Infrared Spectroscopic Investigations of Calcium Oxalate Monohydrate (Whewellite) Dehydration/Rehydration


Abstract
:
1. Introduction
2. Materials and Methods
3. Results
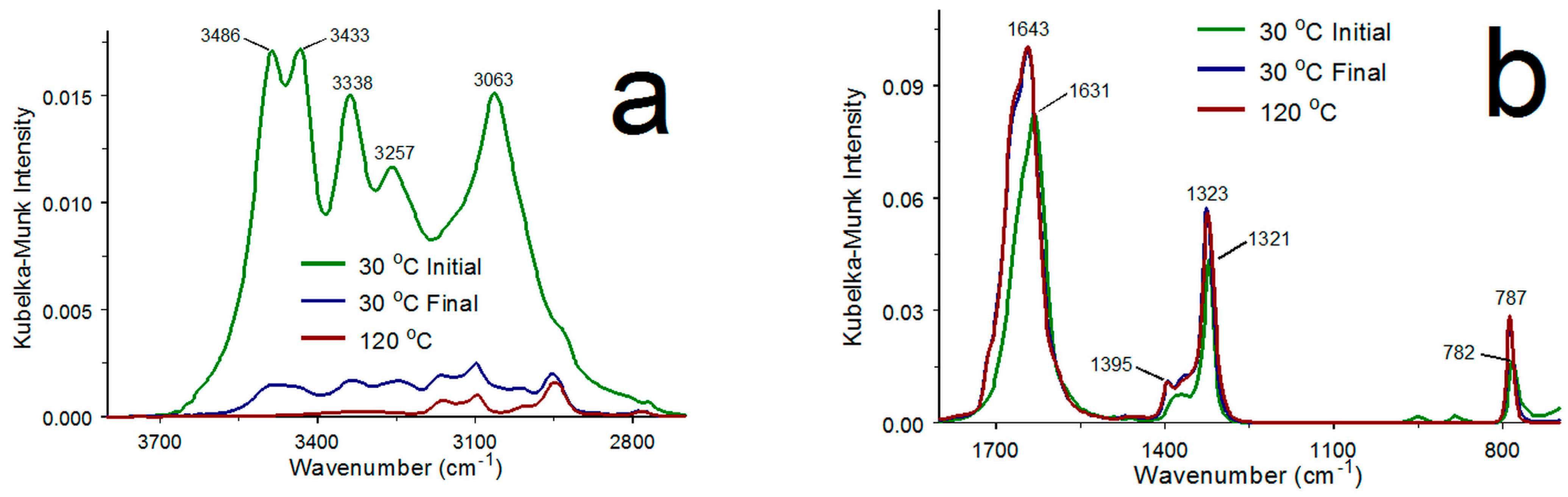
3.1. COM and α-COA Infrared Spectra
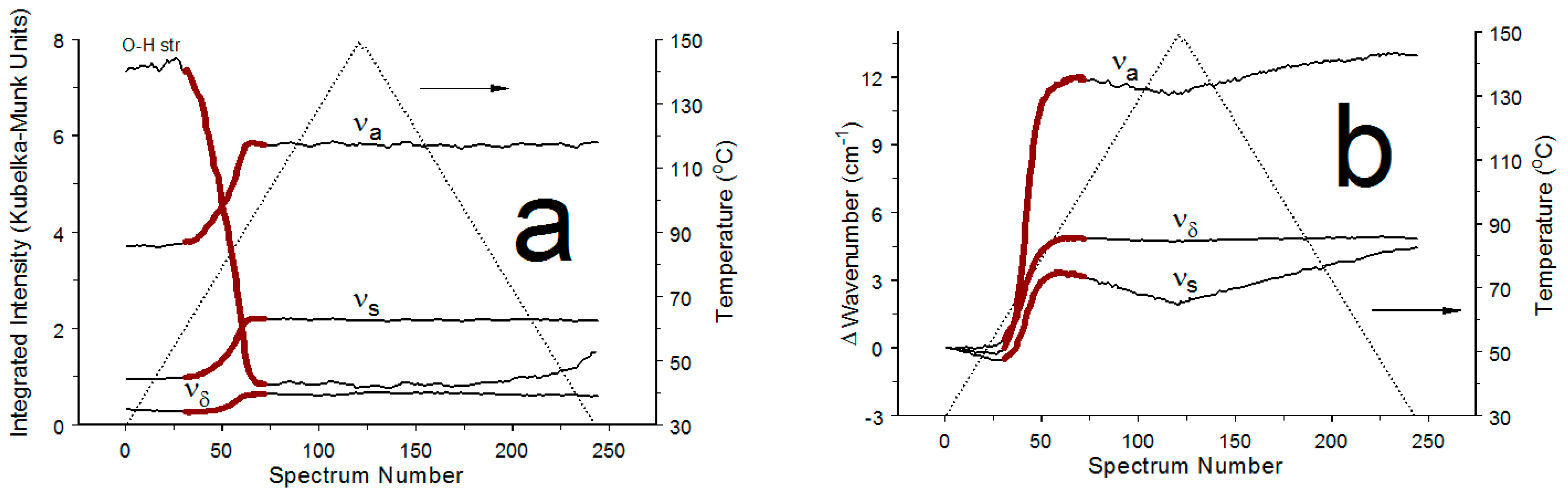
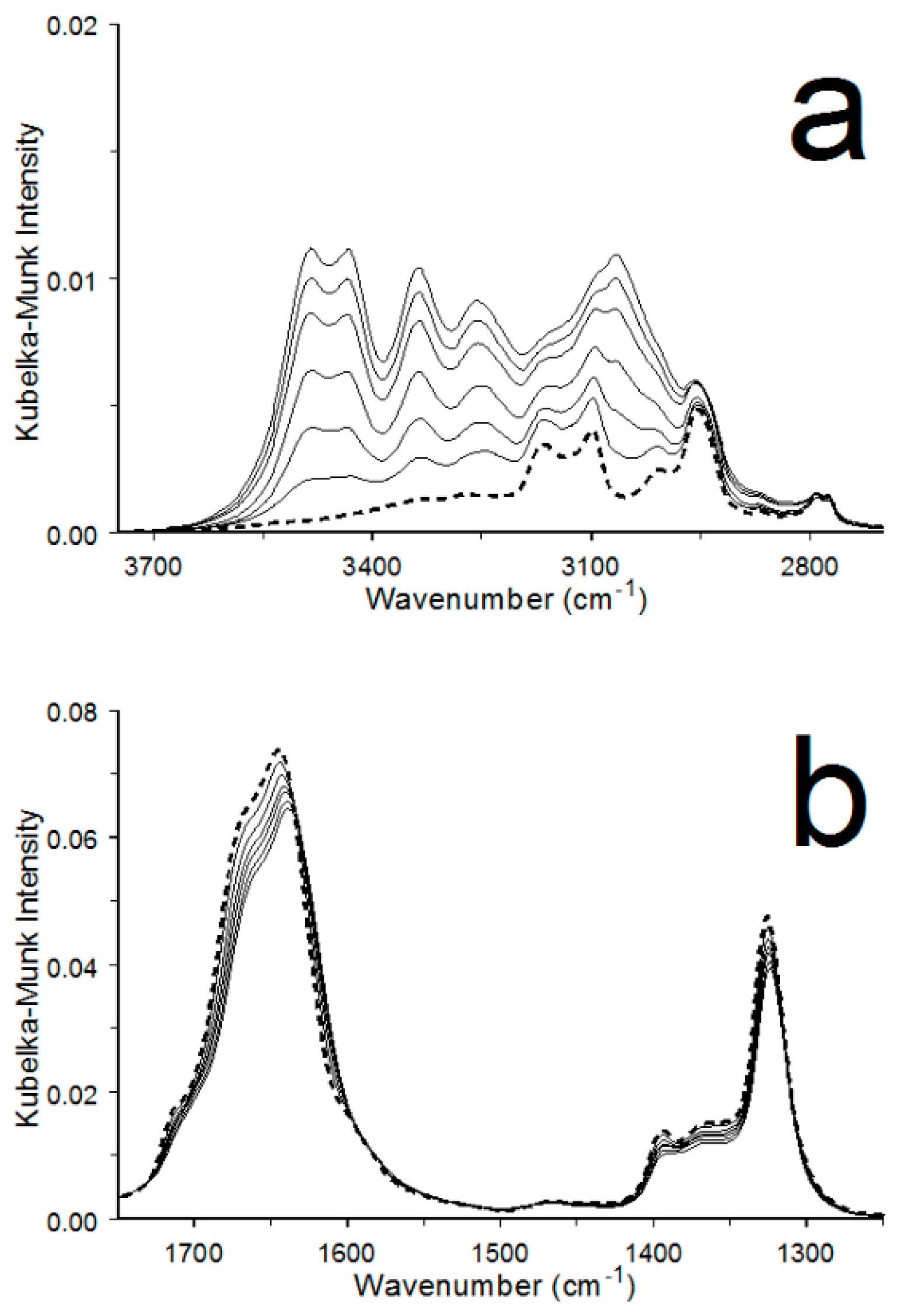
3.2. COM Dehydration
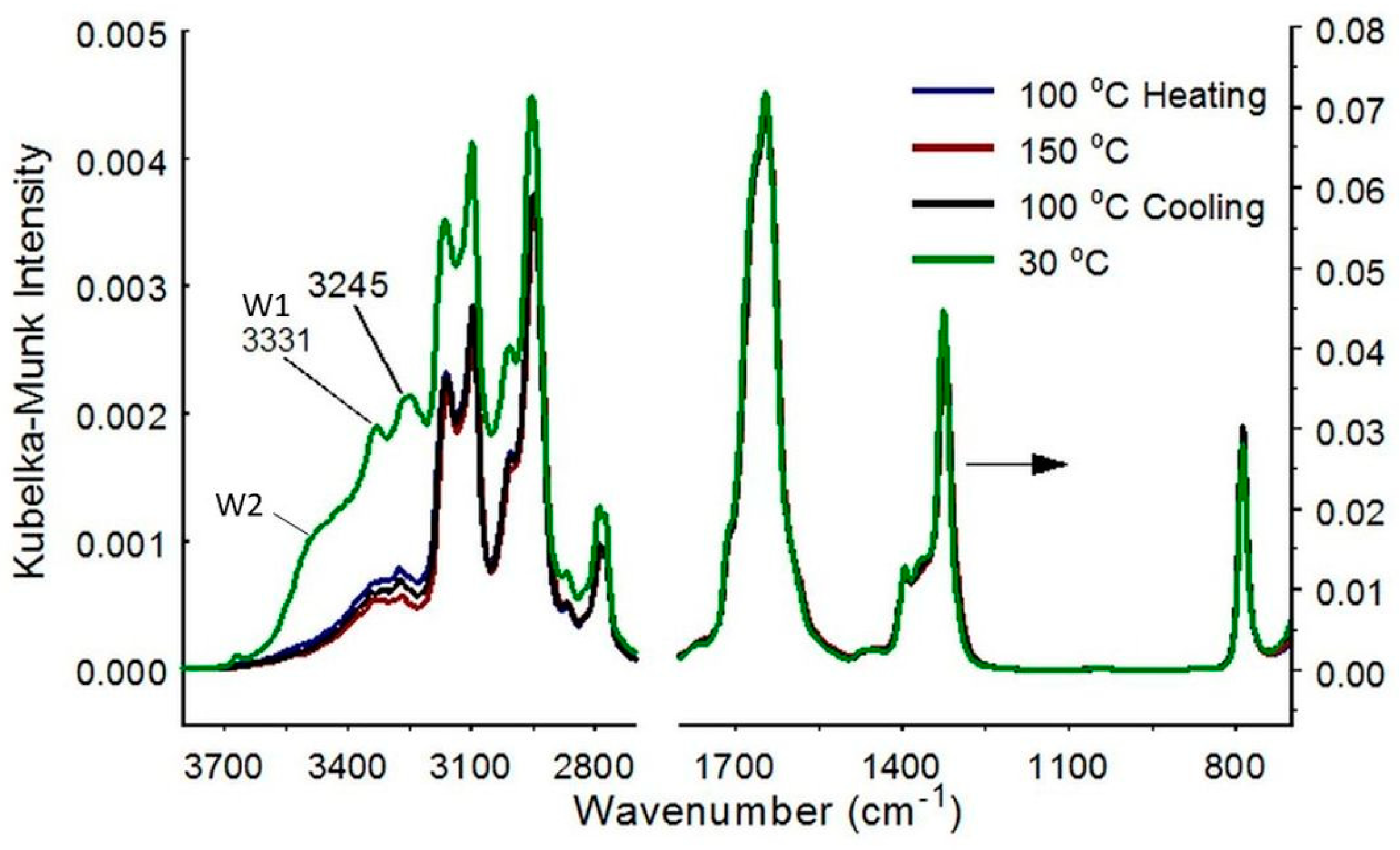
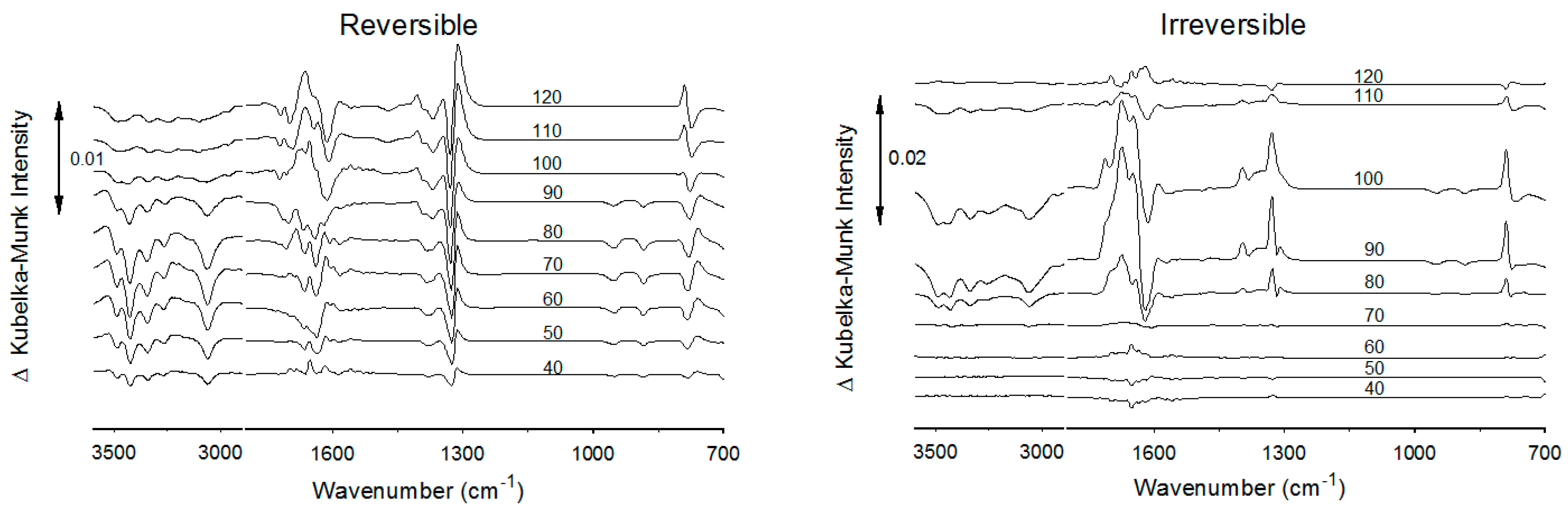
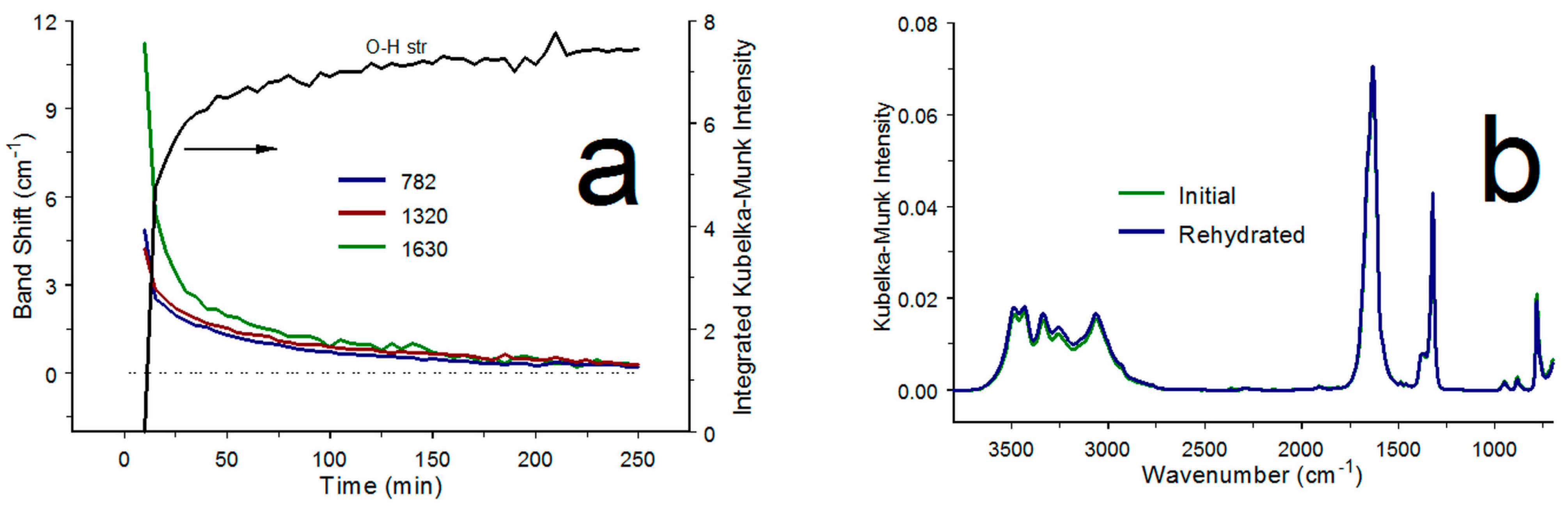
3.3. α-COA Rehydration
4. Discussion
4.1. COM Dehydration Temperature Range
4.2. Distinguishing Dehydration from Crystal Lattice Expansions
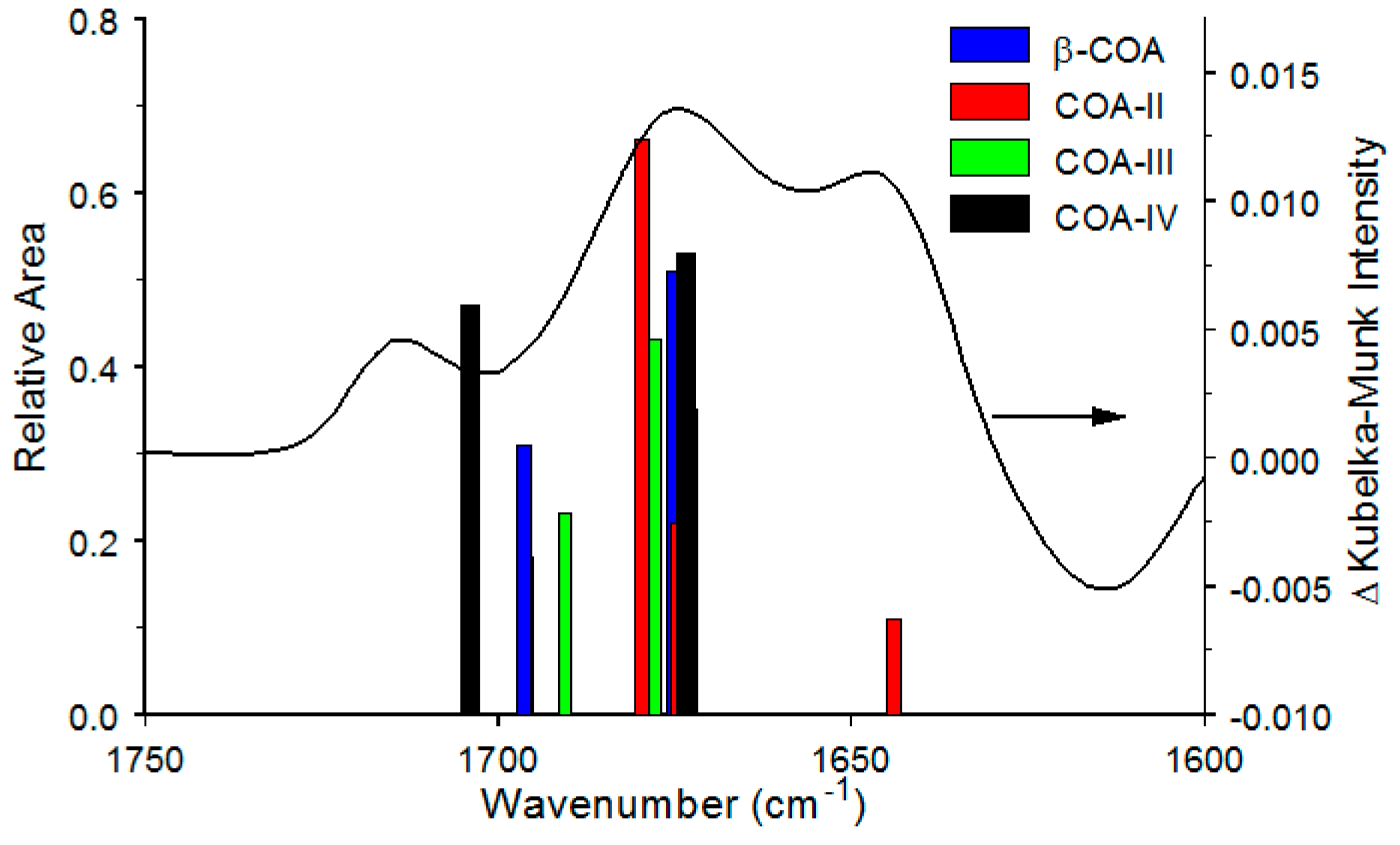
4.3. α-COA Temperature-Dependent Characteristics
4.4. α-COA Hydration
5. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
References
- Nascimento-Dias, B.L.D. Overview about Raman spectroscopy of types of olivine group minerals: A brief review. J. Raman Spectrosc. 2022, 53, 1942–1946. [Google Scholar] [CrossRef]
- Petit, S.; Righi, D.; Madejova, J. Infrared spectroscopy of NH4+-bearing and saturated clay minerals: A review of the study of layer charge. App. Clay Sci. 2006, 34, 22–30. [Google Scholar] [CrossRef]
- Bahfenne, S.; Frost, R.L. A review of the vibrational spectroscopic studies of arsenite, antimonite, and antimonate minerals. Appl. Spectrosc. Rev. 2010, 45, 101–129. [Google Scholar] [CrossRef] [Green Version]
- Armenta, S.; Guardia, M.D.L. Vibrational spectroscopy in soil and sediment analysis. Trends Environ. Anal. Chem. 2014, 2, 43–52. [Google Scholar] [CrossRef]
- Ditler, E.; Luber, S. Vibrational spectroscopy by means of first-principles molecular dynamics simulations. WIREs Comput. Mol. Sci. 2022, 12, e1605. [Google Scholar] [CrossRef]
- Noneman, H.F.; Hollingsworth, M.E.; Singh, J.; White, R.L. A high sensitivity variable temperature infrared spectroscopy investigation of kaolinite structure changes. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2021, 247, 119113. [Google Scholar] [CrossRef]
- Singh, J.; White, R.L. A variable temperature infrared spectroscopy study of NaA zeolite dehydration. Vib. Spectrosc. 2018, 94, 37–42. [Google Scholar] [CrossRef]
- Singh, J.; White, R.L. A variable temperature infrared spectroscopy study of NaY zeolite dehydration. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2020, 231, 118142. [Google Scholar] [CrossRef]
- Tkachenko, Y.; Niedzielski, P. FTIR as a method for qualitative assessment of solid samples in geochemical research: A review. Molecules 2022, 27, 8846. [Google Scholar] [CrossRef]
- White, R.L. Diffuse reflection mid-infrared spectroscopy of neat powders by using a wire mesh “button” sample holder. Anal. Chim. Acta 2020, 1098, 110–116. [Google Scholar] [CrossRef]
- Blum, M.M.; John, H. Historical perspective and modern applications of attenuated total reflectance-Fourier transform infrared spectroscopy (ATR-FTIR). Drug Test. Anal. 2011, 4, 298–302. [Google Scholar] [CrossRef]
- