
Thermal Stability of Calcium Oxalates from CO2 Sequestration for Storage Purposes: An In-Situ HT-XRPD and TGA Combined Study

 , , , ,
, , , , 
Abstract
:
1. Introduction
1.1. Decomposition Pathways
1.2. Previous Kinetic Investigations
2. Materials and Methods
2.1. In-Situ XRPD Experiments
2.2. Thermogravimetric Analysis-Evolved Gas Analysis (TGA-EGA)
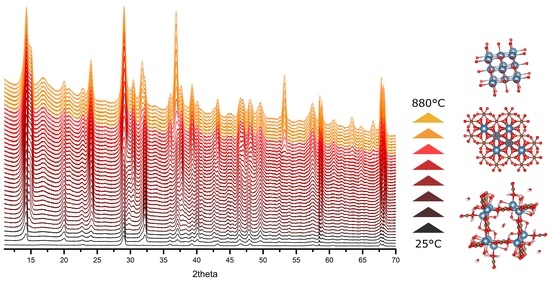
3. Results
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dollimore, D. The thermal decomposition of oxalates. A review. Thermochim. Acta 1987, 117, 331–363. [Google Scholar] [CrossRef]
- Dollimore, D.; Griffiths, D.L. Differential thermal analysis study of various oxalates in oxygen and nitrogen. J. Therm. Anal. Calorim. 1970, 2, 229–250. [Google Scholar] [CrossRef]
- Dollimore, D.; Griffiths, D.L.; Nicholson, D. The thermal decomposition of oxalates. Part II. Thermogravimetric analysis of various oxalates in air and in nitrogen. J. Chem. Soc. 1963, 2617–2623. [Google Scholar] [CrossRef]
- Kutaish, N.; Aggarwal, P.; Dollimore, D. Thermal analysis of calcium oxalate samples obtained by various preparative routes. Thermochim. Acta 1997, 297, 131–137. [Google Scholar] [CrossRef]
- Błażejowski, J.; Zadykowicz, B. Computational prediction of the pattern of thermal gravimetry data for the thermal decomposition of calcium oxalate monohydrate. J. Therm. Anal. Calorim. 2013, 113, 1497–1503. [Google Scholar] [CrossRef] [Green Version]
- Conti, C.; Brambilla, L.; Colombo, C.; Dellasega, D.; Gatta, G.D.; Realini, M.; Zerbi, G. Stability and transformation mechanism of weddellite nanocrystals studied by X-ray diffraction and infrared spectroscopy. Phys. Chem. Chem. Phys. 2010, 12, 14560–14566. [Google Scholar] [CrossRef]
- Conti, C.; Casati, M.; Colombo, C.; Realini, M.; Brambilla, L.; Zerbi, G. Phase transformation of calcium oxalate dihydrate–monohydrate: Effects of relative humidity and new spectroscopic data. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 128, 413–419. [Google Scholar] [CrossRef]
- Simons, E.; Newkirk, A. New studies on calcium oxalate monohydrate: A guide to the interpretation of thermogravimetric measurements. Talanta 1964, 11, 549–571. [Google Scholar] [CrossRef]
- Pastero, L.; Curetti, N.; Ortenzi, M.A.; Schiavoni, M.; Destefanis, E.; Pavese, A. CO2 capture and sequestration in stable Ca-oxalate, via Ca-ascorbate promoted green reaction. Sci. Total Environ. 2019, 666, 1232–1244. [Google Scholar] [CrossRef] [PubMed]
- Pastero, L.; Marengo, A.; Boero, R.; Pavese, A. Non-conventional CO2 sequestration via Vitamin C promoted green reaction: Yield evaluation. J. CO2 Util. 2021, 44, 101420. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Paria, M.; Maiti, H. Preparation of PbTiO3 powder through oxalate precipitation route. Mater. Lett. 1992, 13, 130–134. [Google Scholar] [CrossRef]
- Munson, M.J.; Riman, R.E. Observed phase transformations of oxalate-derived lead monoxide powder. J. Therm. Anal. Calorim. 1991, 37, 2555–2566. [Google Scholar] [CrossRef]
- Andrade, A.; Machado, A.J.S.; Jardim, R. Kinetic study of La2CuO4 formation from an oxalate precursor. Mater. Lett. 1992, 13, 96–101. [Google Scholar] [CrossRef]
- Chen, F.-H.; Tseng, T.-Y. Formation of High-Tc Superconducting Bi-Pb-Sr-Ca-Cu Oxide Films by Spray Pyrolysis of an Oxalate Suspension. J. Am. Ceram. Soc. 1990, 73, 889–892. [Google Scholar] [CrossRef]
- Gurrieri, S.; Siracusa, G.; Calí, R. Thermal decomposition of CaC2O4·H2O. Determination of kinetic parameters by DTG and DTA. J. Therm. Anal. Calorim. 1974, 6, 293–298. [Google Scholar] [CrossRef]
- Frost, R.L.; Weier, M.L. Thermal treatment of whewellite—a thermal analysis and Raman spectroscopic study. Thermochim. Acta 2004, 409, 79–85. [Google Scholar] [CrossRef] [Green Version]
- Hourlier, D. Thermal decomposition of calcium oxalate: Beyond appearances. J. Therm. Anal. Calorim. 2018, 136, 2221–2229. [Google Scholar] [CrossRef]
- Szekely, T.; Varhegyi, G.; Till, F.; Szabo, P.; Jakab, E. The effects of heat and mass transport on the results of thermal decomposition studies: Part 1. The three reactions of calcium oxalate monohydrate. J. Anal. Appl. Pyrolysis 1987, 11, 71–81. [Google Scholar] [CrossRef]
- Price, D.; Dollimore, D.; Fatemi, N.; Whitehead, R. Mass spectrometric determination of kinetic parameters for solid state decomposition reactions. Part 1. Method; calcium oxalate decomposition. Thermochim. Acta 1980, 42, 323–332. [Google Scholar] [CrossRef]
- Kloprogge, T.; Boström, T.E.; Weier, M.L. In situ observation of the thermal decomposition of weddelite by heating stage environmental scanning electron microscopy. Am. Miner. 2004, 89, 245–248. [Google Scholar] [CrossRef]
- Boldyrev, V.V.; Nevyantsev, I.S.; Mikhailov, Y.N.; Khairetdinov, E.F. On the mechanism of thermal decomposition of oxalates. Kinet. Katal. 1970, 11, 367–373. [Google Scholar]
- Kociba, K.J.; Gallagher, P.K. A study of calcium oxalate monohydrate using dynamic differential scanning calorimetry and other thermoanalytical techniques. Thermochim. Acta 1996, 282–283, 277–296. [Google Scholar] [CrossRef]
- McAdie, H.G. Simultaneous Differential Thermal Analysis and Thermogravimetric Analysis Using the Open-Pan Type of Sample Holder. Anal. Chem. 1963, 35, 1840–1844. [Google Scholar] [CrossRef]
- Al-Maskari, N.; McAdams, D.; Reddy, J. Modeling of a biological material nacre: Waviness stiffness model. Mater. Sci. Eng. C 2017, 70, 772–776. [Google Scholar] [CrossRef]
- Barrall, E.M.; Rogers, L.B. Differential thermal analysis of organic samples. Effects of geometry and operating variables. Anal. Chem. 1962, 34, 1101–1106. [Google Scholar] [CrossRef]
- Lee, Y.F.; Dollimore, D. The identification of the reaction mechanism in rising temperature kinetic studies based on the shape of the DTG curve. Thermochim. Acta 1998, 323, 75–81. [Google Scholar] [CrossRef]
- Anderson, E.M.; Ericsson, I. Thermal degradation of organic polymers using different metals as the pyrolysis filament. J. Anal. Appl. Pyrolysis 1981, 3, 35–47. [Google Scholar] [CrossRef]
- Windig, W.; Kistemaker, P.; Haverkamp, J.; Meuzelaar, H. The effects of sample preparation, pyrolysis and pyrolyzate transfer conditions on pyrolysis mass spectra. J. Anal. Appl. Pyrolysis 1979, 1, 39–52. [Google Scholar] [CrossRef]
- Gál, S.; Paulik, F.; Erdey, L.; Bayer, J. Derivatographische untersuchung von calciumoxalathydraten. Period. Polytech. Chem. Eng. 1963, 7, 215–222. [Google Scholar]
- Izatulina, A.R.; Yelnikov, V.Y. Structure, Chemistry and Crystallization Conditions of Calcium Oxalates—The Main Components of Kidney Stones. In Minerals as Advanced Materials I.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 231–239. [Google Scholar] [CrossRef]
- Izatulina, A.R.; Gurzhiy, V.; Krzhizhanovskaya, M.G.; Kuz’Mina, M.A.; Leoni, M.; Frank-Kamenetskaya, O.V. Hydrated Calcium Oxalates: Crystal Structures, Thermal Stability, and Phase Evolution. Cryst. Growth Des. 2018, 18, 5465–5478. [Google Scholar] [CrossRef]
- Walter-Lévy, L.; Laniepce, J. Sur la thermolyse des hydrates de l’oxalate de calcium. C. R. Acad. Sci. Paris 1964, 259, 4686–4688. [Google Scholar]
- Zhao, W.; Sharma, N.; Jones, F.; Raiteri, P.; Gale, J.D.; Demichelis, R. Anhydrous Calcium Oxalate Polymorphism: A Combined Computational and Synchrotron X-ray Diffraction Study. Cryst. Growth Des. 2016, 16, 5954–5965. [Google Scholar] [CrossRef] [Green Version]
- Wojdyr, M. Fityk: A general-purpose peak fitting program. J. Appl. Crystallogr. 2010, 43, 1126–1128. [Google Scholar] [CrossRef]
