISBN 9780727725929. [Google Scholar]
- Skinner, L.B.; Chae, S.R.; Benmore, C.J.; Wenk, H.R.; Monteiro, P.J.M. Nanostructure of Calcium Silicate Hydrates in Cements. Phys. Rev. Lett. 2010, 104, 195502. [Google Scholar] [CrossRef] [PubMed]
- White, C.E. Effects of temperature on the atomic structure of synthetic calcium–silicate–deuterate gels: A neutron pair distribution function investigation. Cem. Concr. Res. 2016, 79, 93–100. [Google Scholar] [CrossRef] [Green Version]
- Cuesta, A.; Zea-Garcia, J.D.; Londono-Zuluaga, D.; de la Torre, A.G.; Santacruz, I.; Vallcorba, O.; Aranda, M.A.G. Synchrotron radiation pair distribution function analysis of gels in cements. Crystals 2017, 7, 317. [Google Scholar] [CrossRef] [Green Version]
- Cuesta, A.; Ichikawa, R.; Londono-Zuluaga, D.; de la Torre, A.G.; Santacruz, I.; Turrillas, X.; Aranda, M.A.G. Aluminum hydroxide gel characterization within a calcium aluminate cement paste by combined Pair Distribution Function and Rietveld analyses. Cem. Concr. Res. 2017, 96, 1–12. [Google Scholar] [CrossRef]
- Cuesta, A.; Zea-Garcia, J.D.; De la Torre, A.G.; Santacruz, I.; Aranda, M.A.G. Synchrotron pair distribution function analyses of ye’elimite-based pastes. Adv. Cem. Res. 2019, 31, 138–146. [Google Scholar] [CrossRef]
- Egami, T.; Billinge, S.J.L. Underneath the Bragg Peaks: Structural Analysis of Complex Materials, 2nd ed.; Elsevier Science, Pergamon: Amsterdam, The Netherlands, 2012; ISBN 9780080971414. [Google Scholar]
- Billinge, S.J.L.; Kanatzidis, M.G. Beyond crystallography: The study of disorder, nanocrystallinity and crystallographically challenged materials with pair distribution functions. Chem. Commun. 2004, 7, 749. [Google Scholar] [CrossRef]
