Abstract
The significant accumulation of Pb from anthropogenic activities threatens environmental ecosystems. In the environment, iron oxides are one of the main carriers of Pb. Thus, the redox cycling of iron oxides, which is due to biotic and abiotic pathways, and which leads to their dissolution or transformation, controls the fate of Pb. However, a knowledge gap exists on the bioreduction in Pb-bearing ferrihydrites, secondary-mineral precipitation, and Pb partitioning during the bioreduction/oxidation/bioreduction cycle. In this study, Pb-bearing ferrihydrite (Fh_Pb) with various Pb/(Fe+Pb) molar ratios (i.e., 0, 2, and 5%) were incubated with the iron-reducing bacterium Shewanella oneidensis MR-1 for 7 days, oxidized for 7 days (atmospheric O2), and bioreduced a second time for 7 days. Pb doping led to a drop in the rate and the extent of the reduction. Lepidocrocite (23–56%) and goethite (44–77%) formed during the first reduction period. Magnetite (72–84%) formed during the second reduction. The extremely-low-dissolved and bioavailable Pb concentrations were measured during the redox cycles, which indicates that the Pb significantly sorbed onto the minerals that were formed. Overall, this study highlights the influence of Pb and redox cycling on the bioreduction of Pb-bearing iron oxides, as well as on the nature of the secondary minerals that are formed.
1. Introduction
Anthropogenic activities have contributed to the release of significant lead (Pb) levels into ecosystems [1]; hence, more than 200 mg kg−1 of Pb is commonly measured in contaminated soils, while these concentrations are lower than 20 mg kg−1 in pristine soils [2,3,4,5,6,7]. The high concentrations of Pb in soils and groundwaters induces adverse effects, including the lower quality of food and land arability, and threatened human and ecosystem health. In this context, the United States Environmental Protection Agency (US EPA) recommends Pb concentrations below 15 µg L−1 in drinking water [8], while the World Health Organization (WHO) sets a maximum permissible Pb level at 100 µg g−1 in agricultural soils [9]. Therefore, understanding the processes that control the fate of Pb in the environment seems to be a requirement for the environment and, overall, for environmental sciences.
Physicochemical processes largely govern the speciation of Pb in the environment. Pb can precipitate as poorly soluble compounds, and it can be complexed by organic matter and/or adsorbed onto carbonates and clays, or onto Mn and Fe oxides [10,11,12,13]. Fe oxides account for up to 5% in the weight of specific fractions of surface soils and sediments, and they exert significant control on the reactivity, speciation, and mobilization of metals and metalloids, such as Pb, Ni, Co, As, and Sb [14,15,16,17,18]. This is due to their high surface areas and the reactive surfaces that they display, which confer significant sorption capacities for organic and inorganic species. Pb interacts with the (hydroxyl) functional groups on the surfaces of Fe oxides [19]. This may lead to the surface adsorption (via complexation), or to the structural incorporation, of the metals into the iron oxide crystal, and/or physical isolation within nanosized pores of iron oxide aggregates [20,21,22,23]. Thus, Pb sequestration by iron (oxyhydr)oxides is one of the most critical geochemical processes that controls the environmental fate, transport, and bioavailability of Pb [24].
Iron oxides undergo redox cycling (biotic and abiotic), depending on the environment’s occurring conditions [25]. The bioreduction of iron oxides results mostly from their use as terminal electron acceptors in several anaerobic subsurface environments by dissimilatory iron-reducing bacteria (DIRB), such as Geobacter sp. and Shewanella sp [26]. These phylogenetically diverse bacteria can couple the oxidation of an organic (e.g., formate, pyruvate, lactate) or inorganic (e.g., H2) electron source to the reduction in Fe(III). Hence, the Fe(II) that is generated is either liberated in solution and/or adsorbed onto mineral surfaces, and it can lead to the precipitation of magnetite (Fe3O4), goethite (α-FeOOH), siderite (FeCO3), or other biogenic minerals [27,28,29,30,31]. These minerals may also form following the abiotic reduction of iron oxides by organic compounds and iron–sulfur minerals (e.g., mackinawite, FeS) [25,32]. On the other hand, the biotic oxidation of iron oxides is catalyzed by diverse bacteria by using O2, NO3, or dissolved Fe(II) species, and Fe(II)-bearing minerals as terminal electron acceptors, such as Gallionella sp. [25,33], while the abiotic oxidation of iron oxides is mainly a function of the pH, O2, and Fe contents [25,34]. Both these chemical and microbial oxidations of iron oxides occur at comparable rates at a circumneutral pH [25]. Given the insolubility of Fe(III) under such conditions, these oxidations result in the formation of Fe(III) minerals. Concomitantly, metals (such as Pb) that are adsorbed onto or coprecipitated with Fe oxides could either be released in solution or partitioned on the solid phases [35,36].
Relatively little is known with regard to the effect of Pb on the microbiologically mediated and abiotic Fe redox reactions, the precipitation of biogenic minerals, and the subsequent redistribution of Pb. To broaden our understanding of Fe and Pb biogeochemical cycles, our study aims to investigate how various Pb concentrations impact the bioreduction and abiotic oxidation of iron oxides and the precipitation of biogenic minerals. To this end, we twice incubated Pb-bearing ferrihydrites with variable molar ratios in the presence of Shewanella oneidensis MR-1, with an intermediary atmospheric O2 oxidation. With this experimental setup, we aimed to study the changes in: (i) the rate and extent of the ferrihydrite bioreduction; (ii) the redistribution of Pb among the liquid and solid phases; and (iii) the nature of the minerals that were formed upon the (a)biotic redox cycling of Fe oxides.
2. Materials and Methods
All the reagents and acids used were of analytical grade and were purchased from Sigma–Aldrich (St. Louis, MO, USA). The solutions were prepared by using 18.2 MΩ cm−1 deionized water. The anaerobic experiments and sample collection were carried out in an anoxic chamber (Coy Laboratory Products Inc., Grass Lake, MI, USA), where a 98% N2 + 2% H2 atmosphere was maintained by the automatized injection of the gas mixture. Trace amounts of oxygen were scavenged by palladium catalysts.
Pb(II)-bearing ferrihydrite synthesis. Ferrihydrite was prepared by the neutralization of 0.4 M FeCl3 (made with 13 g FeCl3 in 200 mL) with 4 M of NaOH (made with 16 g NaOH in 100 mL). For the synthesis of the Pb-bearing ferrihydrites, PbCl2 (8 to 20 mM PbCl2 solutions made with 0.4 to 1.1 g PbCl2 in 200 mL) was added to the ferric salt, before neutralization, at Pb/(Fe+Pb) molar ratios of 0.02 and 0.05 [37]. Hereafter, these samples will be named Fh, Fh_Pb 0.02, and Fh_Pb 0.05 (or the Fh_Pb series). The precipitates obtained were washed free of electrolytes by dialysis (1 nm-pore-size Spectra/Por® 7 dialysis membranes) by using 18.2 MΩ cm−1 deionized water until a steady conductivity <20 µS cm−1 was obtained.
Bioreduction of Pb(II)-bearing ferrihydrites and abiotic oxidation. Shewanella oneidensis MR-1 cells were streaked under aerobic conditions on tryptic soy agar from −80 °C glycerol stocks. They were subcultured once, and the colonies were used to prepare a suspension to inoculate 400 mL of trypticase soy broth (TSB). Cells were grown aerobically to the exponential growth phase in TSB, under agitation, and at 180 rpm and 28 °C. Then, the cells were harvested by centrifugation, washed twice, and concentrated in sterile 0.9% NaCl (70 mL). The bioreduction assays were performed in a mineral medium composed of 22 mM NH4Cl; 1.2 mM KCl; 0.67 mM CaCl2; 1.1 mM MgSO4·7H2O; 1.5 mM NaCl; 0.27 mM MnSO4·H2O; 86 µM ZnCl2; 32.3 µM FeSO4·7H2O; 38.2 µM CoSO4·7H2O; 0.71 mM nitrilotriacetic acid; 9.3 µM Na2MoO4·2H2O; 6.8 µM Na2WO4·2H2O; 9.1 µM NiCl2; 3.6 µM CuSO4·5H2O; 1.9 µM AlK(SO4)2·12H2O; and 1.5 µM H3BO3. This mineral medium was heat-sterilized, purged with filter-sterilized N2, and dispensed into sterile 100 mL serum bottles, in the presence of the Fh and Fh_Pb series (0.5 g Fh), and was crimp-sealed with butyl rubber stoppers. Sodium formate, as the sole electron source, and anthraquinone-2,6-disulfonate (AQDS), as an electron shuttle [38], were sterilized by 0.2 µm filtration, purged with filter-sterilized N2, and introduced in the culture medium under aseptic conditions in order to achieve 35 mM and 100 µM, respectively, in each 80 mL assay. S. oneidensis MR-1 cell suspensions were added to obtain final concentrations of 108 cell mL−1. The initial pH after mixing all the components was adjusted to ca. 7. The assays were incubated in the dark for seven days, in triplicates, at 28 °C. Hereafter, this reduction phase is named Reduction Phase 1 (RP1). All the bottles were then open to laboratory air. They were agitated in the dark at 180 rpm for seven days for oxidation, prior to being switched to anoxic conditions for another cycle of bioreduction with the addition of S. oneidensis MR-1 cells producing the same initial concentration (108 cell mL−1) for seven days. This step is designated Reduction Phase 2 (RP2). The control experiments were cell-free and were otherwise identical to the biotic assays.