- Echigo, T.; Kimata, M.; Kyono, A.; Shimizu, M.; Hatta, T. Re-investigation of the crystal structure of whewellite [Ca(C2O4)·H2O] and the dehydration mechanism of caoxite [Ca(C2O4)·3H2O]. Mineral. Mag. 2005, 69, 77–88. [Google Scholar] [CrossRef]
- Sukarni, S.; Widiono, A.E.; Wulandari, R.; Prasetiyo, A.; Puspitasari, P. Thermogravimetric Study on the Thermal Characteristics of Tetraselmis chuii Microalgae Pyrolysis in the Presence of Titanium dioxide. Key Eng. Mater. 2020, 851, 156–163. [Google Scholar] [CrossRef]
- Coats, A.W.; Redfern, J.P. Kinetic Parameters from Thermogravimetric Data. Nat. Cell Biol. 1964, 201, 68–69. [Google Scholar] [CrossRef]
- Freeman, E.S.; Carroll, B. The Application of Thermoanalytical Techniques to Reaction Kinetics: The Thermogravimetric Evaluation of the Kinetics of the Decomposition of Calcium Oxalate Monohydrate. J. Phys. Chem. 1958, 62, 394–397. [Google Scholar] [CrossRef]
- Doyle, C.D. Kinetic analysis of thermogravimetric data. J. Appl. Polym. Sci. 1961, 5, 285–292. [Google Scholar] [CrossRef]
- Dollimore, D. The application of thermal analysis in studying the thermal decomposition of solids. Thermochim. Acta 1992, 203, 7–23. [Google Scholar] [CrossRef]
- Ebrahimi-Kahrizsangi, R.; Abbasi, M. Evaluation of reliability of Coats-Redfern method for kinetic analysis of non-isothermal TGA. Trans. Nonferr. Met. Soc. China 2008, 18, 217–221. [Google Scholar] [CrossRef]
- Málek, J.; Criado, J.M. Empirical kinetic models in thermal analysis. Thermochim. Acta 1992, 203, 25–30. [Google Scholar] [CrossRef]
- Dollimore, D.; Evans, T.; Lee, Y.; Wilburn, F. Correlation between the shape of a TG/DTG curve and the form of the kinetic mechanism which is applying. Thermochim. Acta 1992, 198, 249–257. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental Evidence | Eatt (kJ/mol) | n (Best Fit) | Possible Reaction Mechanisms | Notes |
|---|---|---|---|---|
| 1st H2O release | 241 | 2 | not determined | The mechanism cannot be proposed due to the low value of αmax obtained from the Coats-Redfern method |
| 2nd H2O release | −22 | 0 | not determined | The mechanism cannot be proposed due to the low value of αmax obtained from the Coats-Redfern method |
| CO release | 118 | 1 | not determined | The mechanism cannot be proposed due to the low value of αmax obtained from the Coats-Redfern method; Overlaps the 1st CO2 release |
| 1st CO2 release | 234 | 2 | 2nd order decay | Overlaps the CO release |
| 2nd CO2 release | 51 | 0 | 1D diffusion | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Curetti, N.; Pastero, L.; Bernasconi, D.; Cotellucci, A.; Corazzari, I.; Archetti, M.; Pavese, A. Thermal Stability of Calcium Oxalates from CO2 Sequestration for Storage Purposes: An In-Situ HT-XRPD and TGA Combined Study. Minerals 2022, 12, 53. https://doi.org/10.3390/min12010053
Curetti N, Pastero L, Bernasconi D, Cotellucci A, Corazzari I, Archetti M, Pavese A. Thermal Stability of Calcium Oxalates from CO2 Sequestration for Storage Purposes: An In-Situ HT-XRPD and TGA Combined Study. Minerals. 2022; 12(1):53. https://doi.org/10.3390/min12010053
Chicago/Turabian StyleCuretti, Nadia, Linda Pastero, Davide Bernasconi, Andrea Cotellucci, Ingrid Corazzari, Maurizio Archetti, and Alessandro Pavese. 2022. "Thermal Stability of Calcium Oxalates from CO2 Sequestration for Storage Purposes: An In-Situ HT-XRPD and TGA Combined Study" Minerals 12, no. 1: 53. https://doi.org/10.3390/min12010053
APA StyleCuretti, N., Pastero, L., Bernasconi, D., Cotellucci, A., Corazzari, I., Archetti, M., & Pavese, A. (2022). Thermal Stability of Calcium Oxalates from CO2 Sequestration for Storage Purposes: An In-Situ HT-XRPD and TGA Combined Study. Minerals, 12(1), 53. https://doi.org/10.3390/min12010053