- Grangeon, S.; Fernandez-Martinez, A.; Baronnet, A.; Marty, N.; Poulain, A.; Elkaïm, E.; Roosz, C.; Gaboreau, S.; Henocq, P.; Claret, F. Quantitative X-ray pair distribution function analysis of nanocrystalline calcium silicate hydrates: A contribution to the understanding of cement chemistry. J. Appl. Crystallogr. 2017, 50, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Cuesta, A.; Zea-Garcia, J.D.; Londono-Zuluaga, D.; De la Torre, A.G.; Santacruz, I.; Vallcorba, O.; Dapiaggi, M.; Sanfélix, S.G.; Aranda, M.A.G. Multiscale understanding of tricalcium silicate hydration reactions. Sci. Rep. 2018, 8, 8544. [Google Scholar] [CrossRef] [Green Version]
- Kupwade-Patil, K.; Boul, P.J.; Rasner, D.K.; Everett, S.M.; Proffen, T.; Page, K.; Ma, D.; Olds, D.; Thaemlitz, C.J.; Büyüköztürk, O. Retarder effect on hydrating oil well cements investigated using in situ neutron/X-ray pair distribution function analysis. Cem. Concr. Res. 2019, 126, 105920. [Google Scholar] [CrossRef]
- Bae, S.; Jee, H.; Suh, H.; Kanematsu, M.; Shiro, A.; Machida, A.; Watanuki, T.; Shobu, T.; Morooka, S.; Geng, G.; et al. Analysis of atomistic structural deformation characteristics of calcium silicate hydrate in 53-year-old tricalcium silicate paste using atomic pair distribution function. Constr. Build. Mater. 2020, 237, 117714. [Google Scholar] [CrossRef]
- Garg, N.; Özçelik, V.O.; Skibsted, J.; White, C.E. Nanoscale ordering and depolymerization of calcium silicate hydrates in the presence of alkalis. J. Phys. Chem. C 2019, 123, 24873–24883. [Google Scholar] [CrossRef]
- Morandeau, A.E.; White, C.E. Role of magnesium-stabilized amorphous calcium carbonate in mitigating the extent of carbonation in alkali-activated slag. Chem. Mater. 2015, 27, 6625–6634. [Google Scholar] [CrossRef]
- Tobler, D.J.; Rodriguez-Blanco, J.D.; Dideriksen, K.; Bovet, N.; Sand, K.K.; Stipp, S.L.S. Citrate effects on amorphous calcium carbonate (ACC) structure, stability, and crystallization. Adv. Funct. Mater. 2015, 25, 3081–3090. [Google Scholar] [CrossRef]