Chemical analyses. The total Fe and Pb concentrations in the initial solids were quantified by using atomic absorption spectroscopy (AAS) after dissolution in 4 M HCl. The AAS limits of quantification were 18 µM Fe and 0.5 µM Pb. At each time point, samples were removed from the bioreduction assay and extracted with 4 M HCl to measure the total Fe (Fetot) and the total Fe(II) (Fe(II)tot) by using the ferrozine method [39]. The limit of quantification was 2 µM. The apparent reduction rate was computed from the first derivative of a nonlinear curve fit (Equation (1)):
where %(Fe(II)tot/Fetot)t is the (Fe(II)tot/Fetot) (%) produced at time t; %(Fe(II)tot/Fetot)max is the maximum %(Fe(II)tot/Fetot) observed at the end of the reduction period (i.e., 7 days); and kobs is the pseudo first-order constant. At the end of the incubation period, 1 mL was withdrawn from the assays, filtered through 0.22 µm pore filters, and acidified in 2 M HCl to measure the Pb in the aqueous phase by AAS.
%(Fe(II)tot/Fetot)t = %Fe(II)tot/Fetot)max × (1 − e−kobs × t)
Culture of biosensors and bioavailable Pb analysis with luminescence measurement. Pb luminescent biosensor cells (E. coli TOP10 pSEVA226 Znt-lux and E. coli TOP10 pSEVA226 J105) were grown overnight under aerobic conditions in a liquid Luria–Bertani (LB) broth, and were supplemented with kanamycin (50 µg mL−1) from −80 °C glycerol stocks. The cells were then harvested by centrifugation, LB-washed, resuspended in a poor metal-complexing medium (GGM adjusted to pH 6.7 with 0.1 M NaOH, Table S1) until DO600nm = 0.2, and stored at 4 °C. For luminescence measurements, 20 µL Pb standard solutions or 0.2 µm filtrated environmental samples, and 180 µL of biosensor cell suspensions, were pipetted into 96-well microplates. Following incubation with orbital shaking (1.8 mm of amplitude and 10 Hz of frequency), luminescence (relative light unit measured as a 10-s integral) and DO600nm were recorded every 20 min for 2 h by using a monochromator spectrofluorometer FLX-Xenius (SAFAS, Monaco). Induction coefficients (ICs) (normalized by the DO600nm) were calculated as follows (Equation (2)):
where RLU is the luminescence value of the sample, and RLUcontrol is the luminescence value of the cells in the solution that contain no inducer (blank). The standard solutions used are presented in SI (Table S2). The limit of quantification was 85 nM Pb.
Characterization of the solid phase. Powder X-ray diffraction (XRD) was performed by using a D8 Bruker diffractometer equipped with a monochromator, a position-sensitive detector, and a Co anode (Kα1 = 0.178897 nm radiation). The diffractograms were recorded in the 3–80° 2θ range, with a 0.03° 2θ step size and a collecting time of 3 s per point, by using sample holders equipped with glass domes, which were filled under a glovebox to maintain the samples under a N2 atmosphere (for reduced samples only). Phase identification was performed by using the software Diffrac Eva and the Crystallography Open Database (COD).
57Fe Mössbauer spectroscopy. Transmission Mössbauer spectroscopy was performed between 298 and 11 K, with a constant-acceleration Mössbauer spectrometer, a 512 multichannel analyzer (Halder Electronic Gmbh, Seehausen, Germany), and a 50 mCi source of 57Co in Rh, maintained at room temperature (RT). Data were obtained from appropriate amounts (10 mg of Fe per cm2) of solid samples to obtain optimal experimental conditions. Analyses were carried out between ambient and 11 K on the ARS cryostat (Advanced Research Systems, Macungie, PA, USA) equipped with a vibration-isolation stand developed in the LCPME laboratory, while the recordings at 4 K were taken on the spectrometer coupled to the Janis Cryostat. Mössbauer spectra were collected in transmission mode. The 50 mCi source of 57Co in the Rh matrix was maintained at room temperature and was mounted at the end of a Mössbauer velocity transducer. The spectrometers were calibrated with a 25 μm foil of α-Fe at RT. Spectra were computer-fitted (Recoil Software, Ottawa University, Ottawa, ON, Canada) with the Voigt-based fitting model for paramagnetic sites that correspond to doublet, which have distributions of quadrupole splittings (QSDs), and for magnetic sites that correspond to sextet, which have a distribution of magnetic hyperfine fields (HFDs), as implemented in the Recoil software.
3. Results
3.1. Pb-Bearing Ferrihydrite Characterization
The concentrations of Fe and Pb within the synthesized Fh_Pb series were measured by using AAS. The concentrations that were obtained were approximately equal to the Pb/(Fe+Pb) molar ratios that were used during the syntheses (i.e., 2 and 5%). The X-ray diffraction patterns obtained for the coprecipitates displayed two broad peaks at 2θ around 41 and 75°, and d-spacings of ca. 0.258 and 0.149 nm, respectively (data not shown). Such peaks are representative of pristine and metal-substituted 2-line ferrihydrite (for example, Sb-containing ferrihydrite) [18,37]. No other ferric phase was obtained, according to the XRD analysis that we performed.
3.2. Bioreduction of the Fh_Pb Series and Concentrations in Solution
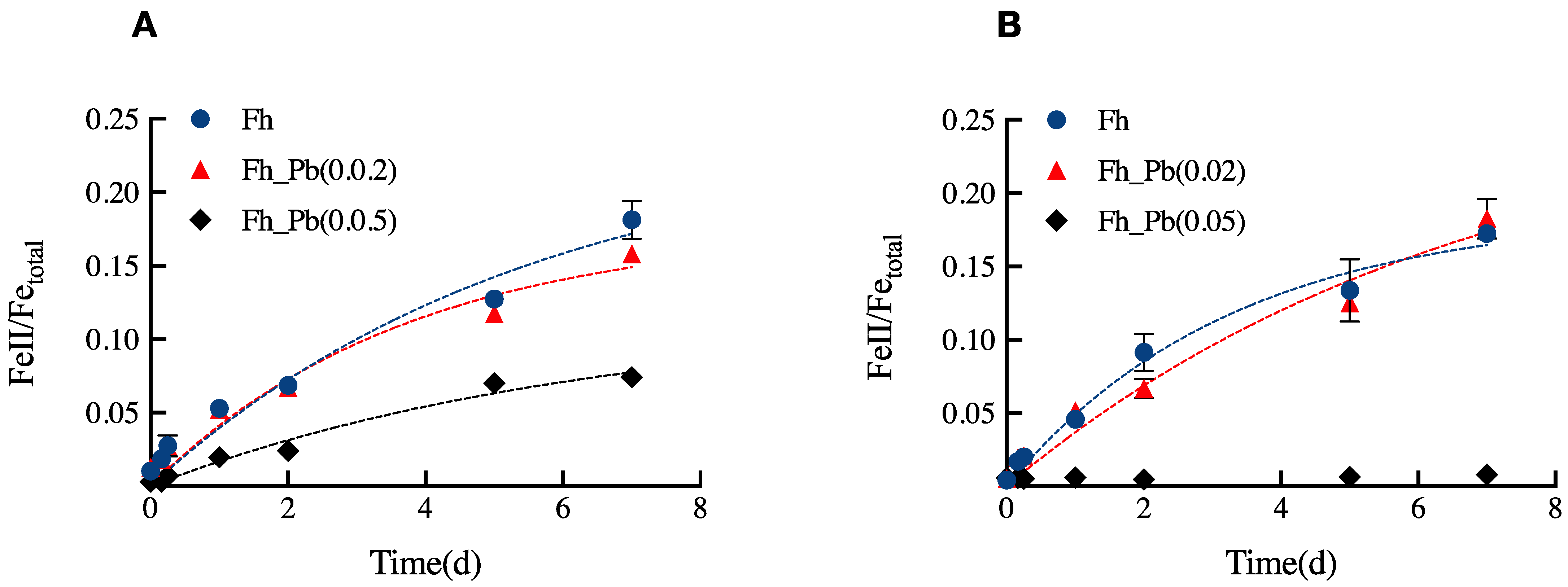
We quantitatively monitored the Fe(III) reduction in the Pb-bearing Fh series upon the two bioreduction periods of 7 days (Figure 1). No measurable Fe(III) reduction took place in the control assays.
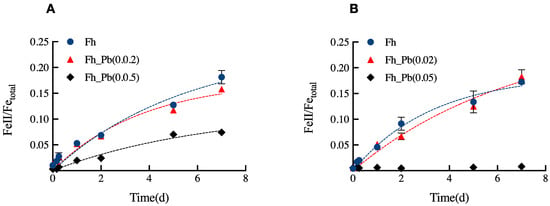
Figure 1.
Concentrations of Fe(II) ions formed in the Fh, Fh_Pb(0.02), and Fh_Pb(0.05) systems during: (A) the RP1, and (B) the RP2 with Shewanella oneidensis MR-1. Error bars indicate the standard deviations of triplicate microbial-reduction assays.
Pb did not inhibit the Fe(III) reduction in both RPs; however, it affected the rate and the extent of the reduction (Figure 1). During RP1, the apparent rate of reduction was of 0.04 ± 0.01 (Fe(II)tot/Fetot) day−1 for assays that were run with Fh and Fh_Pb(0.02), while it decreased to 0.018 ± 0.01 (Fe(II)tot/Fetot) day−1 for the assays that were run with Fh_Pb(0.05) (Figure 1A). The reduction extents were around 18, 16, and 7%, respectively, in the Fh, Fh_Pb(0.02), and Fh_Pb(0.05) systems (Figure 1A).
During RP2, the apparent rates of reduction were of 0.06 ± 0.01 and 0.04 ± 0.01 (Fe(II)tot/Fetot) day−1, respectively, for the Fh and Fh_Pb(0.02) systems. The reduction extent reached around 17 and 18%, respectively, in the pristine Fh and Fh_Pb(0.02) systems. On the contrary, no reduction was determined in the assays that were run with Fh_Pb(0.05), as Fe(II) quantification was below the detection limit, if any occurred in the system (Figure 1B). The concentrations of Fe(II) ions that were measured in solution during the RP1 and RP2 phases were inferior to the ferrozine quantification limits in all of the systems. No reduction was observed in the blank assays (data not shown).
Both the total and bioavailable concentrations of Pb were measured at the end of the RPs, and the oxidation periods were inferior to the measurement methods’ quantification limits (0.5 µM for AAS, and 85 nM for the luminescent biosensors, which represent less than 0.038 and 0.007% Pb, respectively).
3.3. Biogenic-Mineral Characterization
3.3.1. Reduction Phase 1 (RP1)
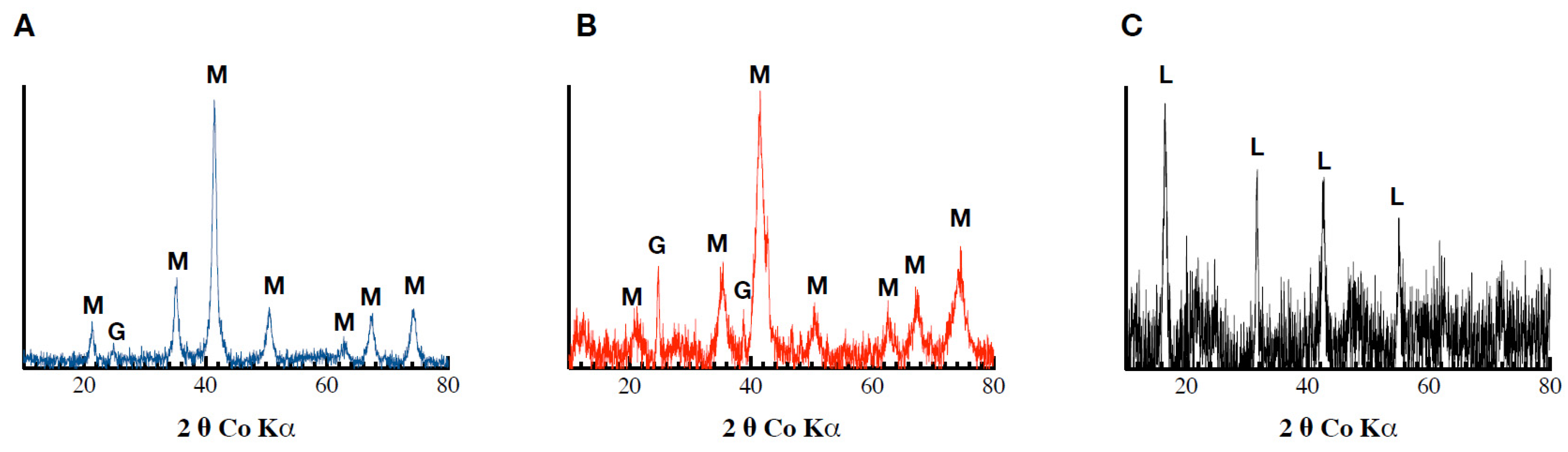
The biogenic minerals that were formed within the Fh, Fh_Pb(0.02), and Fh_Pb(0.05) systems during RP1 displayed X-ray diffraction peaks that had reflections that were characteristic of lepidocrocite and goethite (Figure 2). The low signal-to-noise ratio and the broadening of the peaks in the XRD pattern indicate the low crystallinity of the precipitated phases, or a decrease in the crystallite size. No mineralogical changes were observed in the control assays (data not shown).
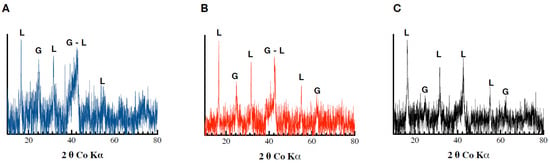
Figure 2.
Diffractograms obtained following Co-XRD measurements of the (A) Fh, (B) Fh_Pb(0.02), and (C) Fh_Pb(0.05) systems after the RP1 with Shewanella oneidensis MR-1.
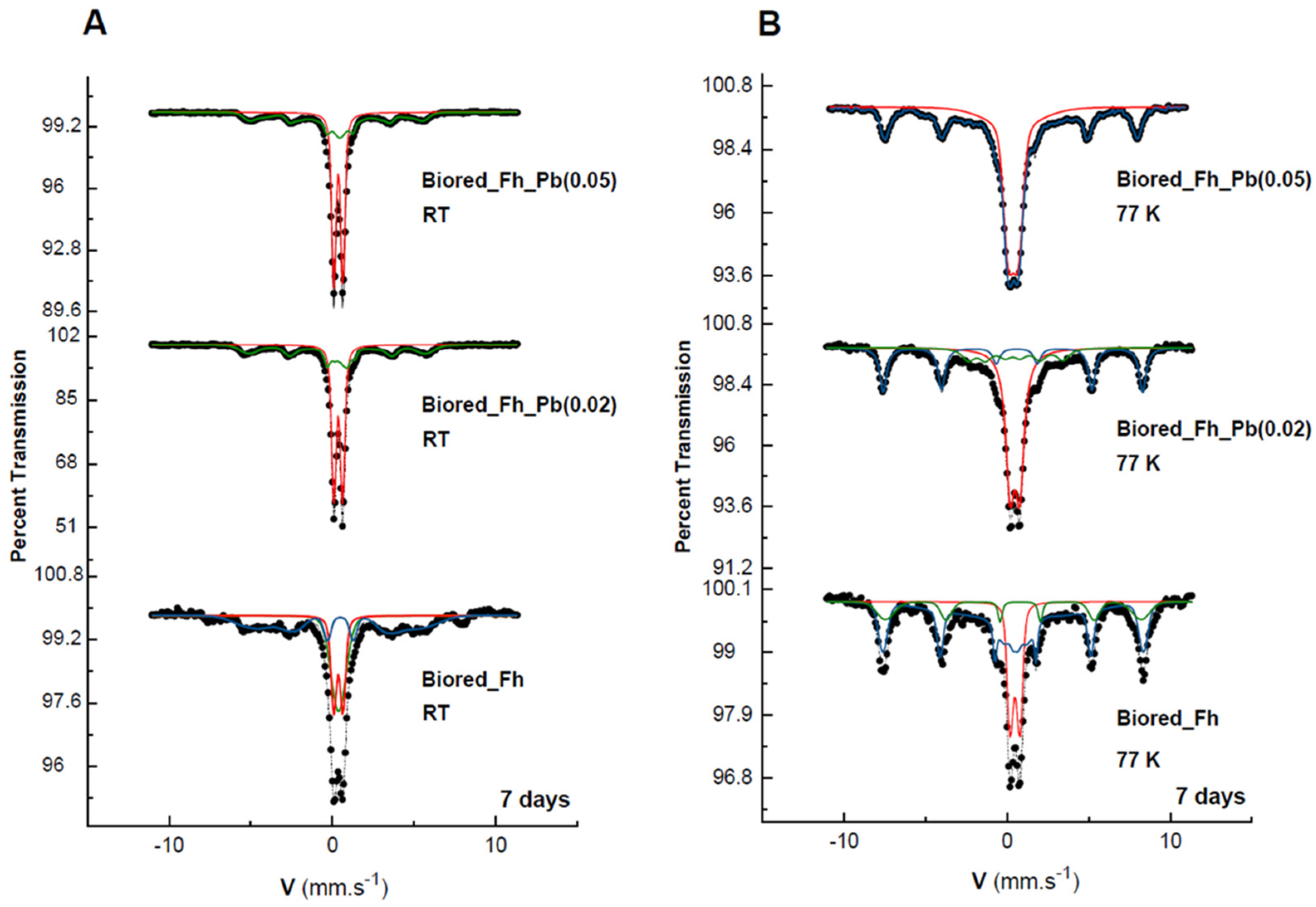
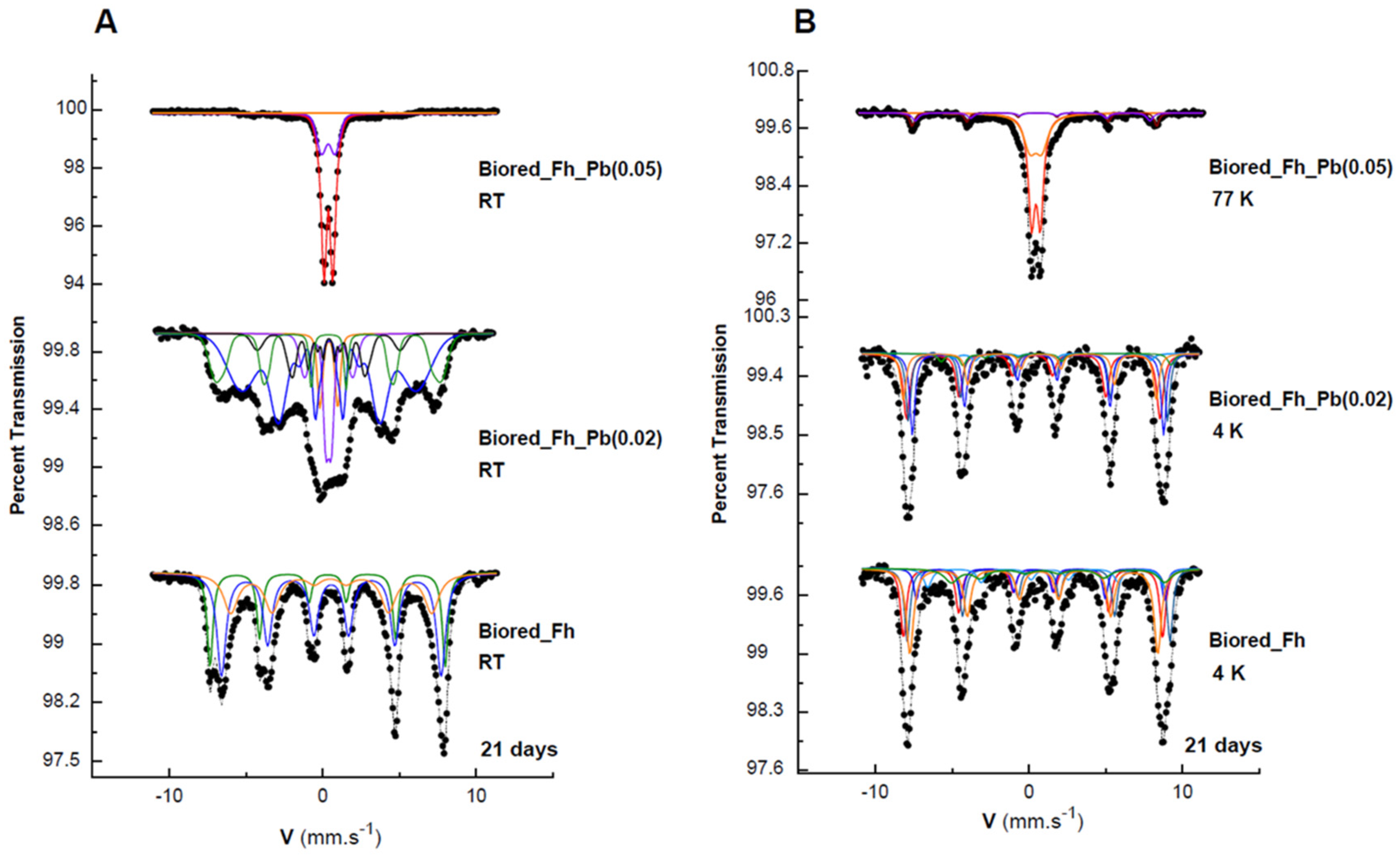
All of the solid biogenic minerals that precipitated from the Fh, Fh_Pb(0.02), and Fh_Pb(0.05) bioreductions were characterized by Mössbauer spectroscopy (Figure 3, Table 1). After RP1, irrespective of the Pb/(Fe+Pb) molar ratio of the initial Fhs, the diffractograms exhibited a mixture of lepidocrocite and goethite. The Mössbauer spectra were measured at 298 and 77 K (Figure 3A,B, respectively), where this last temperature could be necessary to strengthen the identification. Indeed, 77 K is the temperature between those of the magnetic ordering of the two phases so as to distinguish them unambiguously. Nevertheless, one must remain vigilant to the superparamagnetic behavior of the phase-size effect, which can also be a good indication as a signature for the presence of one phase or the other. The Mössbauer spectra of the precipitates that were sampled during the bioreduction of ferrihydrite without Pb (Fh) exhibited one first doublet with CS = 0.37 mm s−1 at room temperature and 0.47 mm s−1 at 77 K, which is typical of lepidocrocite that corresponds to a relative abundance of 23% [40]. The spectrum at 298 K shows that another doublet overlaps that of lepidocrocite, but with larger quadrupole splitting (Δ = 0.67 mm s−1 versus 0.57 mm s−1) and a very broad magnetic sextet with a low hyperfine field. This doublet resembles that of many other paramagnetic Fe3+-bearing species, and it is therefore not applicable for the identification of any mineral. On the other hand, the broad sextet is attributable to goethite, which is usually recognized by its relatively low H (hyperfine field) and its asymmetrically broadened lines. By lowering the temperature at 77 K, the doublet with large Δ = 0.67 mm s−1 disappeared, and we observe the appearance of both sextets, which are better resolved than those obtained at room temperature, but with different hyperfine fields. The transformation during cooling (298 to 77 K) from the doublet with large Δ to the broad magnetic component that has a small hyperfine field corresponds to a part of the goethite population that is in a superparamagnetic phase (nanocrystalline phase). The second sextet, with a larger field (H = 488 kOe), is attributable to a microcrystalline goethite, which indicates the nonuniformity of the particle size within the goethite particles. This distributive behavior, and the reduction in the H, are explained by different effects, such as magnetic collective excitations [41] and magnetic-exchange adjacent particles [42]. The average H depends on the particle size, and it diminishes strongly with increasing temperature.
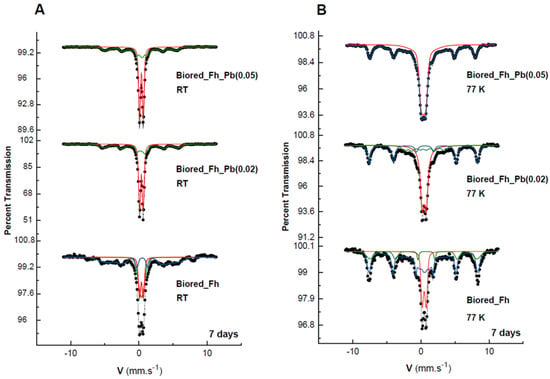
Figure 3.
Mössbauer spectra of the secondary products obtained in the Fh, Fh_Pb(0.02), and Fh_Pb(0.05) systems at: (A) room temperature (RT), and (B) 77 K after the RP1 with Shewanella oneidensis MR-1. Hyperfine parameters that correspond to these spectra are presented in Table 1.

Table 1.
Mössbauer hyperfine parameters of the spectra of the Fh, Fh_Pb(0.02), and Fh_Pb(0.05) secondary minerals obtained after the RP1 with Shewanella oneidensis MR-1, presented in Figure 3. CS: center shift relative to αFe; Δ: quadrupole splitting; ε: quadrupole shift; H: hyperfine magnetic field; and RA: relative area.
In the presence of Pb, the same ferric oxyhydroxide minerals were obtained, with a significant increase in lepidocrocite versus goethite (55%/44%), as can be seen in the Mössbauer spectra. Similar field-reducing effects in the goethite are also observed during the increase in the Pb/(Fe+Pb) molar ratio.
3.3.2. Reduction Phase 2 (RP2)
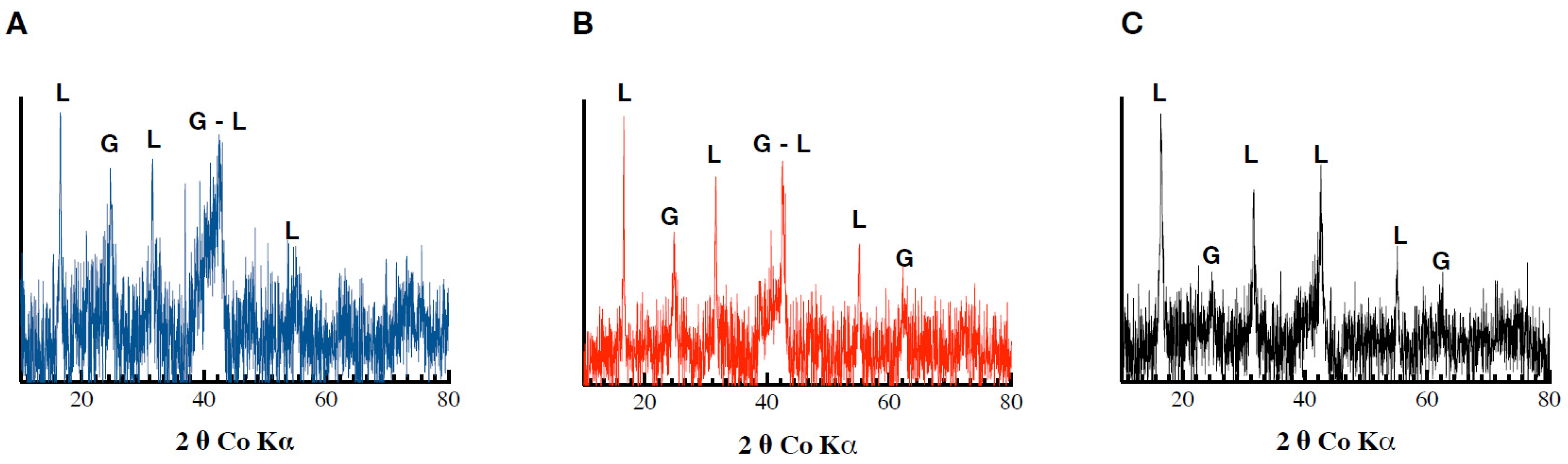
The XRD analyses revealed the precipitation of a mixture of magnetite (Fe3O4) and goethite in the Pb-free assays. The same mixture was also observed in the Fh_Pb(0.02) assays, while no magnetite, but goethite and lepidocrocite, were observed in the Fh_Pb(0.05) assays (Figure 4). These results indicate a mineralogical transformation in the Pb-free and Fh_Pb(0.02) assays, while no significant changes were detected in the Fh_Pb(0.05) assays.
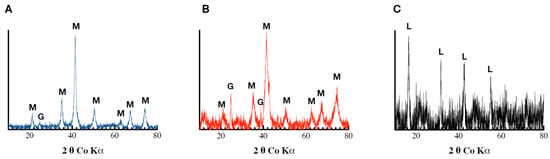
Figure 4.
Diffractograms obtained following Co-XRD measurements of the (A) Fh, (B) Fh_Pb(0.02), and (C) Fh_Pb(0.05) systems after the RP2 with Shewanella oneidensis MR-1.
The Mössbauer spectra at 298 and 77 K of the biogenic-mineral precipitate in the Pb-free assays during RP2 (Figure 5) are significantly different from those observed during RP1, where lepidocrocite and goethite were obtained, as is shown above. During RP2, the spectrum of the biogenic minerals that were formed from Fh display a magnetically ordered system that approaches a probably well-crystallized sample. The total curve can be easily fitted with three resolved subspectra, which result from tetrahedral Fe3+ with H = 475 kOe, CSs = 0.31 mm s−1, and octahedral Fe2.5+ with H = 444 kOe and CS = 0.55 mm s−1, which are both typical of magnetite [40]. Meanwhile, the third sextet (RA = 28%) corresponds to goethite, and it is characterized by its lower hyperfine field at 403 kOe. The 4K spectrum may be described by five sextets that are attributable to magnetite, as it is widely reported in the literature [43,44,45].
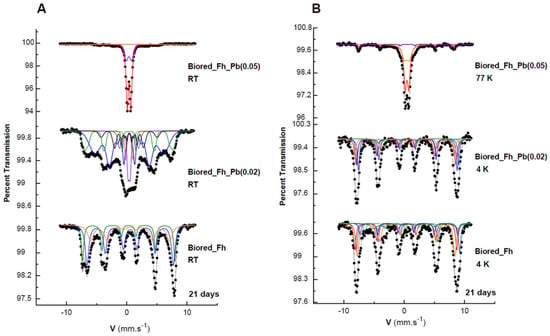
Figure 5.
Mössbauer spectra of the secondary products obtained in the Fh, Fh_Pb(0.02), and Fh_Pb(0.05) systems at: (A) room temperature (RT), and (B) 77 or 4 K after the RP2 with Shewanella oneidensis MR-1. Hyperfine parameters corresponding to these spectra are presented in Table 2.
The 298 K spectrum of the Fh_Pb(0.02) is rather complex (Figure 5). It displays overlap in the central part, and asymmetric sextets that point to a small-particle morphology and doublet that correspond to a phase with superparamagnetic behavior. The analysis at 4 K helped to clarify this complexity, where the shape-line and the deconvolution are similar to that of the biogenic minerals that are precipitated from Fh bioreduction, and it consists of five sextets of magnetite, and one sextet of goethite. As was expected for a poorly crystallized goethite, we observed, at 298 K, and simultaneously, a doublet and sextet that were governed by superparamagnetic relaxation [18,46]. It was that the spectrum at RT evolves drastically when the temperature decreases at 4 K with the collapse of the paramagnetic doublets and their transformation in the magnetic contribution, as was expected with the reduction in the thermal relaxation effects.
When the substitution increases to 0.05, an evolution of the Mössbauer spectra is observed when the temperature decreases from RT to 77 K (Figure 5). At RT, we can practically observe two paramagnetic doublets, which are discriminated by their quadrupole splitting and which correspond to lepidocrocite and goethite with superparamagnetic behavior, as well as a few traces of sextets in the noise. At 77 K, the spectrum shows that a superparamagnetic goethite still appears, as well as a magnetic component for this phase.
4. Discussion
Our data indicate the absence of bioreduction and the precipitation of secondary minerals in the control assays (without bacteria), regardless of the Pb loading during RP1 and RP2. This observation is in line with previous studies that show that the transformation of Fh to a thermodynamically more stable mineral, such as goethite, necessitates an extended period (months or years) under our experimental conditions (i.e., near-neutral pH and room temperature) [47].
At the end of RP1 and RP2, no total Pb and bioavailable Pb were detected in the aqueous phase. Yan et al. [48] have shown a transient release of zinc to the solution during the transformations of zinc–ferrihydrite that is induced by Fe(II). The results suggest that the Pb was either adsorbed (e.g., onto the surface, the structural defects, and the nanopores of biogenic minerals), was structurally incorporated into the biogenic minerals, or was occluded in the aggregate of biogenic minerals at the end of the incubation periods. In addition, the O2-oxidation episode did not change the partitioning of the Pb.
Rate and extent of bioreduction during RP1. Metal-bearing Fh bioreduction has been widely studied to decipher the impact of substituted metals on the reduction processes [48,49,50]. The nature and concentration of the metals modify the Fh particles’ physicochemical properties, which disrupts the bioreduction process, compared to metal-free Fh. Although no previous work has studied the effect of Pb on the bioreduction of Fh, our findings are in line with previous studies that have run with other elements [18,51]. While the Fh_Pb(0.02) reduction rates are comparable to those of Pb-free Fh, the reduction rate decreased by 60% when the Pb loading reached 5%. Similarly, an increase of 13% in aluminum decreased the bioreduction rate by 50% [51]. The same trend was observed in the presence of Sb and Ni, among others [18,52]. Increasing Pb loading may limit the accessibility of Fh surface sites and/or a significant loss of Fe(III) reactive sites, which thereby decreases the rate of reduction [53].
Previous studies have shown that Al- and Sb-bearing Fh displayed more decreased reducibility than metal-free Fh, which thus increases the Fe(II) loading to induce metal-bearing Fh transformation. On the contrary, in our study, a drop of ~60% in the extent of reduction was observed when the Pb/(Fe+Pb) molar ratio increased from 0 to 0.05, relative to pure Fh. Our results suggest that the increase in the Pb in the system reduced the amount of the Fe(II)/Fe(III) molar ratio to trigger the transformation of Fh toward secondary minerals, which indicates the higher lability of the Pb-bearing Fh than that of the Pb-free Fh.
Biogenic-mineral precipitation at the end of RP1. Fh is a thermodynamically metastable solid that gradually converts to more stable and crystallized Fe(III)- or Fe(II)/Fe(III)-bearing phases, such as goethite and magnetite, the latter of which is a mixed-valence iron oxide mineral in the presence of dissolved Fe(II) [54,55]. Fe(II)-catalyzed transformations involve adsorption and electron transfer to structural Fe(III) at the mineral surface. Subsequently, this process leads to the production of reactive Fe(III) species, which facilitates mass transfer, growth, and the precipitation of secondary minerals [56,57], which may halt the extent of the reduction. As was shown previously, factors such as the pH and the ligand concentration may affect the nature and the abundance of these secondary minerals [31,58,59]. During bioreduction, the generation of Fe(II) upon Fe(III) reduction is the initial driving force that catalyzes the Fh transformation; even micromolar concentrations of Fe(II) can trigger the precipitation of substantially more stable minerals, such as goethite and lepidocrocite [60]. In our study, goethite and lepidocrocite were detected, regardless of the Pb loading, as is shown by the XRD and the Mössbauer spectroscopy. The transformation of Fh into more stable phases in the presence of Fe(II) has been observed by several authors. The nature of the secondary mineral that results from the transformation of Fh is related to the Fe(II)/Fe(III) ratios, as is shown by Tronc et al. [61] and Jolivet et al. [62]. At a low Fe(II) concentration (0.67 mM Fe(II) g−1 Fh), the ferrihydrite was transformed into goethite and lepidocrocite [28]. Recently, Sheng et al. [56] showed the formation of labile Fe(III) species that were derived from Fe(II), the concentration of which influences the nature of the secondary minerals. The condensation pathway of labile Fe(III) species by olation and oxolation leads to the formation of lepidocrocite and goethite.
The presence of a foreign element may direct the precipitation of one mineral over another by favoring a given precipitation pathway, or by inhibiting/delaying the transformation of Fh. For instance, Cr(III) substitution inhibited the Fe(II) transformation of schwertmannite, which is a poorly-ordered Fe(III) oxyhydroysulfate [63]. Similarly, the study of Hansl et al. [50] indicates that Al substitution reduces secondary mineralization by preserving Fh. In our study, the presence of Pb did not dictate the nature of the secondary minerals, but it significantly altered their relative proportions, as was shown by the Mössbauer spectroscopy. As the Pb/(Fe+Pb) molar ratio increased from 0 to 0.05, the relative proportion of the goethite decreased from ~77 to ~45%. At the same time, the lepidocrocite increased from ~23 to ~55% (Table 1). Several potential processes may be responsible for driving the observed decrease in gothite and the increase in lepidocrocite. When a solid has several allotropic forms, this is generally the phase of lesser stability and higher solubility (lepidocrocite, in our case), which precipitates first. Lepidocrocite displays a higher solubility (log[(Fe3+).(OH−)3]) = −40.5) and standard-state Gibbs free energy of formation (−408.1 ± 1.4 kJ mol−1) than goethite. This metastable phase is transformed during the ageing of the suspension into a thermodynamically more stable phase (goethite). This process comes from the classical explanation of Ostwald’s rule of stages in its original form, as is presented by Stranski and Totomanov [64]. In the nucleation step, the Gibbs–Duhem equation and the classical equation of the nucleation rate, according to Nielsen and Söhnel [65], show that the size of the germ (or the nuclei) is smaller, and that the nucleation rate of a solid is greater than the solid–solution surface tension is low. Since solubility is inversely proportional to surface tension [65], the precipitation of the lepidocrocite, which is the most soluble phase, is kinetically favored. Because of its solubility and metastability, lepidocrocite is sensitive to aging, which leads to the recrystallization of the more stable phases, such as goethite, according to the spontaneous precipitation mechanism of Furedi–Milhofer [66]. The buildup of lepidocrocite may arise from the inhibitory effect of the Pb on the conversion of lepidocrocite to goethite. Another hypothesis is that Pb interferes with the oloation—oxolation reactions of labile Fe(III) species that favor the accumulation of lepidocrocite to the detriment of goethite.
Rate and extent of bioreduction upon RP2. After the oxidation period, the second phase of bioreduction was initiated by introducing a fresh inoculum. No Fe(II) was detected at the beginning of the incubation, which indicates that the O2 depleted the Fe(II) species in the assays (Figure 1B). During the second reduction phase, although the starting Fe(III) minerals corresponded to a mixture of lepidocrocite and goethite, their relative abundance in each assay was distinct. The reduction rate of the Pb-free and Fh_Pb(0.02) Pb assays did not vary significantly. However, an increase of ~7% in the extent of the reduction was measured in the Fh_Pb(0.02) assay. There does not appear to be a consistent trend in response to the Pb/(Pb+Fe) molar ratio. The initial mineral mixture in the Fh-without-Pb and Fh_Pb(0.02) assays consists of 23% lepidocrocite - 77% goethite, and 51% lepidocrocite - 49% goethite, respectively. Thus, the difference in the extent of the reduction could be explained by the larger lepidocrocite content in Fh_Pb(0.02). Previous studies have shown that lepidocrocite is more readily reduced compared to goethite [54]. The lack of reduction in the Fh_Pb(0.05) assays, despite the same mineral mixture and relative abundance as in Fh_Pb(0.02), indicates the inhibitory effect of Pb. If we assume that lepidocrocite is the bioavailable fraction, the absence of reduction may indicate that Pb blocks Fe(III) sites, or that it diverts the electron flow.
Biogenic-mineral precipitation at the end of RP2. Goethite and magnetite, with distinct relative abundance, precipitated in the Pb-free and Fh_Pb(0.02) assays. Compared to RP1, a disappearance of lepidocrocite, a decrease in goethite, and an appearance of magnetite was observed. The loss of lepidocrocite indicates that this was the main mineral that was transformed by bioreduction, and goethite, to a lesser extent. On the basis of the relative abundance that was established by the Mössbauer analyses (Table 1 and Table 2), one can assess the proportion of minerals that were transformed by bacterial activity. The estimates are based on the assumption that the biogenic magnetite that precipitated could be stochiometric, with the Fe(II) and Fe(III) abundances being 1/3 and 2/3, respectively, and by taking into account the relative abundances that correspond to the best-resolved spectra. During RP1, ~23% of lepidocrocite and ~77% of goethite precipitated in the Pb-free assays. These phases were transformed into ~70% of magnetite and ~30% of goethite during RP2. As the lepidocrocite vanished during RP2, we inferred that it was totally reduced, which led to the formation of ~23% of Fe(II) in the system. The decrease in the goethite implies that ~47% of the goethite was mobilized in the formation of magnetite. As stochiometric magnetite is composed of 1/3 of Fe(II) and 2/3 of Fe(III), there is a need for 23% of Fe(II) (i.e., formed by lepidocrocite reduction) to react with the ~47% of Fe(III) that originated from goethite. The fact that the relative abundance of the goethite decreased from ~77 to ~30% supports our assumption (77% − 30% = 47%).
Table 2.
Mössbauer hyperfine parameters of the spectra of the Fh, Fh_Pb(0.02), and Fh_Pb(0.05) secondary minerals obtained after the RP2 with Shewanella oneidensis MR-1, presented in Figure 5. CS: center shift relative to αFe; Δ: quadrupole splitting; ε: quadrupole shift; H: hyperfine magnetic field; and RA: relative area.
The same approach can be used to explain the phase distribution that was observed in the Fh_Pb(0.02) assays. During RP1, ~51% of lepidocrocite and ~49% of goethite precipitated in the Pb-free assays. These phases were transformed to ~84% of magnetite and ~16% of goethite. Stochiometric magnetite comprises ~28% of Fe(II) and ~56% of Fe(III). As the lepidocrocite vanished at the end of RP2, we assumed it was bioreduced to provide the ~28% of Fe(II) to form the magnetite. The necessary ~56% of Fe(III) originated from the remaining ~23% of lepidocrocite (51% − 28% = 23%) and ~33% of goethite (49% − 33% = 16%).
This approach shows that the lepidocrocite was totally reduced to Fe(II) in the Pb-free assays, while ~55% was reduced to Fe(II) in the Fh_Pb(0.02) assays, and no reduction was achieved in the Fh_Pb(0.05) assays. We infer that Pb may interact with lepidocrocite particles and inhibit the bioreduction and its mineralogical transformation process.
5. Conclusions
This study reports the bioreduction of Pb-bearing Fh during successive Fe redox cycles. The data emphasize that the Fe redox cycling did not impair the bioreduction process, even if the secondary minerals that formed during RP1 and RP2 differed. The precipitation of ferric minerals was observed during RP1, while ferric and Fe(II)/Fe(III) mixed minerals formed during RP2. The significant impact on the bioreduction process was the presence of Pb rather than the Fe redox cycling. Thus, Pb loading affected the Fh bioreduction process and the distribution of biogenic minerals. For example, during RP1, the relative abundance of lepidocrocite increased while, in parallel, that of goethite decreased with the Pb loading. In addition, neither Fe(III) bioreduction nor a mineralogical transformation was observed in the assay with the higher Pb content (Fh_Pb(0.05)).
Although additional research is required to assess this study’s geochemical and environmental implications, the data presented here may be of interest to may improve our understanding of the cycling of metal-bearing Fh in ferruginous nonsulfidic environments. In a redox-dynamic environmental setting, where oxic and anoxic oscillations are established, the redox cycling of iron minerals plays a significant role in the determination of the environmental fate and mobility of the nutrients and contaminants. For instance, due to its large specific surface area, its surface charge, and its high solubility relative to other Fe(III) minerals, Fh has a high sorption capacity for various soil components, such as dissolved carbon, nutrients, and pollutants. However, Fh is a metastable mineral compared to the more stable Fe(III) phases, and it will transform into these phases over time, catalyzed by chemical reductants such as Fe(II), which potentially impact the fate and the transport of the coassociated components in the environment through their retention or release [67]. The results presented in our study show that Pb is not mobilized in the aqueous phase during the transformation of Fh to more crystallized minerals, even after Fe redox cycling. Therefore, we hypothesize that metals that are associated with Fh are not easy mobilized during Fe redox cycling. To fully understand the influence of the microbial Fe cycle and metal mobilization, additional work is warranted to define whether this metal-retention capacity is metal-specific or carrier-specific (iron oxide nature).
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/min12050610/s1, Table S1: Composition of the poor metal-complexing medium (GGM) used for the preparation of the biosensors, Table S2: Pb(NO3)2 standard solutions used for the biosensor analysis, Figure S1: Standard curves obtained by plotting the induction coefficients against (A) all the standard solution concentrations and (B) only the lowest concentrations.
Author Contributions
Conceptualization, A.Z.; methodology, A.Z.; validation, F.M., A.Z., P.B., T.H. and M.A.; formal analysis, F.M., M.A., P.B. and T.H.; investigation, F.M., A.Z. and M.A.; data curation, F.M., A.Z. and M.A.; writing—original draft preparation, F.M., A.Z. and M.A.; writing—review and editing, F.M., A.Z. and M.A.; supervision, A.Z. and M.A. All authors have read and agreed to the published version of the manuscript.
Funding
This work is part of the «SIGMABIO» project, which was funded by the Lorraine University of Excellence (2018).
Acknowledgments
The authors acknowledge Renaud Gley for his support in the XRD analyses.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the analyses or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Singh, N.; Li, J.H. Environmental Impacts of Lead Ore Mining and Smelting. Adv. Mater. Res. 2014, 878, 338–347. [Google Scholar] [CrossRef]
- Gis Sol. Synthèse sur l’état des sols de France. GProupement d’intérêt Scientifique Sur Les Sols. 2011, p. 24. Available online: https://www.gissol.fr/publications/rapports/synthese-resf-869 (accessed on 2 March 2022).
