
Local Structure and Protons in Non-Stoichiometric Pseudo-Cubic Pollucite Mineral by Multinuclear NMR


Abstract
:1. Introduction
2. Materials and Methods
2.1. Specimens
2.2. Chemical Analysis
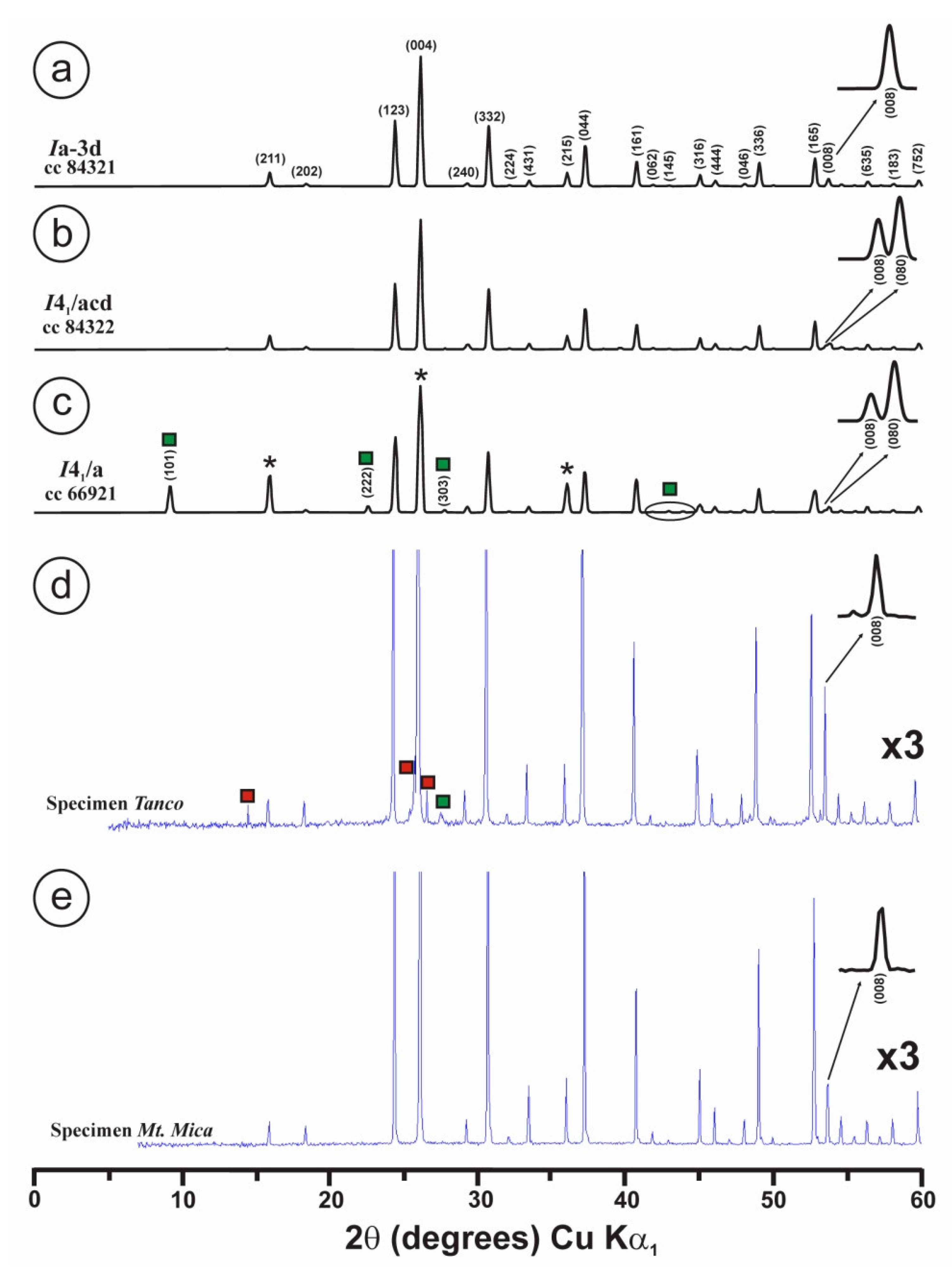
2.3. Powder X-ray Diffraction (XRD)
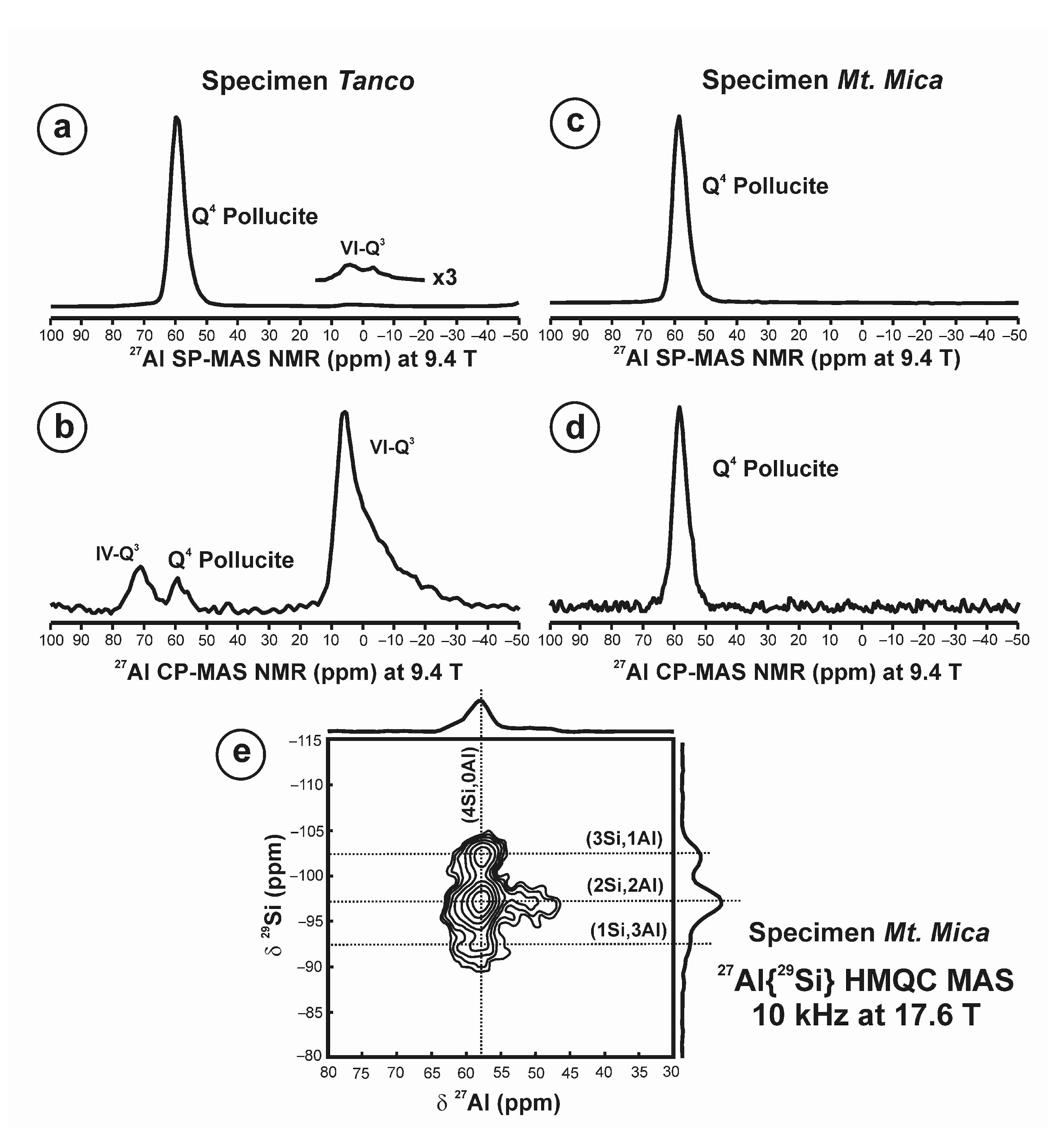
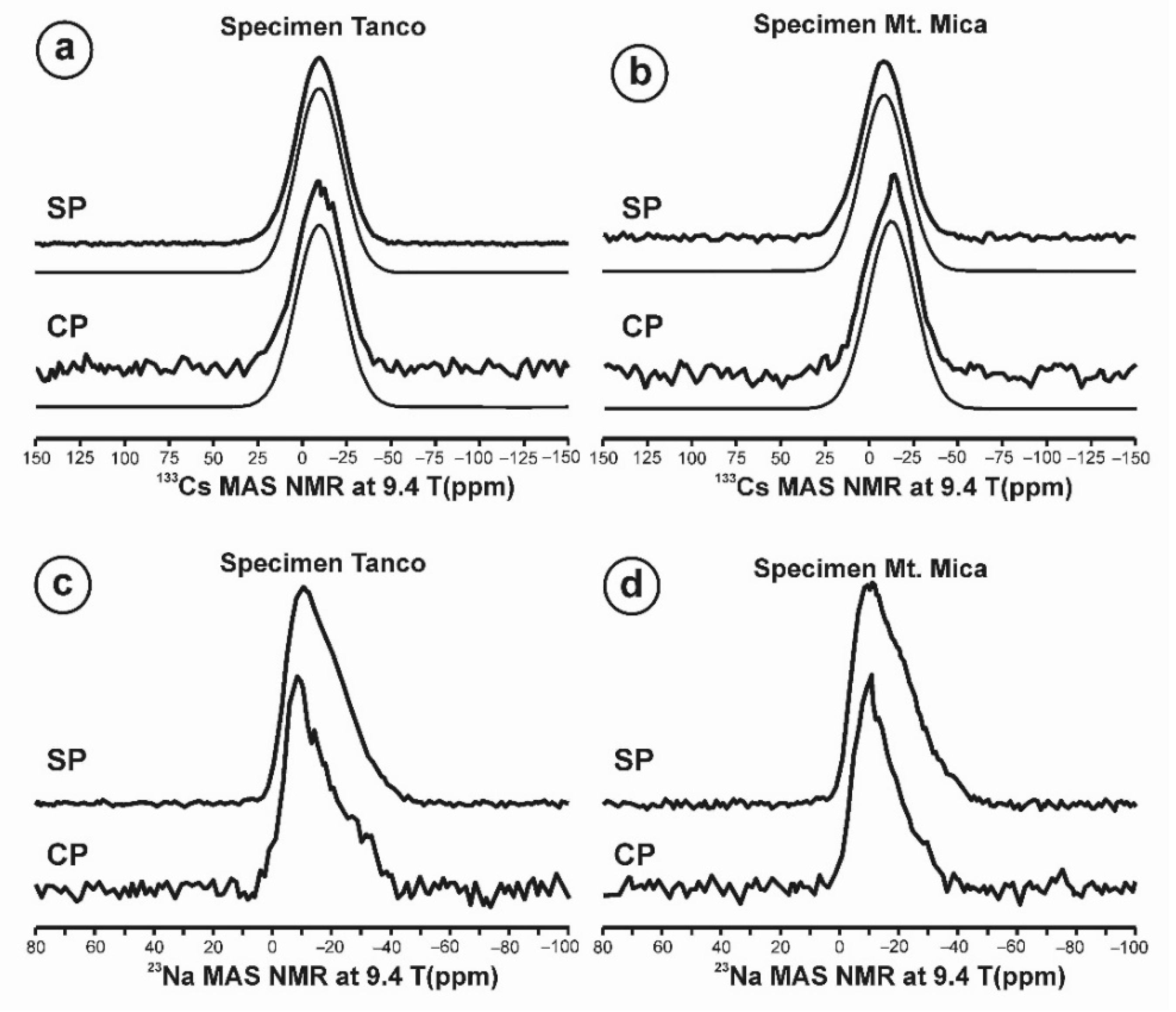
2.4. Nuclear Magnetic Resonance (NMR)
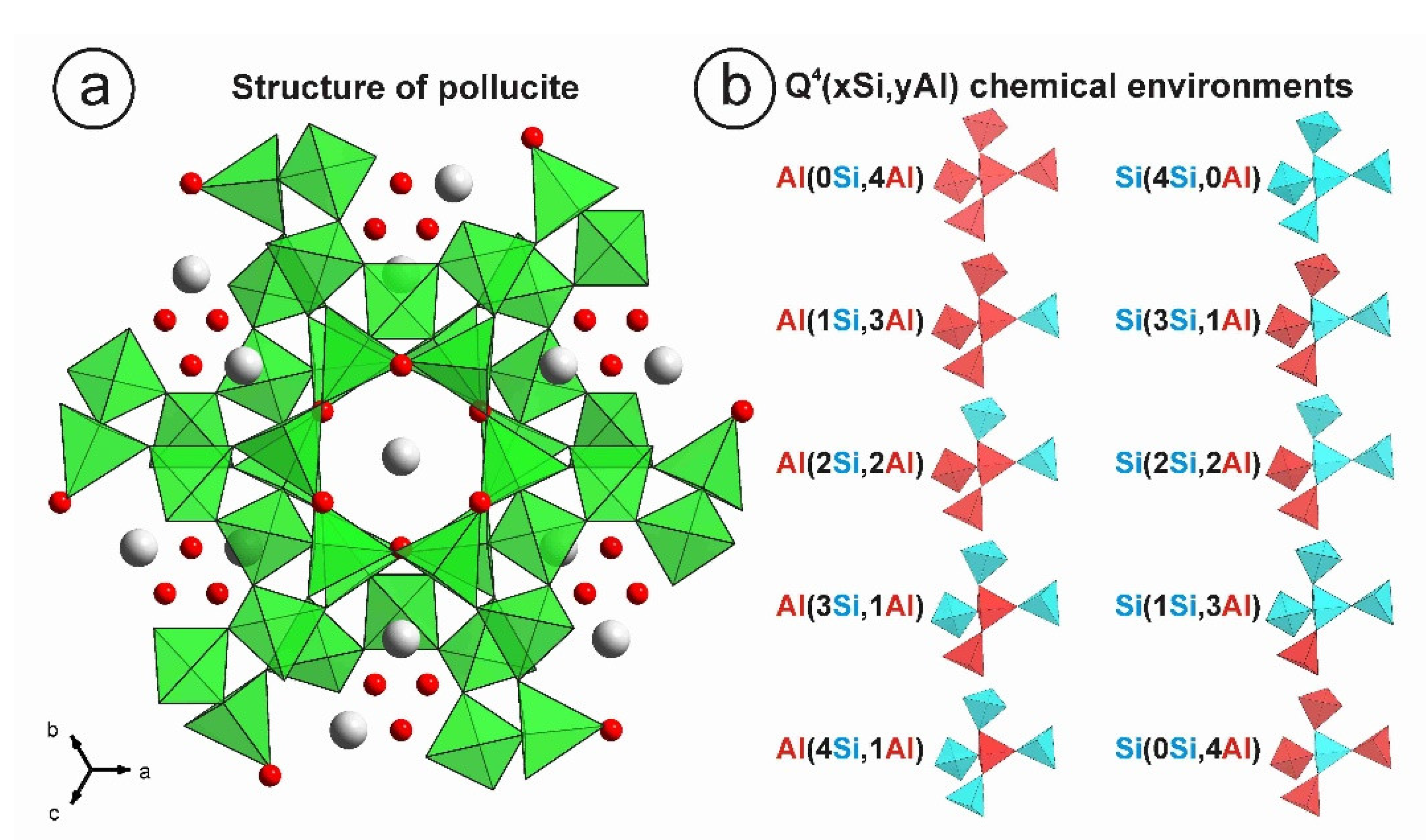
3. Results
3.1. Electron Microprobe Analysis (EMPA)
3.2. Powder XRD Patterns
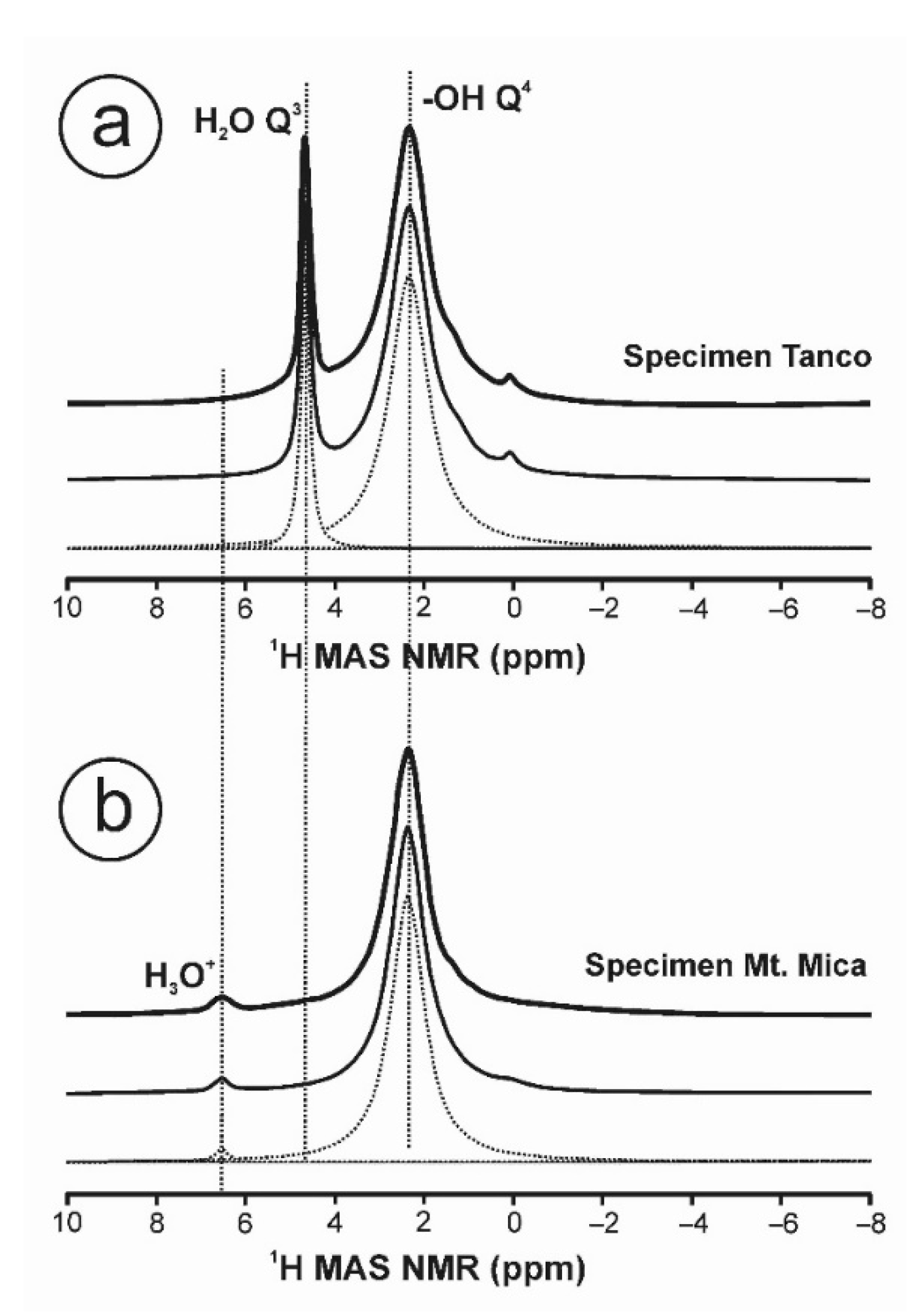
3.3. NMR Spectroscopy
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Coombs, D.S.; Alberti, A.; Armbruster, T.; Artioli, G.; Colella, C.; Galli, E.; Grice, J.D.; Liebau, F.; Mandarino, J.A.; Minato, H.; et al. Recommended nomenclature for zeolite minerals: Report of the Subcommittee on Zeolites of International Mineralogical Association, Commission on new minerals and minerals names. Can. Mineral. 1997, 35, 1571–1606. [Google Scholar]
- Náray-Szabó, S.V. Die struktur des pollucits CsAlSi2O6·xH2O. Z. Krist. 1938, 99, 277–282. [Google Scholar] [CrossRef]
- Taylor, W.H. The structure of analcime (NaAlSi2O6*H2O). In Zeitschrift Fuer Kristallographie, Kristallgeometrie, Kristallphysik, Kristallchemie; 1930; Volume 74, pp. 1–19. [Google Scholar]
- Newman, R.E. Crystal structure and optical properties of pollucite. Am. Mineral. 1967, 52, 1515–1518. [Google Scholar]
