
A Practical Procedure to Determine Natural Radionuclides in Solid Materials from Mining


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
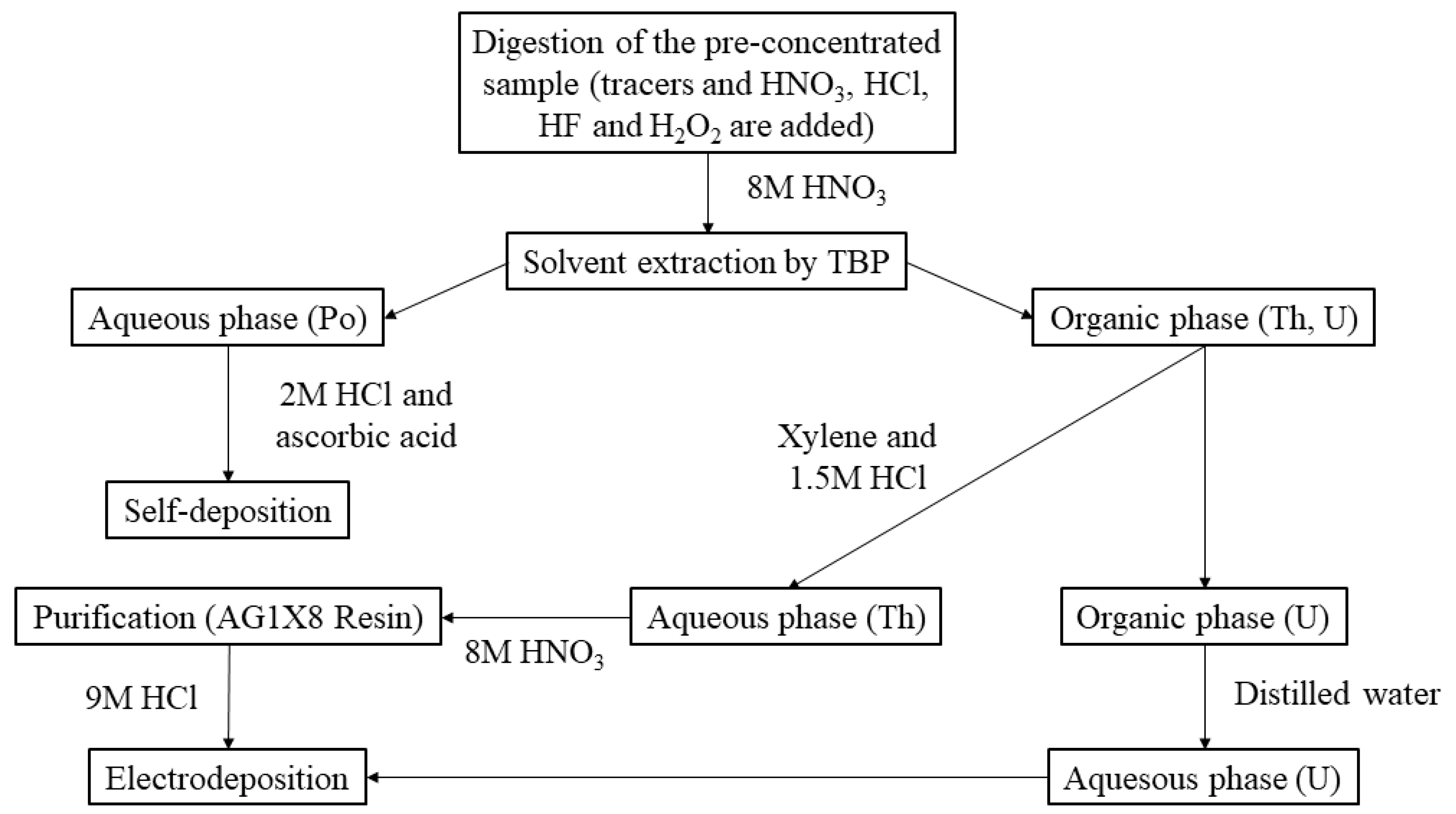
2.2.1. Gamma-ray Spectrometry
2.2.2. Alpha-Particle Spectrometry
3. Results and Discussion
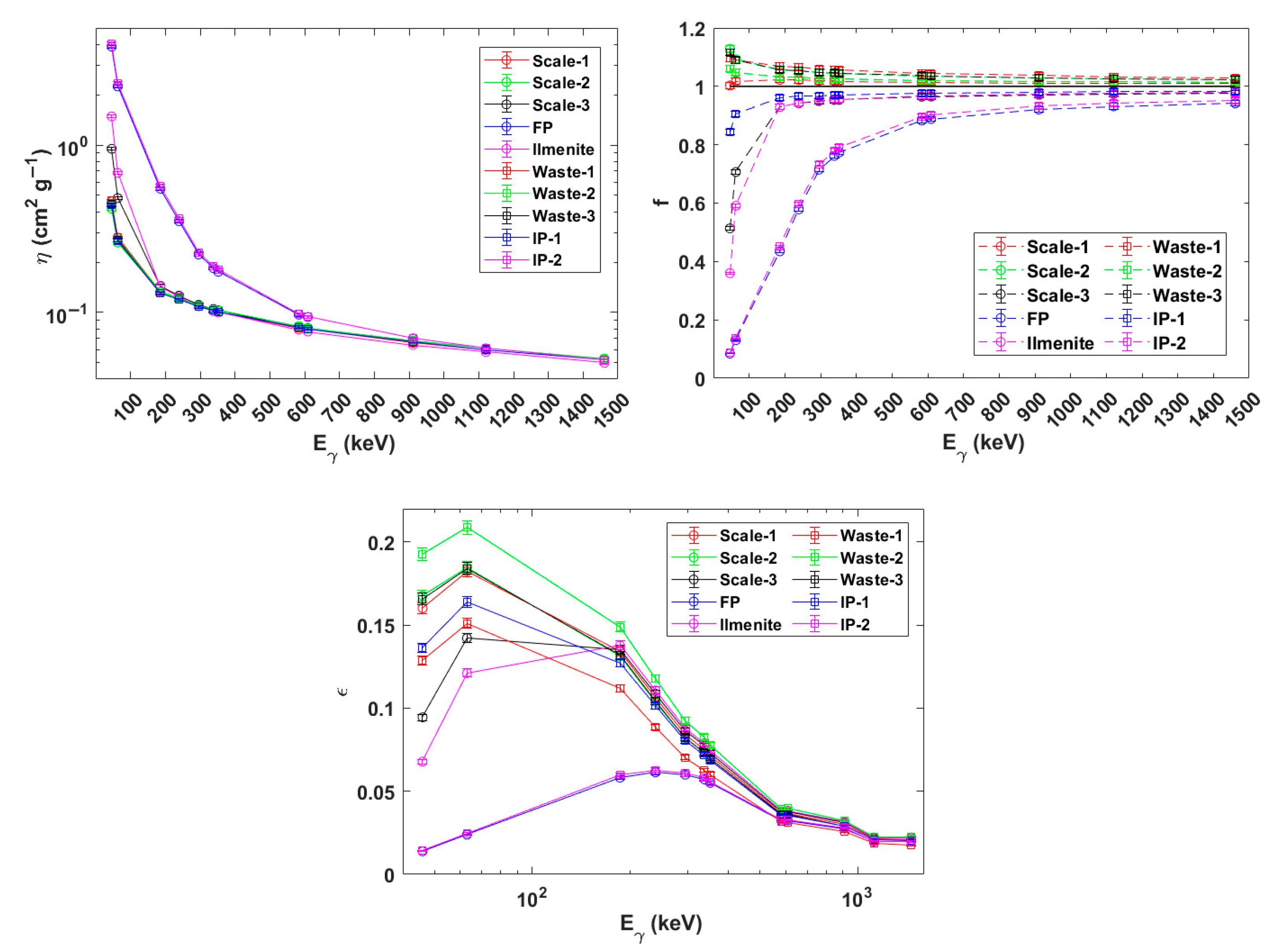
3.1. FEPE Obtention for Problem Samples
3.2. Determination of the Gamma-ray and Alpha-Particle Emitters: Obtention of the RI Index
3.3. Secular Disequilibria between Radionuclides Belonging to the 238U and 232Th Series
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Chambers, D.B. Radiological protection in North American naturally occurring radioactive material industries. Ann. ICRP 2015, 44, 202–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Commission. Council Directive 2013/59/Euratom of 5 December 2013. Laying down Basic Safety Standards for Protection against the Dangers Arising from Exposure to Ionising Radiation. 1983. Available online: https://ec.europa.eu/energy/sites/ener/files/rp_188.pdf (accessed on 5 February 2022).
- Ravisankar, R.; Vanasundari, K.; Chandrasekaran, A.; Rajalakshmi, A.; Suganya, M.; Vijayagopal, P.; Meenakshisundaram, V. Measurement of natural radioactivity in building materials of Namakkal, Tamil Nadu, India using gamma-ray spectrometry. Appl. Radiat. Isot. 2012, 70, 699–704. [Google Scholar] [CrossRef]
- Szabó, Z.; Völgyesi, P.; Nagy, H.É.; Szabó, C.; Kis, Z.; Csorba, O. Radioactivity of natural and artificial building materials—A comparative study. J. Environ. Radioact. 2013, 118, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Krieger, K. NORM contamination—Now you see it, now you don’t. Health Phys. 2005, 89, S20–S21. Available online: http://radonattahoe.com/TENORM.pdf (accessed on 15 February 2022). [CrossRef] [PubMed]
- Innocent, A.J.; Onimisi, M.Y.; Jonah, S.A. Evaluation of Naturally Occurring Radionuclide Materials in Soil Samples Collected From Some Mining Sites in Zamfara State, Nigeria. Br. J. Appl. Sci. Technol. 2013, 3, 684–692. [Google Scholar] [CrossRef]
- Odumo, O.B.; Mustapha, A.O.; Patel, J.P.; Angeyo, H.K. Radiological survey and assessment of associated activity concentration of the naturally occurring radioactive materials (NORM) in the Migori artisanal gold mining belt of southern Nyanza, Kenya. Appl. Radiat. Isot. 2011, 69, 912–916. [Google Scholar] [CrossRef] [PubMed]
- Michalik, B. NORM impacts on the environment: An approach to complete environmental risk assessment using the example of areas contaminated due to mining activity. Appl. Radiat. Isot. 2008, 66, 1661–1665. [Google Scholar] [CrossRef] [PubMed]
- Consejo de Seguridad Nuclear (CSN). Guide for the Control of the Exposure to Natural Radioactive Sources. Available online: https://www.csn.es/documents/10182/896572/GS%2011-02%20Control%20de%20la%20exposici%C3%B3n%20a%20fuentes%20naturales%20de%20radiaci%C3%B3n (accessed on 18 January 2022).
- IAEA. Preparation of Gamma-Ray Spectrometry Reference Materials RGU-1, RGTh-1 and RGK-1. Report-IAEA/RL/148, Vienna. 1987. Available online: https://nucleus.iaea.org/sites/ReferenceMaterials/Shared%20Documents/ReferenceMaterials/Radionuclides/IAEA-RGTh-1/rl_148.pdf (accessed on 7 January 2022).
- Barba-Lobo, A.; Mosqueda, F.; Bolívar, J.P. A general function for determining mass attenuation coefficients to correct self-absorption effects in samples measured by gamma spectrometry. Radiat. Phys. Chem. 2021, 179, 109247. [Google Scholar] [CrossRef]
- Barba-Lobo, A.; San Miguel, E.G.; Lozano, R.L.; Bolívar, J.P. A general methodology to determine natural radionuclides by well-type HPGe detectors. Measurement 2021, 181, 109561. [Google Scholar] [CrossRef]
- Barba-Lobo, A.; Mosqueda, F.; Bolívar, J.P. An upgraded lab-based method to determine natural γ-ray emitters in NORM samples by using Ge detectors. Measurement 2021, 186, 110153. [Google Scholar] [CrossRef]
- The Decay Data Evaluation Project. Available online: http://www.nucleide.org/DDEP_WG/DDEPdata.htm (accessed on 11 January 2022).
- Cutshall, N.H.; Larsen, I.L.; Olsen, C.R. Direct analysis of 210Pb in sediment samples: Self-absorption corrections. Nucl. Instrum. Methods Phys. Res. A 1983, 206, 309–312. [Google Scholar] [CrossRef]
- Agha, A.R.; El-Mongy, S.A.; Kandel, A.E. Assay of uranium isotopic ratios 234U/238U, 235U/238U in bottom sediment samples using destructive and non destructive techniques (Nasser Lake). In Proceedings of the Eighth Nuclear and Particle Physics Conference (NUPPAC-2011), Hurghada, Egypt, 20–24 November 2011; Volume 43, pp. 221–229. Available online: https://inis.iaea.org/search/search.aspx?orig_q=RN:43099476 (accessed on 3 February 2022).
- Holm, E.; Fukai, R. Method for multi-element alpha-spectrometry of actinides and its application to environmental radioactivity studies. Talanta 1977, 24, 659–664. [Google Scholar] [CrossRef]
- El-Daoushy, F.; Olsson, K.; Garcia-Tenorio, R. Accuracies in Po-210 determination for lead-210 dating. Hydrobiologia 1991, 214, 43–52. [Google Scholar] [CrossRef]
- Hurtado, S.; García-Tenorio, R.; García-León, M. 210Pb determination in lead shields for low-level γ-spectrometry applying two independent radiometric techniques. Nucl. Instrum. Methods Phys. Res. Sect. A 2003, 497, 381–388. [Google Scholar] [CrossRef]
- Lee, S.-H.; La Rosa, J.; Gastaud, J.; Povinec, P.P. The development of sequential separation methods for the analysis of actinides in sediments and biological materials using anion-exchange resins and extraction chromatography. J. Radioanal. Nucl. Chem. 2005, 263, 419–425. [Google Scholar] [CrossRef]
- AG1X8 Resin. Available online: https://www.bio-rad.com/es-es/sku/1401441-ag-1-x8-anion-exchange-resin-analytical-grade-100-ndash-200-mesh-chloride-form-500-g?ID=1401441 (accessed on 8 February 2022).
- Cuesta, E.; Barba-Lobo, A.; Lozano, R.L.; San Miguel, E.G.; Mosqueda, F.; Bolívar, J.P. A comparative study of alternative methods for 210Pb determination in environmental samples. Radiat. Phys. Chem. 2022, 191, 109840. [Google Scholar] [CrossRef]
- Kobashi, A.; Sato, J.; Saito, N. Radioactive Disequilibrium with Uranium, Thorium and Radium Isotopes Leached from Euxenite. Radiochim. Acta 1979, 26, 107–111. [Google Scholar] [CrossRef]
- Kratz, J.-V.; Heinrich, K. Nuclear and Radiochemistry: Fundamentals and Applications, 3rd ed.; Wiley: Hoboken, NJ, USA, 2013. [Google Scholar]
- Köhler, M.; Preuβe, W.; Gleisberg, B.; Schäfer, I.; Heinrich, T.; Knobus, B. Comparison of methods for the analysis of 226Ra in water samples. Appl. Radiat. Isot. 2002, 56, 387–392. [Google Scholar] [CrossRef]
- Ebaid, Y.Y.; El-Mongy, S.A.; Allam, K.A. 235U–γ emission contribution to the 186 keV energy transition of 226Ra in environmental samples activity calculations. Int. Congr. Ser. 2005, 1276, 409–411. [Google Scholar] [CrossRef]
- Völgyesi, P.; Kis, Z.; Szabó, Z.; Szabó, C. Using the 186-keV peak for 226Ra activity concentration determination in Hungarian coal-slag samples by gamma-ray spectroscopy. J. Radioanal. Nucl. Chem. 2014, 302, 375–383. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Radionuclide | All Materials | Sludges from Petroleum and Gas Industries |
|---|---|---|
| U-238 (sec) incl. U-235 (sec) | 0.5 | 5 |
| Natural U | 5 | 100 |
| Th-230 | 10 | 100 |
| Ra-226+ | 0.5 | 5 |
| Pb-210+ | 5 | 100 |
| Po-210 | 5 | 100 |
| U-235 (sec) | 1 | 10 |
| U-235+ | 5 | 50 |
| Pa-231 | 5 | 50 |
| Ac-227+ | 1 | 10 |
| Th-232 (sec) | 0.5 | 5 |
| Th-232 | 5 | 100 |
| Ra-228+ | 1 | 10 |
| Th-228+ | 0.5 | 5 |
| K-40 | 5 | 100 |
| ρ, Zi | Scale-1 | Scale-2 | Scale-3 | FP | Ilmenite | Waste-1 | Waste-2 | Waste-3 | IP-1 | IP-2 |
|---|---|---|---|---|---|---|---|---|---|---|
| ρ | 1.32 | 1.00 | 2.66 | 3.69 | 2.73 | 1.08 | 1.07 | 0.98 | 2.16 | 3.30 |
| O | 48.00 | 49.00 | 42.80 | 39.00 | 33.00 | 50.80 | 53.78 | 54.30 | 44.45 | 40.7 |
| Na | 2.00 | 3.00 | 0.59 | 3.59 | 2.00 | 0.59 | 1.69 | |||
| Mg | 6.00 | 9.00 | 0.62 | 3.62 | 5.00 | 0.62 | 1.50 | |||
| Al | 10.00 | 12.00 | 1.50 | 3.00 | 10.00 | 5.50 | 2.20 | |||
| Si | 14.20 | 9.00 | 30.27 | 25.68 | 10.00 | 32.68 | 32.40 | |||
| P | 0.70 | 0.90 | 0.08 | 2.75 | 0.70 | 0.08 | ||||
| S | 10.00 | 12.00 | 2.00 | 10.00 | ||||||
| K | 0.80 | 0.60 | 8.31 | 0.88 | 0.22 | 0.09 | 1.97 | |||
| Ca | 7.30 | 4.50 | 1.16 | 13.50 | 4.16 | 7.30 | 2.97 | 2.20 | 15.30 | |
| Ti | 0.45 | 30.00 | 0.45 | 0.45 | 0.50 | |||||
| Mn | 1.00 | 0.06 | 1.60 | 1.00 | 0.06 | 0.09 | ||||
| Fe | 10.67 | 37.00 | 1.47 | 2.67 | 13.00 | |||||
| Cu | 2.00 | |||||||||
| Ba | 1.00 | |||||||||
| W | 47.50 | 44.00 | ||||||||
| Pb | 0.50 |
| RN | Eγ, Eα (keV) | Scale-1 | Scale-2 | Scale-3 | FP | Ilmenite |
|---|---|---|---|---|---|---|
| a (Bq·kg−1) | a (Bq·kg−1) | a (Bq·kg−1) | a (Bq·kg−1) | a (Bq·kg−1) | ||
| 238U * | 4150 and 4200 | 4.7(8) | 2.9(9) | 8.5(1.9) | 1140(30) | 27(3) |
| 234Th | 63.29 | <18 | <15 | <18 | 1380(140) | 96(8) |
| 234U * | 4722.4 and 4774.6 | 6.3(3) | 4.4(4) | 9.4(1.8) | 1130(30) | 38(3) |
| 230Th * | 4620.5 and 4687.0 | <3 | <2 | 40(10) | 1170(50) | 88(4) |
| 226Ra | 185.96 (non-sec. eq.) | 8658(254) | 2962(95) | 43,725(1015) | 932(110) | 113(8) |
| 185.96 (sec. eq.) | 4906(336) | 1695(118) | 24,998(1015) | 1194(82) | 106(6) | |
| 226Ra(214Pb) | 295.22 | 8744(201) | 2965(69) | 50,973(2044) | 1256(29) | 112(5) |
| 351.93 | 8607(197) | 2911(67) | 52,635(2109) | 1291(30) | 111(5) | |
| 226Ra(214Bi) | 609.31 | 8770(203) | 2911(68) | 56,612(2272) | 1304(30) | 111(5) |
| 1120.29 | 8825(230) | 3021(84) | 58,996(2406) | 1340(35) | 136(9) | |
| 210Pb | 46.54 | 352(53) | 22(12) | <50 | 399(67) | 139(12) |
| 210Po * | 5330 | 644(18) | 38(3) | 1852(27) | 1440(120) | 65(6) |
| 232Th * | 3947.2 and 4012.3 | 0.4(1.4) | <1.5 | 14(6) | 34(6) | 357(11) |
| 228Ra(228Ac) | 338.42 | 3829(651) | 1997(340) | 30,719(1242) | 34(8) | 521(22) |
| 911.16 | 3884(105) | 2043(56) | 34,619(1398) | 27(3) | 511(21) | |
| 228Th(212Pb) | 238.63 | 3082(69) | 520(13) | 15,827(635) | 26.8(1.6) | 506(20) |
| 228Th(208Tl) | 583.19 | 3047(71) | 513(15) | 17,886(728) | 29(2) | 483(20) |
| 40K | 1460.83 | 251(60) | 183(30) | 2600(206) | 4(7) | 32(5) |
| RN | Eγ, Eα (keV) | Waste-1 | Waste-2 | Waste-3 | IP-1 | IP-2 |
|---|---|---|---|---|---|---|
| a (Bq·kg−1) | a (Bq·kg−1) | a (Bq·kg−1) | a (Bq·kg−1) | a (Bq·kg−1) | ||
| 238U * | 4150 and 4200 | 2.2(1.4) | 7.2(8) | 22,300(400) | 865(20) | 2510(60) |
| 234Th | 63.29 | <15 | <14 | 20,300(1500) | 1290(110) | 2210(190) |
| 234U * | 4722.4 and 4774.6 | 4.0(1.4) | 6.4(8) | 22,350(410) | 846(20) | 2560(60) |
| 230Th * | 4620.5 and 4687.0 | 9(3) | <1.5 | 179(9) | 895(22) | 2590(90) |
| 226Ra | 185.96 (non-sec. eq.) | 4117(96) | 1514(55) | 4216(1169) | 2675(120) | 1544(179) |
| 185.96 (sec. eq.) | 2356(96) | 863(61) | 10,226(689) | 2147(145) | 1989(136) | |
| 226Ra(214Pb) | 295.22 | 4065(163) | 1507(36) | 179(5) | 2488(57) | 2020(46) |
| 351.93 | 4002(161) | 1509(35) | 211(5) | 2546(58) | 2097(48) | |
| 226Ra(214Bi) | 609.31 | 4116(166) | 1536(37) | 116(4) | 2569(59) | 2113(49) |
| 1120.29 | 4172(173) | 1528(46) | 109(7) | 2665(67) | 2178(56) | |
| 210Pb | 46.54 | <15 | <20 | 216(14) | 704(21) | 1411(110) |
| 210Po * | 5330 | 32(1) | 3.7(8) | 52(4) | 682(21) | 1610(230) |
| 232Th * | 3947.2 and 4012.3 | 3.4(1.9) | <1.5 | 5(2) | 16(2) | 98(12) |
| 228Ra(228Ac) | 338.42 | 3074(125) | 1159(198) | 24(5) | 55(11) | 86(16) |
| 911.16 | 3017(123) | 1169(33) | 16(3) | 47(5) | 81(5) | |
| 228Th(212Pb) | 238.63 | 464(19) | 34(2) | 7.9(1.9) | 45.1(1.8) | 84(3) |
| 228Th(208Tl) | 583.19 | 448(20) | 34(6) | 10(3) | 47(4) | 80(4) |
| 40K | 1460.83 | 275(24) | 69(22) | 28(8) | 615(21) | 44(11) |
| Sample | RI |
|---|---|
| Soil | 0.250(15) |
| Scale-1 | 23(2) |
| Scale-2 | 7.8(5) |
| Scale-3 | 149(9) |
| FP | 3.7(2) |
| Ilmenite | 1.88(12) |
| Waste-1 | 1.05(7) |
| Waste-2 | 2.20(15) |
| Waste-3 | 2.20(11) |
| IP-1 | 6.0(3) |
| IP-2 | 6.0(3) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barba-Lobo, A.; Gázquez, M.J.; Bolívar, J.P. A Practical Procedure to Determine Natural Radionuclides in Solid Materials from Mining. Minerals 2022, 12, 611. https://doi.org/10.3390/min12050611
Barba-Lobo A, Gázquez MJ, Bolívar JP. A Practical Procedure to Determine Natural Radionuclides in Solid Materials from Mining. Minerals. 2022; 12(5):611. https://doi.org/10.3390/min12050611
Chicago/Turabian StyleBarba-Lobo, Alejandro, Manuel Jesús Gázquez, and Juan Pedro Bolívar. 2022. "A Practical Procedure to Determine Natural Radionuclides in Solid Materials from Mining" Minerals 12, no. 5: 611. https://doi.org/10.3390/min12050611