- Gutiérrez, M.; Mickus, K.; Camacho, L.M. Abandoned PbZn Mining Wastes and Their Mobility as Proxy to Toxicity: A Review. Sci. Total Environ. 2016, 565, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, L.; Probst, A.; Probst, J.L.; Ulrich, E. Heavy Metal Distribution in Some French Forest Soils: Evidence for Atmospheric Contamination. Sci. Total Environ. 2003, 312, 195–219. [Google Scholar] [CrossRef] [Green Version]
- Kabata-Pendias, A.; Pendias, H. Trace Elements in Soils and Plants; CRC Press: Boca Raton, FL, USA, 2000; ISBN 978-0-429-19112-1. [Google Scholar]
- National Academies Press. National Academies of Sciences, Engineering, and Medicine Investigative Strategies for Lead-Source Attribution at Superfund Sites Associated with Mining Activities; National Academies Press: Washington, DC, USA, 2017; ISBN 978-0-309-46559-5. [Google Scholar]
- Villen-Guzman, M.; Garcia-Rubio, A.; Paz-Garcia, J.M.; Vereda-Alonso, C.; Gomez-Lahoz, C.; Rodriguez-Maroto, J.M. Aging Effects on the Mobility of Pb in Soil: Influence on the Energy Requirements in Electroremediation. Chemosphere 2018, 213, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.; Griswold, W. Health Effects of Heavy Metals. Environ. Sci. Technol. Briefs Citiz. 2009, 15, 1–6. [Google Scholar]
- Chiroma, T.M.; Ebewele, R.O.; Hymore, F.K. Comparative Assessement Of Heavy Metal Levels in Soil, Vegetables and Urban Grey Waste Water Used for Irrigation in Yola and Kano. Int. Refereed J. Eng. Sci. 2014, 3, 1–9. [Google Scholar]
- Bradl, H.B. Adsorption of Heavy Metal Ions on Soils and Soils Constituents. J. Colloid Interface Sci. 2004, 277, 1–18. [Google Scholar] [CrossRef]
- Fleming, M.; Tai, Y.; Zhuang, P.; McBride, M.B. Extractability and Bioavailability of Pb and As in Historically Contaminated Orchard Soil: Effects of Compost Amendments. Environ. Pollut 2013, 177, 90–97. [Google Scholar] [CrossRef] [Green Version]
- Jensen, P.E.; Ottosen, L.M.; Pedersen, A.J. Speciation of Pb in Industrially Polluted Soils. Water Air Soil Pollut 2006, 170, 359–382. [Google Scholar] [CrossRef]
- Liu, G.; Yu, Z.; Liu, X.; Xue, W.; Dong, L.; Liu, Y. Aging Process of Cadmium, Copper, and Lead under Different Temperatures and Water Contents in Two Typical Soils of China. J. Chem. 2020, 2020, e2583819. [Google Scholar] [CrossRef]
- Journet, E.; Balkanski, Y.; Harrison, S.P. A New Data Set of Soil Mineralogy for Dust-Cycle Modeling. Atmos. Chem. Phys. 2014, 14, 3801–3816. [Google Scholar] [CrossRef] [Green Version]
- Erbs, J.J.; Berquó, T.S.; Reinsch, B.C.; Lowry, G.V.; Banerjee, S.K.; Penn, R.L. Reductive Dissolution of Arsenic-Bearing Ferrihydrite. Geochim. Cosmochim. Acta 2010, 74, 3382–3395. [Google Scholar] [CrossRef]
- Frierdich, A.J.; Luo, Y.; Catalano, J.G. Trace Element Cycling through Iron Oxide Minerals during Redox-Driven Dynamic Recrystallization. Geology 2011, 39, 1083–1086. [Google Scholar] [CrossRef]
- Zachara, J.M.; Fredrickson, J.K.; Smith, S.C.; Gassman, P.L. Solubilization of Fe(III) Oxide-Bound Trace Metals by a Dissimilatory Fe(III) Reducing Bacterium. Geochim. Cosmochim. Acta 2001, 65, 75–93. [Google Scholar] [CrossRef] [Green Version]
- Zegeye, A.; Carteret, C.; Mallet, M.; Billet, D.; Ferté, T.; Chang, C.S.; Hauet, T.; Abdelmoula, M. Effect of Sb on Precipitation of Biogenic Minerals during the Reduction of Sb-Bearing Ferrihydrites. Geochim. Cosmochim. Acta 2021, 309, 96–111. [Google Scholar] [CrossRef]
- Cornell, R.M.; Schwertmann, U. The Iron Oxides: Structure, Properties, Reactions, Occurrences and Uses, 2nd, Completely Revised and Extended Edition; Wiley: Hoboken, NJ, USA, 2003. [Google Scholar]
- Davranche, M.; Bollinger, J.-C. Release of Metals from Iron Oxyhydroxides under Reductive Conditions: Effect of Metal/Solid Interactions. J. Colloid Interface Sci. 2000, 232, 165–173. [Google Scholar] [CrossRef] [Green Version]
- Hesterberg, D.; Duff, M.C.; Dixon, J.B.; Vepraskas, M.J. X-ray Microspectroscopy and Chemical Reactions in Soil Microsites. J. Environ. Qual. 2011, 40, 667–678. [Google Scholar] [CrossRef]
- Lu, P.; Nuhfer, N.T.; Kelly, S.; Li, Q.; Konishi, H.; Elswick, E.; Zhu, C. Lead Coprecipitation with Iron Oxyhydroxide Nano-Particles. Geochim. Cosmochim. Acta 2011, 75, 4547–4561. [Google Scholar] [CrossRef] [Green Version]
- Seda, N.N.; Koenigsmark, F.; Vadas, T.M. Sorption and Coprecipitation of Copper to Ferrihydrite and Humic Acid Organomineral Complexes and Controls on Copper Availability. Chemosphere 2016, 147, 272–278. [Google Scholar] [CrossRef] [Green Version]
- Hassellöv, M.; von der Kammer, F. Iron Oxides as Geochemical Nanovectors for Metal Transport in Soil-River Systems. Elements 2008, 4, 401–406. [Google Scholar] [CrossRef]
- Ionescu, D.; Heim, C.; Polerecky, L.; Thiel, V.; de Beer, D. Biotic and Abiotic Oxidation and Reduction of Iron at Circumneutral PH Are Inseparable Processes under Natural Conditions. Geomicrobiol. J. 2015, 32, 221–230. [Google Scholar] [CrossRef]
- Bleam, W.F. Chapter 8—Redox Chemistry. In Soil and Environmental Chemistry; Bleam, W.F., Ed.; Academic Press: Boston, MA, USA, 2012; pp. 321–370. ISBN 978-0-12-415797-2. [Google Scholar]
- Han, R.; Liu, T.; Li, F.; Li, X.; Chen, D.; Wu, Y. Dependence of Secondary Mineral Formation on Fe(II) Production from Ferrihydrite Reduction by Shewanella Oneidensis MR-1. ACS Earth Space Chem. 2018, 2, 399–409. [Google Scholar] [CrossRef]
- Hansel, C.M.; Benner, S.G.; Fendorf, S. Competing Fe(II)-Induced Mineralization Pathways of Ferrihydrite. Environ. Sci. Technol. 2005, 39, 7147–7153. [Google Scholar] [CrossRef] [PubMed]
- Hansel, C.M.; Benner, S.G.; Neiss, J.; Dohnalkova, A.; Kukkadapu, R.K.; Fendorf, S. Secondary Mineralization Pathways Induced by Dissimilatory Iron Reduction of Ferrihydrite under Advective Flow. Geochim. Cosmochim. Acta 2003, 67, 2977–2992. [Google Scholar] [CrossRef] [Green Version]
- Pallud, C.; Kausch, M.; Fendorf, S.; Meile, C. Spatial Patterns and Modeling of Reductive Ferrihydrite Transformation Observed in Artificial Soil Aggregates. Environ. Sci. Technol. 2010, 44, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Zachara, J.M.; Kukkadapu, R.K.; Fredrickson, J.K.; Gorby, Y.A.; Smith, S.C. Biomineralization of Poorly Crystalline Fe(III) Oxides by Dissimilatory Metal Reducing Bacteria (DMRB). Geomicrobiol. J. 2002, 19, 179–207. [Google Scholar] [CrossRef]
- Cheng, D.; Yuan, S.; Liao, P.; Zhang, P. Oxidizing Impact Induced by Mackinawite (FeS) Nanoparticles at Oxic Conditions Due to Production of Hydroxyl Radicals. Environ. Sci. Technol. 2016, 50, 11646–11653. [Google Scholar] [CrossRef]
- Miot, J.; Etique, M. Formation and Transformation of Iron-Bearing Minerals by Iron(II)-Oxidizing and Iron(III)-Reducing Bacteria. In Iron Oxides; Wiley-Blackwell: Hoboken, NJ, USA, 2016; pp. 53–98. ISBN 978-3-527-69139-5. [Google Scholar]
- Jones, A.M.; Griffin, P.J.; Collins, R.N.; Waite, T.D. Ferrous Iron Oxidation under Acidic Conditions—The Effect of Ferric Oxide Surfaces. Geochim. Cosmochim. Acta 2014, 145, 1–12. [Google Scholar] [CrossRef]