- Beger, R.M. The crystal structure and chemical composition of pollucite. Z. Krist. 1969, 129, 280–302. [Google Scholar] [CrossRef]
- Frank-Kamenetskaya, O.V.; Rozhdestvenskaya, I.V.; Bannova, I.I.; Kostitsyna, A.V.; Kaminskaya, T.N.; Gordienko, V.V. Dissymmetrization of crystal structures of sodium pollucites. Crystallogr. Rep. 1995, 40, 645–654. [Google Scholar]
- Palmer, D.C.; Dove, M.T.; Ibberson, R.M.; Powell, B.M. Structural behavior, crystal chemistry, and phase transitions in substituted leucite: High-resolution neutron powder diffraction studies. Am. Mineral. 1997, 82, 16–29. [Google Scholar] [CrossRef]
- Gatta, G.D. On the crystal structure and crystal chemistry of pollucite, (Cs,Na)16Al16Si32O96·nH2O: A natural microporous material of interest in nuclear technology. Am. Mineral. 2009, 94, 1560–1568. [Google Scholar] [CrossRef]
- Yanase, I.; Kobayashi, H.; Shibasaki, Y.; Mitamura, T. Tetragonal-to-cubic structural phase transition in pollucite by low-temperature X-ray powder diffraction. J. Am. Ceram. Soc. 1997, 80, 2693–2695. [Google Scholar] [CrossRef]
- Presser, V.; Klouzkova, A.; Mrazova, M.; Kohoutkova, M.; Berthold, C. Micro-Raman spectroscopy on analcime and pollucite in comparison to X-ray diffraction. J. Raman Spectrosc. 2008, 39, 587–592. [Google Scholar] [CrossRef]
