

Comparison of the Analytical Performance of Inductively Coupled Plasma Mass Spectrometry and Instrumental Neutron Activation Analysis Techniques in the Elemental Analysis of Coal Fly Ash

Abstract
:1. Introduction
- To study the impact of its storage/use on the environment.
2. Materials and Methods
2.1. Samples and Reagents
2.2. Apparatus
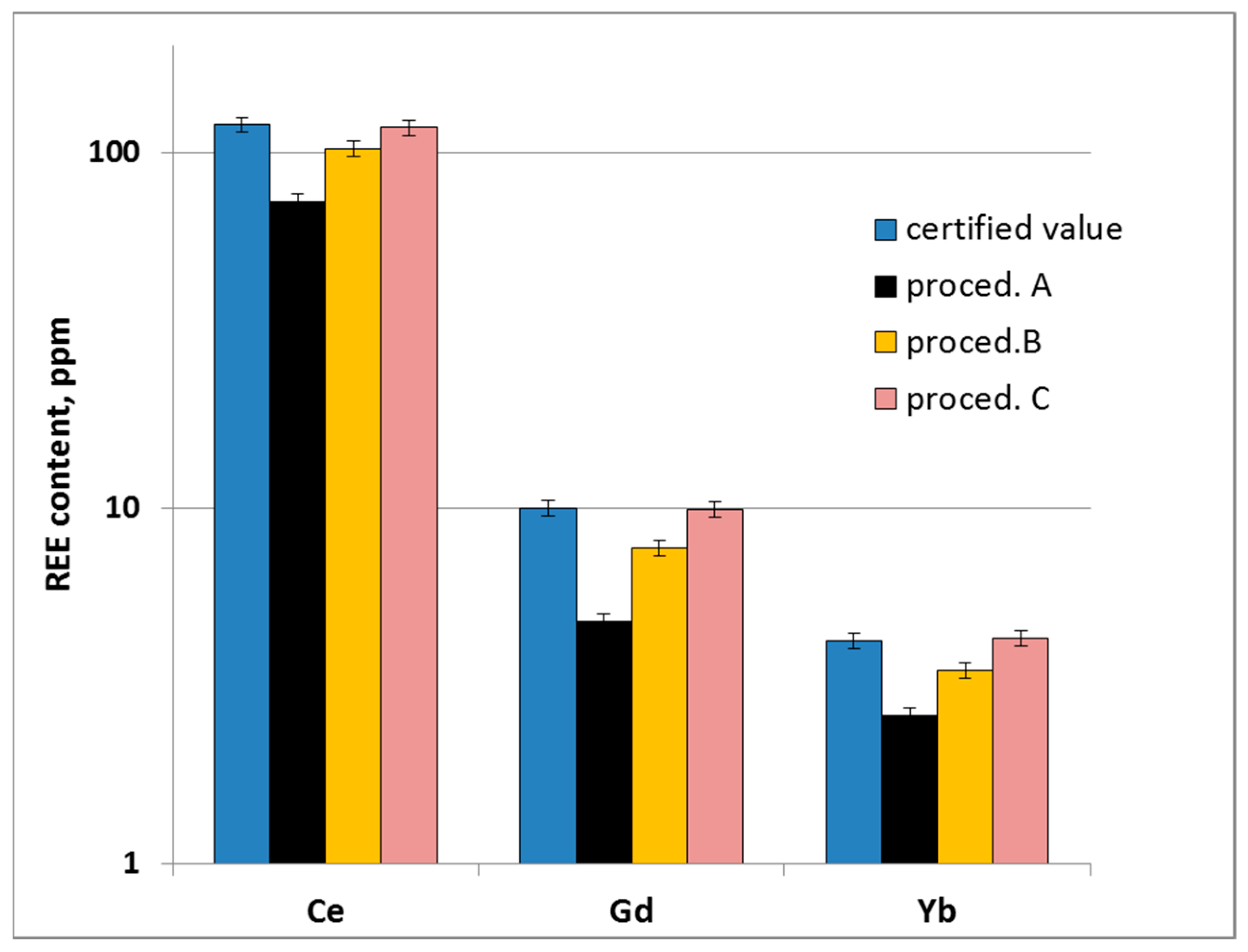
2.3. Sample Digestion Optimization
2.4. Neutron Activation Analysis
2.5. Microwave-Assisted Digestion and ICP-MS Measurements
2.6. Leaching Experiments
3. Results and Discussion
3.1. Optimization of the Sample Microwave Digestion
3.2. Analysis of Certified Reference Material CTA-FFA-1
3.2.1. Estimation of the Uncertainty Budget for Ash Analysis by ICP-MS and INAA
INAA
ICP-MS
- Preparation of samples and standards;
- Measurement by ICP-MS technique.