- Tobler, D.J.; Rodriguez Blanco, J.D.; Sørensen, H.O.; Stipp, S.L.S.; Dideriksen, K. Effect of pH on Amorphous Calcium Carbonate Structure and Transformation. Cryst. Growth Des. 2016, 16, 4500–4508. [Google Scholar] [CrossRef]
- Fauth, F.; Peral, I.; Popescu, C.; Knapp, M. The New Material Science Powder Diffraction beamline at ALBA Synchrotron; Cambridge University Press: Cambridge, UK, 2013; Volume 28, pp. S360–S370. [Google Scholar]
- Larson, A.C.; von Dreele, R.B. General structure analysis system (GSAS). Los Alamos Natl. Lab. Rep. LAUR 2004, 748, 86–748. [Google Scholar]
- Thompson, P.; Cox, D.E.; Hastings, J.B. Rietveld Refinement of Debye-Scherrer Synchrotron X-ray data from A1203. J. Appl. Crystallogr. 1987, 20, 79–83. [Google Scholar] [CrossRef] [Green Version]
- Finger, L.W.; Cox, D.E.; Jephcoat, A.P. Correction for powder diffraction peak asymmetry due to axial divergence. J. Appl. Crystallogr. 1994, 27, 892–900. [Google Scholar] [CrossRef]
- Juhas, P.; Davis, T.; Farrow, C.L.; Billinge, S.J.L. PDFgetX3: A rapid and highly automatable program for processing powder diffraction data into total scattering pair distribution functions. J. Appl. Crystallogr. 2013, 46, 560–566. [Google Scholar] [CrossRef] [Green Version]
- Farrow, C.L.; Juhas, P.; Liu, J.W.; Bryndin, D.; Božin, E.S.; Bloch, J.; Proffen, T.; Billinge, S.J.L. PDFfit2 and PDFgui: Computer programs for studying nanostructure in crystals. J. Phys. Condens. Matter 2007, 19, 335219. [Google Scholar] [CrossRef] [Green Version]
- Jeong, I.K.; Proffen, T.; Mohiuddin-Jacobs, F.; Billinge, S.J.L. Measuring correlated atomic motion using X-ray diffraction. J. Phys. Chem. C 1999, 103, 921–924. [Google Scholar] [CrossRef]