- Fleischer, M.; Ksanda, C.J. Dehydration of pollucite. Am. Mineral. 1940, 25, 666–672. [Google Scholar]
- Ashbrook, S.E.; Whittle, K.R.; Le Pollès, L.; Farnan, I. Disorder and dynamics in pollucite from 133Cs and 27Al NMR. J. Am. Ceram. Soc. 2005, 88, 1575–1583. [Google Scholar] [CrossRef]
- Teertstra, D.K.; Sherriff, B.L.; Xu, Z.; Černý, P. MAS and DOR NMR study of Al-Si order in the analcime-pollucite series. Can. Mineral. 1994, 32, 69–80. [Google Scholar]
- Simon, A.; Köhler, J.; Keller, P.; Weitkamp, J.; Buchholz, A.; Hunger, M. Phase transformation of zeolites Cs,Na-Y and Cs,Na-X impregnated with cesium hydroxide. Microporous Mesoporous Mater. 2004, 68, 143–150. [Google Scholar] [CrossRef]
- Takaishi, T.; Yamazaki, S.; Yamazaki, A. Al–Si ordering in the framework of leucite and Cs-exchanged leucite. Phys. Chem. Chem. Phys. 2002, 4, 3078–3084. [Google Scholar] [CrossRef]
- Teertstra, D.K.; Lahti, S.I.; Alviola, R.; Černý, P. Pollucite and its alteration in Finnish pegmatites. Geol. Surv. Finl. Bull. 1993, 368, 15–22. [Google Scholar]
- Cerný, P.; Simpson, F.M. The Tanco Pegmatite at Bernic Lake, Manitoba; X. Pollucite. Can. Mineral. 1978, 16, 325–333. [Google Scholar]
- Cerný, P. Alteration of pollucite in some pegmatites of southeastern Manitoba. Can. Mineral. 1978, 16, 89–95. [Google Scholar]
- Magnenet, C.; Massiot, D.; Klur, I.; Coutures, J.P. Characterization of aluminosilicate on rutile surface by high resolution solid state NMR. J. Mater. Sci. 2000, 35, 115–121. [Google Scholar] [CrossRef]
- Florian, P.; Veron, E.; Green, T.F.G.; Yates, J.R.; Massiot, D. Elucidation of the Al/Si ordering in gehlenite Ca2Al2SiO7 by combined 29Si and 27Al NMR spectroscopy/quantum chemical calculations. Chem. Mater. 2012, 24, 4068–4079. [Google Scholar] [CrossRef]
- Massiot, D.; Fayon, F.; Capron, M.; King, I.; Le Calvé, S.; Alonso, B.; Durand, J.O.; Bujoli, B.; Gan, Z.H.; Hoatson, G. Modelling one and two-dimensional solid-state NMR spectra. Magn. Reson. Chem. 2002, 40, 70–76. [Google Scholar] [CrossRef]
- Lowenstein, W. The distribution of aluminum in the tetrahedral of silicat es and aluminates. Am. Mineral. 1954, 39, 92–96. [Google Scholar]
- Klinowski, J.; Ramdas, S.; Thomas, J.M. A re-examination of Si, Al ordering in zeolite NaX and NaY. J. Chem. Soc. Faraday Trans. 1982, 78, 1025–1050. [Google Scholar] [CrossRef]
- Sánchez-Muñoz, L.; Sanz, J.; Sobrados, J.; Gan, Z. Medium-range-order in disordered K-feldspars by multinuclear NMR. Am. Mineral. 2013, 98, 2115–2131. [Google Scholar] [CrossRef]
- Sánchez-Muñoz, L.; Sanz, J.; Sobrados, I.; Gan, Z.H.; Santos, J.I. Medium-range order in crystal structures of minerals. In Applications of NMR Spectroscopy in the Solid State; Consejo Superior de Investigaciones Científicas (CSIC): Madrid, Spain, 2019; Volume 47, pp. 103–131. [Google Scholar]