3.3. Elemental Content of Ash Samples
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Gopinathan, P.; Santosh, M.S.; Dileepkumar, V.G.; Subramani, T.; Reddy, R.; Masto, R.E.; Maity, S. Geochemical, mineralogical and toxicological characteristics of coal fly ash and its environmental impacts. Chemosphere 2022, 307, 135710. [Google Scholar] [CrossRef]
- Sandeep, P.; Maity, S.; Mishra, S.; Chaudhary, D.P.; Dusane, C.B.; Pillai, A.S.; Vinod Kumar, A. Estimation of rare earth elements in Indian coal fly ashes for recovery feasibility as a secondary source. J. Hazard. Mater. Advan. 2023, 10, 100257. [Google Scholar] [CrossRef]
- Lu, X.; Liu, B.; Zhang, Q.; Wen, Q.; Wang, S.; Xiao, K.; Zhang, S. Recycling of Coal Fly Ash in Building Materials: A Review. Minerals 2023, 13, 25. [Google Scholar] [CrossRef]
- Taggart, R.K.; Hower, J.C.; Dwyer, G.S.; Hsu-Kim, H. Trends in the rare earth element content of US-based coal combustion fly ashes. Environ. Sci. Technol. 2016, 50, 5919–5926. [Google Scholar] [CrossRef]
- Available online: https://ec.europa.eu/eurostat/statistics-explained/index.php?title=Coal_production_and_consumption_statistics (accessed on 6 November 2023).
- Wasielewski, R.; Radko, T. Problem zagospodarowania odpadów z palenisk domowych. Ecol. Eng. Environ. Technol. 2018, 19, 36–44. [Google Scholar] [CrossRef]
- Coal from Colombia, Mozambique, South Africa … Despite Embargo, Imports of Raw Material to Poland Grow. Available online: https://gospodarka.dziennik.pl/news/artykuly/8523681,embargo-wegiel-polska-rosja-ukraina.html (accessed on 24 September 2023).
- Waste Classification Defined by the Regulation of the Ministry of Climate (in Polish). Available online: https://isap.sejm.gov.pl/isap.nsf/DocDetails.xsp?id=WDU20200000010 (accessed on 6 November 2023).