- Jeong, I.K.; Heffner, R.H.; Graf, M.J.; Billinge, S.J.L. Lattice dynamics and correlated atomic motion from the atomic pair distribution function. Phys. Rev. B Condens. Matter Mater. Phys. 2003, 67, 104301. [Google Scholar] [CrossRef] [Green Version]
- Cuesta, A.; de la Torre, A.G.; Losilla, E.R.; Peterson, V.K.; Rejmak, P.; Ayuela, A.; Frontera, C.; Aranda, M.A.G. Structure, atomistic simulations, and phase transition of stoichiometric yeelimite. Chem. Mater. 2013, 25, 1680–1687. [Google Scholar] [CrossRef] [Green Version]
- Cuesta, A.; Aranda, M.A.G.; Sanz, J.; de la Torre, A.G.; Losilla, E.R. Mechanism of stabilization of dicalcium silicate solid solution with aluminium. Dalt. Trans. 2014, 43, 2176–2182. [Google Scholar] [CrossRef] [Green Version]
- Cuesta, A.; Álvarez-Pinazo, G.; Sanfélix, S.G.; Peral, I.; Aranda, M.A.G.; De la Torre, A.G. Hydration mechanisms of two polymorphs of synthetic ye’elimite. Cem. Concr. Res. 2014, 63, 127–136. [Google Scholar] [CrossRef] [Green Version]
- Zea-Garcia, J.D.; De la Torre, A.G.; Aranda, M.A.G.; Cuesta, A. A Comparative study of experimental configurations in synchrotron pair distribution function. Materials 2019, 12, 1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kangni-Foli, E.; Poyet, S.; Le Bescop, P.; Charpentier, T.; Bernachy-Barbé, F.; Dauzères, A.; L’Hôpital, E.; d’Espinose de Lacaillerie, J.-B. Carbonation of model cement pastes: The mineralogical origin of microstructural changes and shrinkage. Cem. Concr. Res. 2021, 144, 106446. [Google Scholar] [CrossRef]
- White, C.E.; Daemen, L.L.; Hartl, M.; Page, K. Intrinsic differences in atomic ordering of calcium (alumino) silicate hydrates in conventional and alkali-activated cements. Cem. Concr. Res. 2015, 67, 66–73. [Google Scholar] [CrossRef] [Green Version]