- Werner-Zwanziger, U.; Paterson, A.L.; Zwanziger, J.W. The Czjzek distribution in solid-state NMR: Scaling properties of central and satellite transitions. J. Non-Cryst. Solids 2020, 550, 120383. [Google Scholar] [CrossRef]
- Yesinowski, J.P.; Eckert, H.; Rossman, G.R. Characterization of hydrous species in minerals by high-speed 1H MAS-NMR. J. Am. Chem. 1988, 110, 1367–1375. [Google Scholar] [CrossRef]
- Xue, X.; Kanzaki, M. High-pressure δ-Al (OH)3 and δ-AlOOH phases and isostructural hydroxides/oxyhydroxides: New structural insights from high-resolution 1H and 27Al NMR. J. Phys. Chem. B 2007, 111, 13156–13166. [Google Scholar] [CrossRef]
- Xue, X.; Kanzaki, M. Proton Distributions and Hydrogen Bonding in Crystalline and Glassy Hydrous Silicates and Related Inorganic Materials: Insights from High-Resolution Solid-State Nuclear Magnetic Resonance Spectroscopy. J. Am. Ceram. Soc. 2009, 92, 2803–2830. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analysis Nº | 1 | 2 | 3 | 4 | 5 | Av | 6 | 7 | 8 | 9 |
|---|---|---|---|---|---|---|---|---|---|---|
| Specimen | Pollucite Tanco | Alterations/Veining | ||||||||
| SiO2 | 46.29 | 46.43 | 44.09 | 46.89 | 46.89 | 46.12 | 67.19 | 65.02 | 62.83 | 62.13 |
| Al2O3 | 17.42 | 17.41 | 17.49 | 17.09 | 17.37 | 17.36 | 28.29 | 28.29 | 18.68 | 17.78 |
| Na2O | 2.52 | 2.17 | 1.01 | 1.98 | 1.97 | 1.93 | 0.06 | 0.04 | 0.07 | 0.01 |
| K2O | - | - | - | - | - | - | 0.01 | 0.01 | 12.7 | 5.71 |
| Cs2O | 34.37 | 34.15 | 39.49 | 34.24 | 33.62 | 35.17 | 0.00 | 0.00 | 1.24 | 0.56 |
| Total | 100.60 | 100.16 | 102.08 | 100.20 | 99.85 | 100.58 | 95.53 | 93.36 | 95.52 | 86.19 |
| Si | 2.55 | 2.56 | 2.52 | 2.58 | 2.57 | 2.56 | 8.09 | 7.93 | 8.27 | 8.56 |
| Al | 1.13 | 1.13 | 1.18 | 1.11 | 1.12 | 1.13 | 3.87 | 4.07 | 2.9 | 2.89 |
| Na | 0.24 | 0.23 | 0.11 | 0.21 | 0.21 | 0.20 | 0.01 | 0.01 | 0.02 | 0.00 |
| Cs | 0.81 | 0.80 | 0.96 | 0.80 | 0.79 | 0.83 | 0.00 | 0.00 | 0.07 | 0.03 |
| K | - | - | - | - | - | - | - | 2.13 | 1.0 | |
| O | 6 | 6 | 6 | 6 | 6 | 6 | 18 | 18 | 18 | 18 |
| Si/Al | 2.26 | 2.27 | 2.14 | 2.32 | 2.29 | 2.26 | 2.09 | 1.95 | 2.85 | 2.96 |
| Analysis | 1 | 2 | 3 | Av. |
|---|---|---|---|---|
| Specimens | Pollucite Mt. Mica | |||
| SiO2 | 46.48 | 46.14 | 46.61 | 46.41 |
| Al2O3 | 16.42 | 17.43 | 17.61 | 17.17 |
| Na2O | 1.42 | 1.44 | 1.51 | 1.46 |
| Cs2O | 36.50 | 36.43 | 36.63 | 36.52 |
| K2O | 0.03 | 0.06 | 0.03 | 0.04 |
| Total | 100.85 | 101.5 | 102.36 | 101.60 |
| Si | 2.81 | 2.77 | 2.78 | 2.79 |
| Al | 1.17 | 1.23 | 1.23 | 1.21 |
| Na | 0.17 | 0.17 | 0.18 | 0.17 |
| Cs | 0.95 | 0.93 | 0.94 | 0.94 |
| O | 6 | 6 | 6 | 6 |
| Si/Al | 2.40 | 2.25 | 2.26 | 2.31 |
| RD | LD | Cs-Leucite [15] | Specimen Tanco | Specimen Mt. Mica | ||||
|---|---|---|---|---|---|---|---|---|
| 29Si SP-MAS | 29Si SP-MAS | 29Si SP-MAS | ||||||
| c.g. (ppm) | - | −97.9 | −98.4 | |||||
| Sites | A | A | δ | A | δ | A | δ | A |
| Q40 (4Si,0Al) | 22.67 | 9.83 | −104.4 | 3.8 | −107.6 | 5.9 | −107.9 | 6.4 |
| Q41 (3Si,1Al) | 40.74 | 30.91 | −100.0 | 22.1 | −102.6 | 31.9 | −103.3 | 31.0 |
| Q42 (2Si,2Al) | 27.47 | 36.47 | −95.5 | 42.7 | −97.8 | 50.8 | −98.3 | 50.9 |
| Q43 (1Si,3Al) | 8.22 | 19.08 | −91.3 | 27.7 | −93.2 | 11.4 | −93.5 | 11.7 |
| Q44 (0Si,4Al) | 0.92 | 3.75 | −87.8 | 3.7 | - | - | - | - |
| Specimen Tanco 29Si CP-MAS | ||||||
|---|---|---|---|---|---|---|
| 8 ms | 4 ms | 2 ms | ||||
| c.g. total spectrum (ppm) | −94.4 | −94.3 | −93.7 | |||
| c.g. pollucite (ppm) | −99.6 | −100.0 | −100.9 | |||
| Ratio Q3/Q4 | ~0.60 | ~0.72 | ~1.04 | |||
| Sites | δ | A | δ | A | δ | A |
| Q40 (4Si,0Al) | −107.3 | 6.3 | −107.0 | 4.6 | −107.3 | 16.4 |
| Q41 (3Si,1Al) | −102.5 | 39.2 | −102.6 | 42.1 | −102.1 | 38.7 |
| Q42 (2Si,2Al) | −97.6 | 48.2 | −97.9 | 50.1 | −97.5 | 44.9 |
| Q43 (1Si,3Al) | −91.9 | 6.3 | −91.9 | 4.1 | - | - |
| Q44 (0Si,4Al) | - | - | - | - | - | - |
| Q41/Q42 ratio | 0.81 | 0.84 | 0.86 | |||
| Specimen Mt. Mica 29Si CP-MAS | ||||||
|---|---|---|---|---|---|---|
| 8 ms | 2 ms | 1 ms | ||||
| c.g. (ppm) | −99.6 | −99.8 | −101.1 | |||
| Sites | δ | A | δ | A | δ | A |
| Q40 (4Si,0Al) | −107.5 | 7.8 | −107.1 | 8.7 | −107.4 | 14.6 |