- Satayeva, A.; Baimenov, A.; Azat, S.; Zhantikeyev, U.; Seisenova, A.; Tauanov, Z. Review on coal fly ash generation and utilization for resolving mercury contamination issues in Central Asia: Kazakhstan. Environ. Rev. 2022, 30, 418–437. [Google Scholar] [CrossRef]
- Santos, A.C.; Cruz, C.; Font, E.; French, D.; Guedes, A.; Moreira, K.; Sant’Ovaia, H.; Vieira, B.J.C.; Waerenborgh, J.C.; Valentim, B. Physicochemical Properties of Fe-Bearing Phases from Commercial Colombian Coal Ash. Minerals 2023, 13, 1055. [Google Scholar] [CrossRef]
- Pöykiö, R.; Manskinen, K.; Nurmesniemi, H.; Dahl, O. Comparison of Trace Elements in Bottom Ash and Fly Ash from a Large-Sized (77 MW) Multi-Fuel Boiler at the Power Plant of a Fluting Board Mill, Finland. Energy Explor. Exploit. 2011, 29, 217–234. [Google Scholar] [CrossRef]
- Pan, J.; Zhao, X.; Zhou, C.; Yang, F.; Ji, W. Study on Solvent Extraction of Rare Earth Elements from Leaching Solution of Coal Fly Ash by P204. Minerals 2022, 12, 1547. [Google Scholar] [CrossRef]
- Abd Manan, T.S.B.; Beddu, S.; Mohd Kamal, N.L.; Mohamad, D.; Itam, Z.; Khan, T.; Machmudah, A.; Dutykh, D.; Mohtar, W.H.M.W.; Jusoh, H.; et al. Ecological Risk Indicators for Leached Heavy Metals from Coal Ash Generated at a Malaysian Power Plant. Sustainability 2021, 13, 10222. [Google Scholar] [CrossRef]
- Kalembkiewicz, J.; Sitarz-Palczak, E. Efficiency of leaching tests in the context of the influence of the fly ash on the environment. J. Ecol. Eng. 2015, 16, 67–80. [Google Scholar] [CrossRef]
- Banerjee, R.; Chakladar, S.; Mohanty, A.; Chattopadhyay, S.K.; Chakravarty, S. Leaching characteristics of rare earth elements from coal ash using organosulphonic acids. Miner. Eng. 2022, 185, 107664. [Google Scholar] [CrossRef]
- Pan, J.; Hassas, B.V.; Rezaee, M.; Zhou, C.; Sarma, V. Recovery of rare earth elements from coal fly ash through sequential chemical roasting, water leaching, and acid leaching processes. J. Clean. Product 2021, 284, 124725. [Google Scholar] [CrossRef]
- Eze, C.P.; Fatoba, O.; Madzivire, G.; Ostrovnaya, T.M.; Petrik, L.F.; Frontasyeva, M.V.; Nechaev, A.N. Elemental composition of fly ash: A comparative study using nuclear and related analytical techniques. Chem. Didact. Ecol. Metrol. 2013, 18, 19–28. [Google Scholar] [CrossRef]
- Wilschefski, S.C.; Baxter, M.R. Inductively Coupled Plasma Mass Spectrometry: Introduction to Analytical Aspects. Clin. Biochem. Rev. 2019, 40, 115–133. [Google Scholar] [CrossRef]
- Bulska, E.; Wagner, B. Quantitative aspects of inductively coupled plasma mass spectrometry. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2016, 374, 20150369. [Google Scholar] [CrossRef]
- Hamidatou, L.; Slamene, H.; Akhal, T.; Zouranen, B. Concepts, instrumentation and techniques of neutron activation analysis. Imaging Radioanal. Tech. Interdiscip. Res. Fundam. Cut. Edge Appl. 2013, 141–178. [Google Scholar] [CrossRef]
- Kafala, S.I.; MacMahon, T.D. Comparison of neutron activation analysis methods. J. Radioanal. Nucl. Chem. 2007, 271, 507–516. [Google Scholar] [CrossRef]
- Lim, J.M.; Jeong, J.H.; Lee, J.H. Instrumental neutron activation analysis of coal and its combustion residues from a power plant. J. Radioanal. Nucl. Chem. 2013, 298, 201–208. [Google Scholar] [CrossRef]
- Chung, Y.S.; Moon, J.H.; Kim, S.H.; Kang, S.H.; Kim, Y.J. Determination of the elemental composition of the bottom ash of a municipal incinerator by instrumental neutron activation analysis. J. Radioanal. Nucl. Chem. 2007, 271, 339–344. [Google Scholar] [CrossRef]
- Rietz, B.; Heydorn, K.; Pritzl, G. Determination of aluminium in fish tissues by means of INAA and ICP-MS. J. Radioanal. Nucl. Chem. 1997, 216, 113–116. [Google Scholar] [CrossRef]
- Freitas, M.C.; Pacheco, A.M.G.; Dionísio, I.; Sarmento, S.; Baptista, M.S.; Vasconcelos, M.T.S.D.; Cabral, J.P. Multianalytical determination of trace elements in atmospheric biomonitors by k0-INAA, ICP-MS and AAS. Nucl. Instrum. Methods Phys. Res. B 2006, 564, 733–742. [Google Scholar] [CrossRef]
- Bédard, L.P.; Barnes, S.J. A comparison of the capacity of FA-ICP-MS and FA-INAA. J. Radioanal. Nucl. Chem. 2002, 254, 319–329. [Google Scholar] [CrossRef]
- Wallis, N.J.; Kamenov, G.D. Challenges in the Analysis of Heterogeneous Pottery by LA–ICP–MS: A Comparison with INAA. Archaeometry 2013, 55, 893–909. [Google Scholar] [CrossRef]
- Bedregal, P.; Ubillus, M.; Cáceres-Rivero, C.; Olivera, P.; Garay, R.; Rojas, J.; Zafra, R.; Urdanivia, R. Determination of atmospheric aerosol components in an urban area to evaluate the air quality and identify the sources of contamination. J. Radioanal. Nucl. Chem. 2022, 332, 3507–3514. [Google Scholar] [CrossRef]
- Weilert, T.M.; Ray, C.L.; Gawenis, J.A.; Brockman, J.D. Neutron activation analysis and ICP-MS for provenance of honey collected from American Midwest region. J. Radioanal. Nucl. Chem. 2022, 331, 4971–4981. [Google Scholar] [CrossRef]
- Ebihara, M.; Hayano, K.; Shirai, N. Determination of trace rare earth elements in rock samples including meteorites by ICP-MS coupled with isotope dilution and comparison methods. Anal. Chim. Acta 2020, 1101, 81–89. [Google Scholar] [CrossRef]
- Yagob, T.; van de Wiel, A.; Bode, P.; Demir, A.; van der Wagt, B.; Krystek, P.; Wolterbeek, B. Measurement of the enriched stable isotope 58Fe in iron related disorders-comparison of INAA and MC-ICP-MS. J. Trace Elem. Med. Biol. 2019, 53, 77–83. [Google Scholar] [CrossRef]
- Chajduk, E.; Polkowska-Motrenko, H. Application of ICP-MS, INAA and RNAA to the determination of some “difficult” elements in infant formulas. J. Radioanal. Nucl. Chem. 2017, 311, 1347–1353. [Google Scholar] [CrossRef]
- Chajduk, E.; Polkowska-Motrenko, H. The use of HPLC-NAA and HPLC-ICP-MS for the speciation of As in infant food. Food Chem. 2019, 292, 129–133. [Google Scholar] [CrossRef]
- Chajduk, E.; Kalbarczyk, P.; Dudek, J.; Pyszynska, M.; Bojanowska-Czajka, A.; Samczyński, Z. Development of Analytical Procedures for Chemical Characterization of Substrates for the Production of TRISO Coated Particles as Nuclear Fuel in High Temperature Gas-Cooled Reactors. Sustainability 2020, 12, 7221. [Google Scholar] [CrossRef]
- Ugurlu, A. Leaching characteristics of fly ash. Environ. Geol. 2004, 46, 890–895. [Google Scholar] [CrossRef]
- Kolker, A.; Scott, C.; Hower, J.C.; Vazquez, J.A.; Lopano, C.L.; Dai, S. Distribution of rare earth elements in coal combustion fly ash, determined by SHRIMP-RG ion microprobe. Int. J. Coal Geol. 2017, 184, 1–10. [Google Scholar] [CrossRef]
- Wang, Z.; Dai, S.; Zou, J.; French, D.; Graham, I.T. Rare earth elements and yttrium in coal ash from the Luzhou power plant in Sichuan, Southwest China: Concentration, characterization and optimized extraction. Int. J. Coal Geol. 2019, 203, 1–14. [Google Scholar] [CrossRef]
- Baranowska, I. Handbook of trace analysis. In Handbook of Trace Analysis: Fundamentals and Applications, 1st ed.; Springer: Basel, Switzerland, 2016. [Google Scholar]
- Hossain, M.; Karmakar, D.; Begum, S.N.; Ali, S.Y.; Patra, P.K. Recent trends in the analysis of trace elements in the field of environmental research: A review. Microchem. J. 2021, 165, 106086. [Google Scholar] [CrossRef]
- Gaudino, S.; Galas, C.; Belli, M.; Barbizzi, S.; de Zorzi, P.; Jaćimović, R.; Jeran, Z.; Pati, A.; Sansone, U. The role of different soil sample digestion methods on trace elements analysis: A comparison of ICP-MS and INAA measurement results. Accredit. Qual. Assur. 2007, 12, 84–93. [Google Scholar] [CrossRef]
- Conrey, R.M.; Bailey, D.G.; Singer, J.W.; Wagoner, L.J.; Parfitt, B.; Hay, J.; Keh, O.; Chang, Z.; Huang, S. Combined use of multiple external and internal standards in LA-ICP-MS analysis of bulk geological samples using lithium borate fused glass. Geochem. Explor. Env. A. 2023, 23, geochem2023-001. [Google Scholar] [CrossRef]
- Yu, Z.; Norman, M.D.; Robinson, P. Major and trace element analysis of silicate rocks by XRF and laser ablation ICP-MS using lithium borate fused glasses: Matrix effects, instrument response and results for international reference materials. Geostand. Newsl. 2003, 27, 67–89. [Google Scholar] [CrossRef]
- ASTM D6357-11; Test Methods for Determination of Trace Elements in Coal, Coke, & Combustion Residues from Coal Utilization Processes by Inductively Coupled Plasma Atomic Emission, Inductively Coupled Plasma Mass, & Graphite Furnace Atomic Absorption Spectrometry. ASTM: West Conshohocken, PA, USA, 2019.
- ASTM D6357-21b; Standard Test Methods for Determination of Trace Elements in Coal, Coke, and Combustion Residues from Coal Utilization Processes by Inductively Coupled Plasma Atomic Emission Spectrometry, Inductively Coupled Plasma Mass Spectrometry, and Graphite Furnace Atomic Absorption Spectrometry. ASTM: West Conshohocken, PA, USA, 2021.
- Saiki, M.; Alves, E.R.; Jaluul, O. Determination of trace elements in scalp hair of an elderly population by neutron activation analysis. J. Radioanal. Nucl. Chem. 2008, 276, 53–57. [Google Scholar] [CrossRef]
- van der Veen, A.; Cox, M.; Possolo, A. GUM guidance on developing and using measurement models. Accredit. Qual. Assur. 2022, 27, 295–297. [Google Scholar] [CrossRef]
- IAEA-TECDOC-1401: Quantifying Uncertainty in Nuclear Analytical Measurements; IAEA: Vienna, Austria, 2004.
- Greenberg, R.R.; Bode, P.; Fernandes, E.A.D.N. Neutron activation analysis: A primary method of measument. Spectrochim. Acta B At. Spectrosc. 2011, 66, 193–241. [Google Scholar] [CrossRef]
- Balla, M.; Molnár, Z.; Kőrös, Á. Uncertainty budget and validation of NAA using Reference Materials. J. Radioanal. Nucl. Chem. 2004, 259, 395–400. [Google Scholar] [CrossRef]
- Cho, H.; Dasari, K.B.; Jaćimović, R.; Zeisler, R.; Sharp, N.E.; Kim, S.H.; Sun, G.M.; Yim, Y.H. Application of the INAA methods for KRISS infant formula CRM analysis: Standardization of INAA at KRISS. J. Radioanal. Nucl. Chem. 2019, 322, 1537–1547. [Google Scholar] [CrossRef]
- Barwick, V.J.; Ellison, S.L.R.; Fairman, B. Estimation of uncertainties in ICP-MS analysis: A practical methodology. Anal. Chim. Acta 1999, 394, 281–291. [Google Scholar] [CrossRef]
- Thompson, R.L.; Bank, T.; Roth, E.; Granite, E. Resolution of rare earth element interferences in fossil energy by-product samples using sector-field ICP-MS. Fuel 2016, 185, 94–101. [Google Scholar] [CrossRef]
- Yenisoy-Karakaş, S. Estimation of uncertainties of the method to determine the concentrations of Cd, Cu, Fe, Pb, Sn and Zn in tomato paste samples analysed by high resolution ICP-MS. Food Chem. 2012, 132, 1555–1561. [Google Scholar] [CrossRef]
- Levine, R.M.; Brininstool, M.; Wallace, G.J. The mineral industry of Kazakhstan. In Minerals Yearbook 2009; USGS Publications Warehouse: Reston, VA, USA, 2001. [Google Scholar]
- Akinyemi, S.A.; Bohórquez, F.; Islam, N.; Saikia, B.K.; Sampaio, C.H.; Crissien, T.J.; Silva, L.F. Petrography and geochemistry of exported Colombian coals: Implications from correlation and regression analyses. Energy Geosci. 2021, 2, 201–210. [Google Scholar] [CrossRef]
- Smołka-Danielowska, D.; Jabłońska, M.; Godziek, S. The Influence of Hard Coal Combustion in Individual Household Furnaces on the Atmosphere Quality in Pszczyna (Poland). Minerals 2021, 11, 1155. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element | Certified Value | ICP-MS | INAA |
|---|---|---|---|
| Al | 14.87 ± 0.39 | 15.5 ± 1.0 | - |
| As | 53.6 ± 2.7 | 56.1 ± 8.4 | 54.7 ± 4.2 |
| Ba | 835 ± 56 | 860 ± 64 | 850 ± 50 |
| Ce | 120 ± 7 | 110 ± 11 | 127 ± 16 |
| Co | 39.9 ± 1.7 | 42.5 ± 2.0 | 38.5 ± 0.8 |
| Cr | 156 ± 8 | 140 ± 17 | 165 ± 16 |
| Cs | 48.2 ± 2.6 | 50.1 ± 3.2 | 50.0 ± 2.6 |
| Cu | 158 ± 9 | 167 ± 15 | 153 ± 12 |
| Dy | 9.09 ± 1.45 | 8.02 ± 0.80 | - |
| Er | 4.52 ± 1.12 | 4.15 ± 0.42 | - |
| Eu | 2.39 ± 0.06 | 2.18 ± 0.22 | 2.38 ± 0.01 |
| Fe | 4.89 ± 0.14 | 4.82 ± 0.30 | 4.83 ± 0.24 |
| Gd | 10.0 ± 2.6 | 8.7 ± 0.9 | 9.2 ± 0.9 |
| Hf | 6.0 ± 0.45 | 6.2 ± 0.6 | 5.4 ± 0.5 |
| La | 60.7 ± 4.0 | 57.1 ± 5.5 | 63.7 ± 2.0 |
| Lu | 0.658 ± 0.043 | 0.572 ± 0.06 | 0.630 ± 0.090 |
| Mn | 1066 ± 41 | 1073 ± 50 | - |
| Na | 2.19 ± 0.08 | 2.28 ± 0.15 | 2.13 ± 0.15 |
| Nd | 56.8 ± 3.7 | 52.1 ± 5.3 | 54.6 ± 5.5 |
| Pb | 369 ± 19 | 378 ± 18 | - |
| Rb | 185 ± 5 | 178 ± 9 | 186 ± 9 |
| Sb | 17.6 ± 2.5 | 15.8 ± 1.6 | 16.2 ± 1.0 |
| Sc | 24.2 ± 1.1 | 25.2 ± 1.7 | 24.0 ± 0.2 |
| Si | 22.48 ± 0.92 | 21.60 ± 2.00 | - |
| Sm | 10.9 ± 0.6 | 9.8 ± 1.0 | 11.1 ± 0.5 |
| Tb | 1.38 ± 0.14 | 1.26 ± 0.13 | 1.41 ± 0.10 |
| Th | 29.4 ± 0.7 | 29.5 ± 0.9 | 29.2 ± 1.4 |
| Tm | 0.705 ± 0.200 | 0.92 ± 0.22 | - |
| U | 15.1 ± 0.8 | 14.9 ± 0.4 | 14.6 ± 1.2 |
| Yb | 4.24 ± 0.19 | 4.00 ± 0.43 | 4.30 ± 0.30 |
| Zn | 569 ± 58 | 580 ± 41 | 545 ± 28 |
| Ion | Reagent 1 | Reagent 2 | Reagent 3 |
|---|---|---|---|
| K+, µg/g | 129 | 1800 | 76 |
| SO42−, µg/g | 475 | 11,800 | 4790 |
| Mn, ng/g | 9.8 | 28.4 | 2.72 |
| Poland | Columbia | Kazakhstan | |
|---|---|---|---|
| As | 20.4 | 65.2 | 15.1 |
| Ba | 1422 | 1751 | 1185 |
| Cd | 2.08 | 2.53 | 2.58 |
| Co | 36.3 | 20.3 | 32.5 |
| Cr | 137 | 101 | 86.5 |
| Cs | 14.2 | 12.7 | 8.2 |
| Cu | 91.9 | 57.1 | 353 |
| Hf | 6.2 | 17.2 | 28.3 |
| Mn | 564 | 450 | 1070 |
| Ni | 93.1 | 77.8 | 105 |
| Pb | 44.8 | 63.6 | 85.7 |
| Sb | 8.08 | 7.13 | 6.3 |
| Sc | 87.2 | 35.1 | 60.5 |
| ∑La-Gd | 264 | 394 | 125 |
| ∑Tb-Lu | 21.7 | 34.2 | 10.2 |
| U | 9.38 | 13.2 | 24.4 |
| Th | 14.7 | 30.5 | 55.6 |
| V | 293 | 354 | 380 |
| Zn | 275 | 285 | 875 |
| Element | % of Elution |
|---|---|
| As | 35 ± 5 |
| Cd | <5 |
| Co | <5 |
| Cr | <10 |
| Cu | <5 |
| Fe | <5 |
| Mn | <5 |
| Mo | 45 ± 10 |
| Ni | <5 |
| Sb | 55 ± 10 |
| Se | 45 ± 10 |
| Zn | <5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chajduk, E.; Kalbarczyk, P. Comparison of the Analytical Performance of Inductively Coupled Plasma Mass Spectrometry and Instrumental Neutron Activation Analysis Techniques in the Elemental Analysis of Coal Fly Ash. Minerals 2023, 13, 1484. https://doi.org/10.3390/min13121484
Chajduk E, Kalbarczyk P. Comparison of the Analytical Performance of Inductively Coupled Plasma Mass Spectrometry and Instrumental Neutron Activation Analysis Techniques in the Elemental Analysis of Coal Fly Ash. Minerals. 2023; 13(12):1484. https://doi.org/10.3390/min13121484
Chicago/Turabian StyleChajduk, Ewelina, and Paweł Kalbarczyk. 2023. "Comparison of the Analytical Performance of Inductively Coupled Plasma Mass Spectrometry and Instrumental Neutron Activation Analysis Techniques in the Elemental Analysis of Coal Fly Ash" Minerals 13, no. 12: 1484. https://doi.org/10.3390/min13121484