- Ohara, K.; Tominaka, S.; Yamada, H.; Takahashi, M.; Yamaguchi, H.; Utsuno, F.; Umeki, T.; Yao, A.; Nakada, K.; Takemoto, M.; et al. Time-resolved pair distribution function analysis of disordered materials on beamlines BL04B2 and BL08W at SPring-8. J. Synchrotron Radiat. 2018, 25, 1627–1633. [Google Scholar] [CrossRef] [PubMed]
- Biswas, R.K.; Khan, P.; Mukherjee, S.; Mukhopadhyay, A.K.; Ghosh, J.; Muraleedharan, K. Study of short range structure of amorphous Silica from PDF using Ag radiation in laboratory XRD system, RAMAN and NEXAFS. J. Non. Cryst. Solids 2018, 488, 1–9. [Google Scholar] [CrossRef]
- Borges, P.H.R.; Costa, J.O.; Milestone, N.B.; Lynsdale, C.J.; Streatfield, R.E. Carbonation of CH and C-S-H in composite cement pastes containing high amounts of BFS. Cem. Concr. Res. 2010, 40, 284–292. [Google Scholar] [CrossRef]
- Cuesta, A.; De la Torre, A.G.; Santacruz, I.; Trtik, P.; da Silva, J.C.; Diaz, A.; Holler, M.; Aranda, M.A.G. Chemistry and mass density of aluminum hydroxide gel in eco-cements by ptychographic X-ray computed tomography. J. Phys. Chem. C 2017, 121, 3044–3054. [Google Scholar] [CrossRef]
- Thiery, M.; Villain, G.; Dangla, P.; Platret, G. Investigation of the carbonation front shape on cementitious materials: Effects of the chemical kinetics. Cem. Concr. Res. 2007, 37, 1047–1058. [Google Scholar] [CrossRef]
- Lothenbach, B.; Durdzinski, P.; de Weerdt, K. Thermogravimetric analysis. In A Practical Guide to Microstructural Analysis of Cementitious Materials; Scrivener, K., Snellings, R., Lothenbach, B., Eds.; CRC Press: Boca Raton, FL, USA, 2016; pp. 177–211. ISBN 9781498738675. [Google Scholar]
- Shah, V.; Scrivener, K.L.; Bhattacharjee, B.; Bishnoi, S. Changes in microstructure characteristics of cement paste on carbonation. Cem. Concr. Res. 2018, 109, 184–197. [Google Scholar] [CrossRef]
- Thiery, M.; Dangla, P.; Belin, P.; Habert, G.; Roussel, N. Carbonation kinetics of a bed of recycled concrete aggregates: A laboratory study on model materials. Cem. Concr. Res. 2013, 46, 50–65. [Google Scholar] [CrossRef]
- Koga, N.; Kasahara, D.; Kimura, T. Aragonite crystal growth and solid-state aragonite-calcite transformation: A physico-geometrical relationship via thermal dehydration of included water. Cryst. Growth Des. 2013, 13, 2238–2246. [Google Scholar] [CrossRef]
- Chakoumakos, B.C.; Pracheil, B.M.; Koenigs, R.P.; Bruch, R.M.; Feygenson, M. Empirically testing vaterite structural models using neutron diffraction and thermal analysis. Sci. Rep. 2016, 6, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Golovastikov, N.I.; Matveeva, R.G.; Belov, N.V. Crystal structure of the tricalcium silicate (CaOSi2)3=C3S. Kristallografiya 1975, 20, 721–729. [Google Scholar]
- Mumme, W.G.; Hill, R.J.; Bushnell-Wye, G.; Segnit, E.R. Rietveld crystal structure refinements, crystal chemistry and calculated powder diffraction data for the polymorphs of dicalcium silicate and related phases. Neues Jahrb. Fuer Mineral. 1995, 169, 35–68. [Google Scholar]
- Chaix-Pluchery, O.; Pannetier, J.; Bouillot, J.; Niepce, J.C. Structural prereactional transformations in Ca(OH)2. J. Solid State Chem. 1987, 67, 225–234. [Google Scholar] [CrossRef]
- Goetz-Neunhoeffer, F.; Neubauer, J. Refined ettringite (Ca6Al2(SO4)3(OH)12∙26HO) structure for quantitative X-ray diffraction analysis. Powder Diffr. 2006, 21, 4–11. [Google Scholar] [CrossRef]
- Maslen, E.N.; Streltsov, V.A.; Streltsova, N.R.; Ishizawa, N. Electron density and optical anisotropy in rhombohedral carbonates. III. Synchrotron X-ray studies of CaCO3, MgCO3 and MnCO3. Acta Crystallogr. Sect. B 1995, 51, 929–939. [Google Scholar] [CrossRef] [Green Version]
- Kamhi, S.R. On the structure of vaterite CaCO3. Acta Crystallogr. 1963, 16, 770–772. [Google Scholar] [CrossRef]
- Pokroy, B.; Fieramosca, J.S.; von Dreele, R.B.; Fitch, A.N.; Caspi, E.N.; Zolotoyabko, E. Atomic structure of biogenic aragonite. Chem. Mater. 2007, 19, 3244–3251. [Google Scholar] [CrossRef]
- Bezou, C.; Nonat, A.; Mutin, J.-C.; Christensen, A.N.; Lehmann, M.S. Investigation of the Crystal Structure of γ-CaSO4, CaSO4 0.5H2O, and CaSO4·0.6H2O by Powder Diffraction Methods. J. Solid State Chem. 1995, 117, 165–176. [Google Scholar] [CrossRef]
- François, M.; Renaudin, G.; Evrard, O. A cementitious compound with composition 3CaO.Al2O3-CaCO3.11H2O. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1998, 54, 1214–1217. [Google Scholar] [CrossRef] [Green Version]
- Saalfeld, H.; Wedde, M. Refinement of the crystal structure of gibbsite, Al(OH)3. Z. fur Krist. New Cryst. Struct. 1974, 139, 129–135. [Google Scholar] [CrossRef]
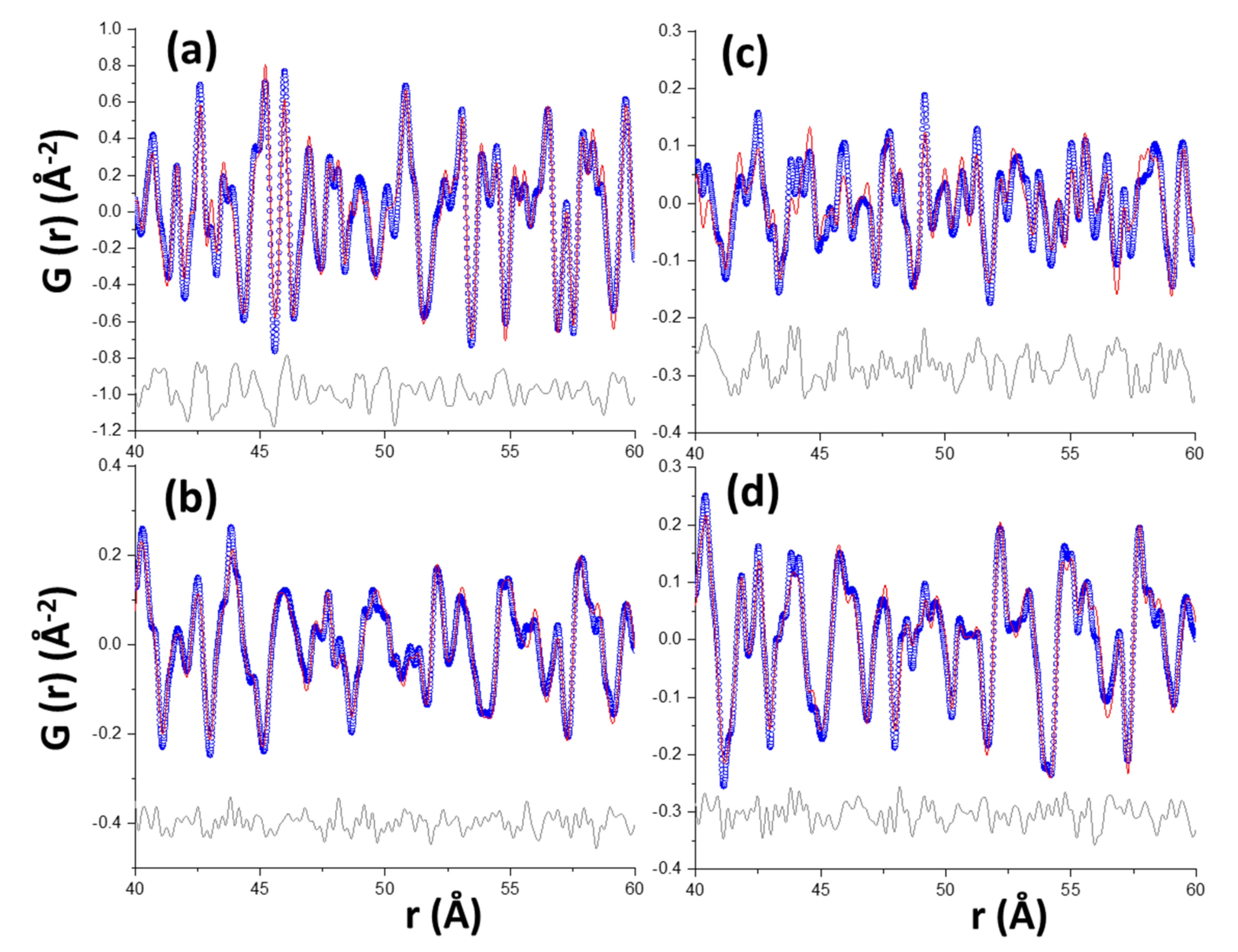

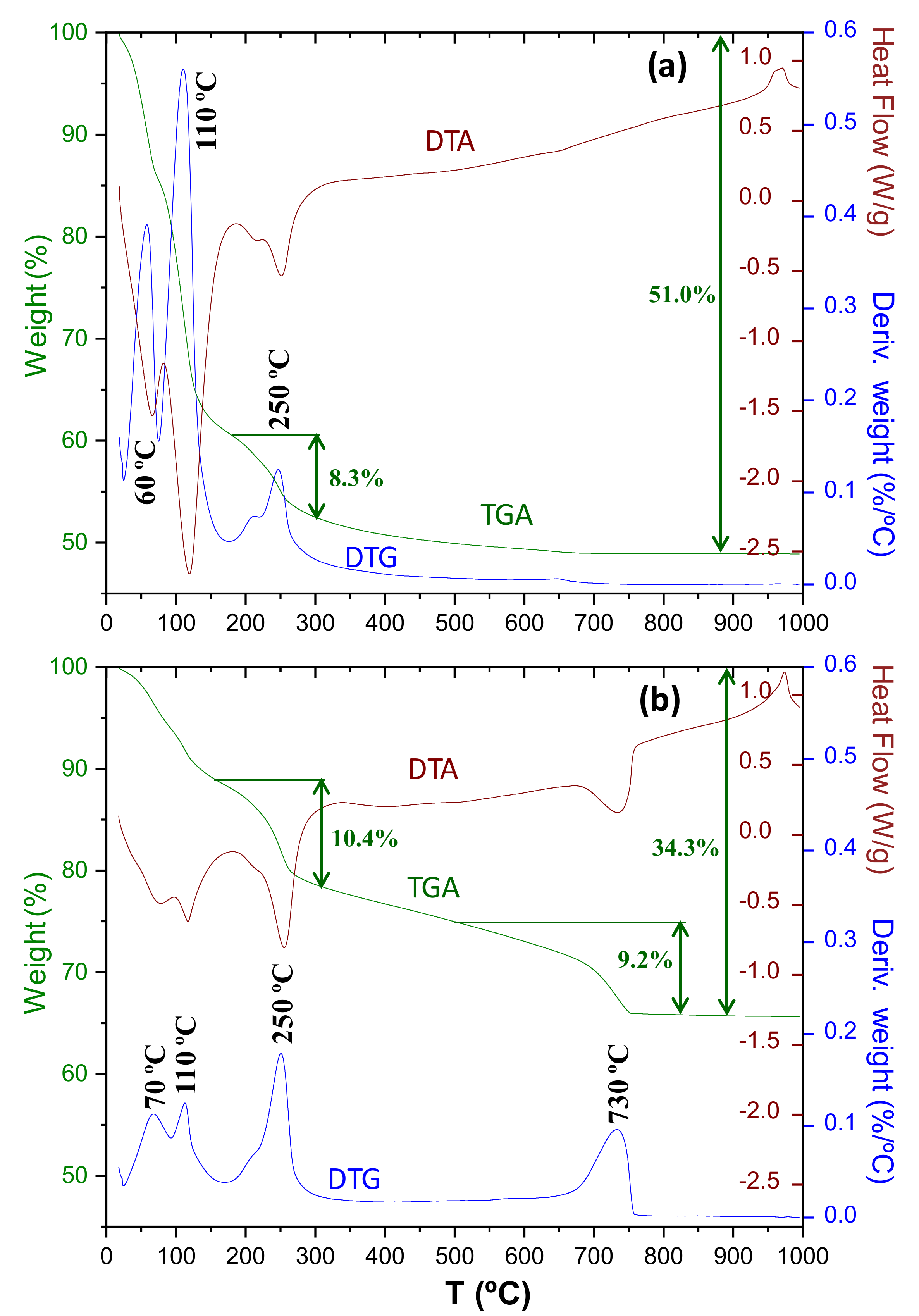

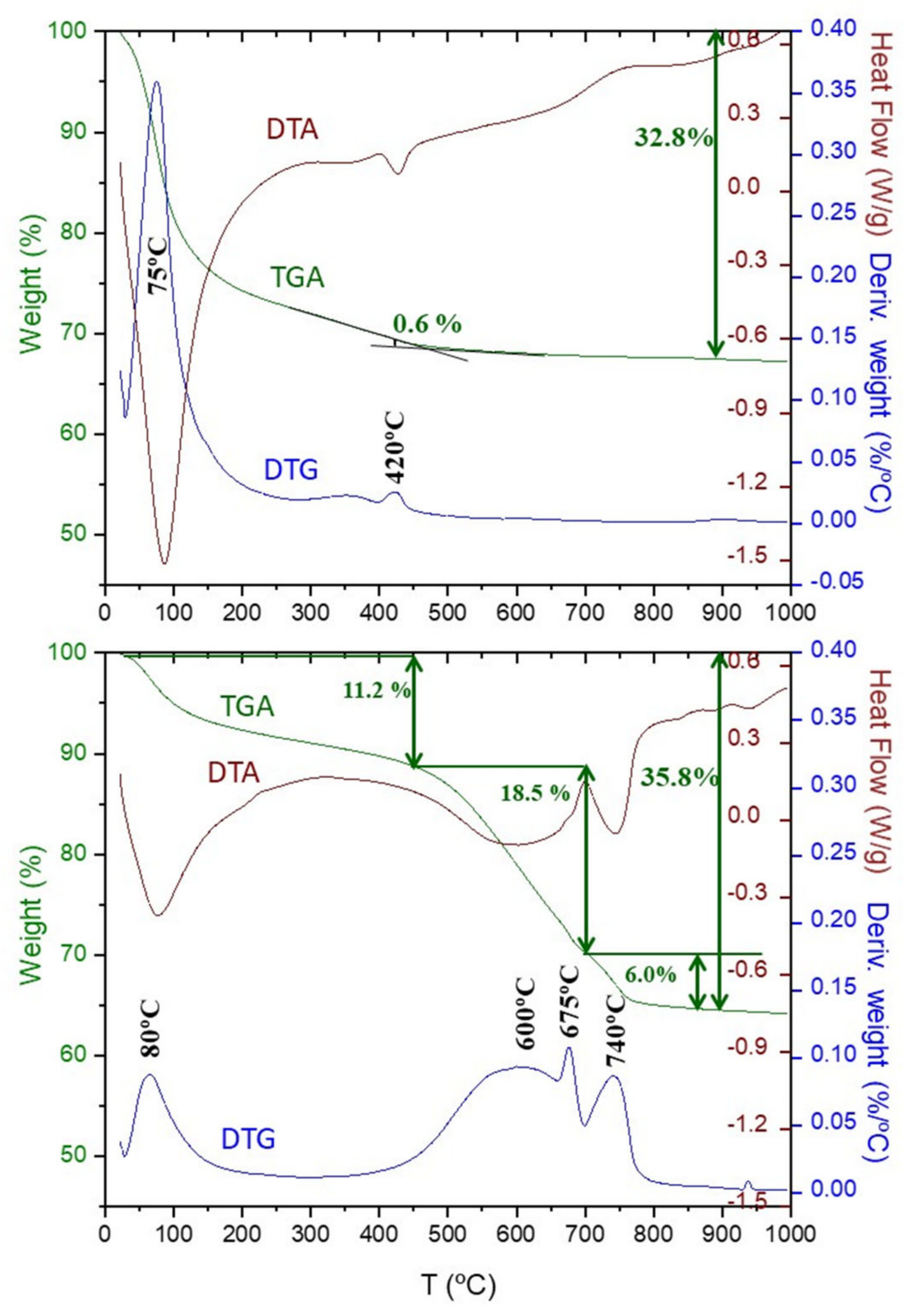
| Component | Chemical Formula | Carbonation Reaction (in H2O Presence) | Reaction |
|---|---|---|---|
| Portlandite | Ca(OH)2 | Ca(OH)2 + CO2 → CaCO3 # + H2O | (1) |
| C-S-H-gel * | (CaO)xSiO2.nH2O * | (CaO)1.8SiO2.4H2O + 1.8CO2 → 1.8CaCO3 + SiO2.nH2O + yH2O | (2) |
| Ettringite, AFt | Ca6Al2(SO4)3(OH)12·26H2O | AFt + 3CO2 → 3CaCO3 + 3CaSO4·mH2O $ + 2Al(OH)3 + zH2O | (3) |
| Gibbsite | Al(OH)3 | Al(OH)3 + CO2 → stable? | (4) |
| Materials | Dv,10 | Dv,50 | Dv,90 | ρ (g/cm3) | BET (m2/g) |
|---|---|---|---|---|---|
| o-C4A3 | 1.3 | 7.1 | 22.1 | 2.61(1) | 1.1(1) |
| C | 1.5 | 10.1 | 42.6 | 2.93(1) | 2.6(1) |
| t-C3S | 0.8 | 4.6 | 11.0 | 3.15(1) | 1.9(1) |
| β-C2S | 1.0 | 6.5 | 35.7 | 3.22(1) | 1.7(1) |
| Hydration Reactions, With the Amount of Products in [g/mol] | Reaction | ||
|---|---|---|---|
| Ca4[Al6O12]SO4 + 2CaSO4 + 38·H2O | → | Ca6Al2(SO4)3(OH)12·26H2O [1255.1] + 4 Al(OH)3 [312.0] | (5) |
| Ca3SiO5 + 5.2 H2O | → | (CaO)1.8SiO2.4H2O [233.1] + 1.2 Ca(OH)2 [88.9] | (6) |
| Ca2SiO4 + 4.2 H2O | → | (CaO)1.8SiO2.4H2O [233.1] + 0.2 Ca(OH)2 [14.8] | (7) |
| Phase (wt%) | Y-A_hyd | Y-A_carb | C3S_hyd | C3S_carb | C2S_hyd | C2S_carb |
|---|---|---|---|---|---|---|
| t-C3S | - | - | 6.7(1) | - | - | - |
| β-C2S | - | - | - | - | 55.0(3) | 8.1(1) |
| γ-C2S | - | - | - | - | 28.7(4) | - |
| CH | - | - | 93.3(4) | 0.6(1) | 8.7(2) | - |
| AFt | 100.0 | - | - | - | - | - |
| Calcite | - | - | - | 26.8(9) | 2.2(2) | 43.1(1) |
| Vaterite | - | - | - | 34.1(3) | - | 5.9(4) |
| Aragonite | - | 23.4(2) | - | 38.5(3) | - | 42.9(1) |
| Bassanite | - | 38.6(1) | - | - | - | - |
| Monocarbonate * | - | - | - | - | 5.4(5) | - |
| nano-Gibbsite | - | 37.9(9) | - | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuesta, A.; De la Torre, A.G.; Aranda, M.A.G. X-ray Total Scattering Study of Phases Formed from Cement Phases Carbonation. Minerals 2021, 11, 519. https://doi.org/10.3390/min11050519
Cuesta A, De la Torre AG, Aranda MAG. X-ray Total Scattering Study of Phases Formed from Cement Phases Carbonation. Minerals. 2021; 11(5):519. https://doi.org/10.3390/min11050519
Chicago/Turabian StyleCuesta, Ana, Angeles G. De la Torre, and Miguel A. G. Aranda. 2021. "X-ray Total Scattering Study of Phases Formed from Cement Phases Carbonation" Minerals 11, no. 5: 519. https://doi.org/10.3390/min11050519
APA StyleCuesta, A., De la Torre, A. G., & Aranda, M. A. G. (2021). X-ray Total Scattering Study of Phases Formed from Cement Phases Carbonation. Minerals, 11(5), 519. https://doi.org/10.3390/min11050519