| Q41 (3Si,1Al) | −102.3 | 36.5 | −102.1 | 38.8 | −102.6 | 40.6 |
| Q42 (2Si,2Al) | −97.5 | 47.9 | −97.7 | 46.1 | −97.7 | 44.8 |
| Q43 (1Si,3Al) | −92.4 | 7.8 | −92.5 | 6.5 | - | - |
| Q44 (0Si,4Al) | - | - | - | - | - | - |
| Q41/Q42 ratio | 0.76 | 0.84 | 0.91 | |||
| Specimen Tanco | Specimen Mt. Mica | |||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 27Al SP-MAS | 27Al CP-MAS | 27Al SP-MAS | 27Al CP-MAS | ||||
| Site | c.g. | A | c.g. | A | c.g. | A | c.g. | A |
| VI Q3 | 0.94 | 2.4 | −1.91 | 83.9 | - | - | - | - |
| IV Q3 | - | - | 71.15 | 10.2 | - | - | - | - |
| IV Q4 | 58.9 | 97.6 | 58.9 | 5.9 | 58.0 | 100.0 | 58.3 | 100.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Muñoz, L.; Santos, J.-I.; Simmons, W.B.; Florian, P. Local Structure and Protons in Non-Stoichiometric Pseudo-Cubic Pollucite Mineral by Multinuclear NMR. Minerals 2022, 12, 1181. https://doi.org/10.3390/min12101181
Sánchez-Muñoz L, Santos J-I, Simmons WB, Florian P. Local Structure and Protons in Non-Stoichiometric Pseudo-Cubic Pollucite Mineral by Multinuclear NMR. Minerals. 2022; 12(10):1181. https://doi.org/10.3390/min12101181
Chicago/Turabian StyleSánchez-Muñoz, Luis, José-Ignacio Santos, William B. Simmons, and Pierre Florian. 2022. "Local Structure and Protons in Non-Stoichiometric Pseudo-Cubic Pollucite Mineral by Multinuclear NMR" Minerals 12, no. 10: 1181. https://doi.org/10.3390/min12101181