- Vu, H.P.; Shaw, S.; Brinza, L.; Benning, L.G. Partitioning of Pb(II) during Goethite and Hematite Crystallization: Implications for Pb Transport in Natural Systems. Appl. Geochem. 2013, 39, 119–128. [Google Scholar] [CrossRef]
- Vu, H.P.; Shaw, S.; Brinza, L.; Benning, L.G. Crystallization of Hematite (α-Fe2O3) under Alkaline Condition: The Effects of Pb. Cryst. Growth Des. 2010, 10, 1544–1551. [Google Scholar] [CrossRef]
- Schwertmann, U.; Cornell, R.M. Iron Oxides in the Laboratory: Preparation and Characterization; John Wiley & Sons: Hoboken, NJ, USA, 2000; ISBN 978-3-527-29669-9. [Google Scholar]
- Watanabe, K.; Manefield, M.; Lee, M.; Kouzuma, A. Electron Shuttles in Biotechnology. Curr. Opin. Biotechnol. 2009, 20, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Viollier, E.; Inglett, P.W.; Hunter, K.; Roychoudhury, A.N.; Van Cappellen, P. The Ferrozine Method Revisited: Fe(II)/Fe(III) Determination in Natural Waters. Appl. Geochem. 2000, 15, 785–790. [Google Scholar] [CrossRef]
- Murad, E.; Johnston, J.H. Iron Oxides and Oxyhydroxides. In Mössbauer Spectroscopy Applied to Inorganic Chemistry; Springer: New York, NY, USA, 1987; pp. 507–582. [Google Scholar]
- Mørup, S.; Topsøe, H. Mössbauer Studies of Thermal Excitations in Magnetically Ordered Microcrystals. Appl. Phys. 1976, 11, 63–66. [Google Scholar] [CrossRef]
- Mørup, S.; Bo Madsen, M.; Franck, J.; Villadsen, J.; Koch, C.J.W. A New Interpretation of Mössbauer Spectra of Microcrystalline Goethite: “Super-Ferromagnetism” or “Super-Spin-Glass” Behaviour? J. Magn. Magn. Mater. 1983, 40, 163–174. [Google Scholar] [CrossRef]
- Srivastava, C.M.; Shringi, S.N.; Babu, M.V. Mössbauer Study of the Low-Temperature Phase of Magnetite. Phys. Status Solidi 1981, 65, 731–735. [Google Scholar] [CrossRef]
- Vandenberghe, R.E.; Barrero, C.A.; da Costa, G.M.; Van San, E.; De Grave, E. Mössbauer Characterization of Iron Oxides and (Oxy)Hydroxides: The Present State of the Art. Hyperfine Interact. 2000, 126, 247–259. [Google Scholar] [CrossRef]
- Daou, T.J.; Pourroy, G.; Bégin-Colin, S.; Grenèche, J.M.; Ulhaq-Bouillet, C.; Legaré, P.; Bernhardt, P.; Leuvrey, C.; Rogez, G. Hydrothermal Synthesis of Monodisperse Magnetite Nanoparticles. Chem. Mater. 2006, 18, 4399–4404. [Google Scholar] [CrossRef]
- Janot, C.; Chabanel, M.; Herzog, E. Étude d’une limonite par effet Mössbauer. Bull. De Minéralogie 1968, 91, 166–171. [Google Scholar] [CrossRef]
- Schwertmann, U.; Stanjek, H.; Becher, H.-H. Long-Term in Vitro Transformation of 2-Line Ferrihydrite to Goethite/Hematite at 4, 10, 15 and 25 °C. Clay Miner. 2004, 39, 433–438. [Google Scholar] [CrossRef]
- Yan, J.; Frierdich, A.J.; Catalano, J.G. Impact of Zn Substitution on Fe(II)-Induced Ferrihydrite Transformation Pathways. Geochim. Cosmochim. Acta 2022, 320, 143–160. [Google Scholar] [CrossRef]
- Burton, E.D.; Hockmann, K.; Karimian, N.; Johnston, S.G. Antimony Mobility in Reducing Environments: The Effect of Microbial Iron(III)-Reduction and Associated Secondary Mineralization. Geochim. Cosmochim. Acta 2019, 245, 278–289. [Google Scholar] [CrossRef]
- Hansel, C.M.; Learman, D.R.; Lentini, C.J.; Ekstrom, E.B. Effect of Adsorbed and Substituted Al on Fe (II)-Induced Mineralization Pathways of Ferrihydrite. Geochim. Cosmochim. Acta 2011, 75, 4653–4666. [Google Scholar] [CrossRef]
- Ekstrom, E.B.; Learman, D.R.; Madden, A.S.; Hansel, C.M. Contrasting Effects of Al Substitution on Microbial Reduction of Fe(III) (Hydr)Oxides. Geochim. Cosmochim. Acta 2010, 74, 7086–7099. [Google Scholar] [CrossRef]
- Fredrickson, J.K.; Zachara, J.M.; Kukkadapu, R.K.; Gorby, Y.A.; Smith, S.C.; Brown, C.F. Biotransformation of Ni-Substituted Hydrous Ferric Oxide by an Fe(III)-Reducing Bacterium. Environ. Sci. Technol. 2001, 35, 703–712. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhu, M.; Lan, S.; Ginder-Vogel, M.; Liu, F.; Feng, X. Formation and Secondary Mineralization of Ferrihydrite in the Presence of Silicate and Mn(II). Chem. Geol. 2015, 415, 37–46. [Google Scholar] [CrossRef]
- Usman, M.; Abdelmoula, M.; Hanna, K.; Grégoire, B.; Faure, P.; Ruby, C. FeII Induced Mineralogical Transformations of Ferric Oxyhydroxides into Magnetite of Variable Stoichiometry and Morphology. J. Solid State Chem. 2012, 194, 328–335. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Barnard, A.S. Naturally Occurring Iron Oxide Nanoparticles: Morphology, Surface Chemistry and Environmental Stability. J. Mater. Chem. A 2013, 1, 27–42. [Google Scholar] [CrossRef]
- Sheng, A.; Liu, J.; Li, X.; Qafoku, O.; Collins, R.N.; Jones, A.M.; Pearce, C.I.; Wang, C.; Ni, J.; Lu, A. Labile Fe (III) from Sorbed Fe (II) Oxidation Is the Key Intermediate in Fe (II)-Catalyzed Ferrihydrite Transformation. Geochim. Cosmochim. Acta 2020, 272, 105–120. [Google Scholar] [CrossRef]
- Sheng, A.; Li, X.; Arai, Y.; Ding, Y.; Rosso, K.M.; Liu, J. Citrate Controls Fe (II)-Catalyzed Transformation of Ferrihydrite by Complexation of the Labile Fe (III) Intermediate. Environ. Sci. Technol. 2020, 54, 7309–7319. [Google Scholar] [CrossRef]
- Kukkadapu, R.K.; Zachara, J.M.; Fredrickson, J.K.; Kennedy, D.W. Biotransformation of Two-Line Silica-Ferrihydrite by a Dissimilatory Fe(III)-Reducing Bacterium: Formation of Carbonate Green Rust in the Presence of Phosphate1 1Associate Editor: D. J. Vaughan. Geochim. Cosmochim. Acta 2004, 68, 2799–2814. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Zheng, S.; Qiu, H.; Cao, C.; Tang, X.; Hao, L.; Liu, F.; Li, J. Ferrihydrite Reduction and Vivianite Biomineralization Mediated by Iron Reducing Bacterium Shewanella Oneidensis MR-4. Acta Microbiologica Sinica 2018, 58, 573–583. [Google Scholar]
- Yee, N.; Shaw, S.; Benning, L.G.; Nguyen, T.H. The Rate of Ferrihydrite Transformation to Goethite via the Fe (II) Pathway. Am. Mineral. 2006, 91, 92–96. [Google Scholar] [CrossRef]
- Tronc, E.; Belleville, P.; Jolivet, J.P.; Livage, J. Transformation of Ferric Hydroxide into Spinel by Iron(II) Adsorption. Langmuir 1992, 8, 313–319. [Google Scholar] [CrossRef]
- Jolivet, J.P.; Belleville, P.; Tronc, E.; Livage, J. Influence of Fe(II) on the Formation of the Spinel Iron Oxide in Alkaline Medium. Clays Clay Miner. 1992, 40, 531–539. [Google Scholar] [CrossRef]
- Choppala, G.; Burton, E.D. Chromium(III) Substitution Inhibits the Fe(II)-Accelerated Transformation of Schwertmannite. PLoS ONE 2018, 13, e0208355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stranski, I.N.; Totomanov, D. Die Ostwaldsche Stufenregel. Die Nat. 1932, 20, 905. [Google Scholar] [CrossRef]
- Nielsen, A.E.; Söhnel, O. Interfacial Tensions Electrolyte Crystal-Aqueous Solution, from Nucleation Data. J. Cryst. Growth 1971, 11, 233–242. [Google Scholar] [CrossRef]
- Furedi-Milhofer, H. Spontaneous Precipitation from Electrolytic Solutions. Pure Appl. Chem. 1981, 53, 2041–2055. [Google Scholar] [CrossRef] [Green Version]
- Muehe, E.M.; Obst, M.; Hitchcock, A.; Tyliszczak, T.; Behrens, S.; Schröder, C.; Byrne, J.M.; Michel, F.M.; Krämer, U.; Kappler, A. Fate of Cd during Microbial Fe(III) Mineral Reduction by a Novel and Cd-Tolerant Geobacter Species. Environ. Sci. Technol. 2013, 47, 14099–14109. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).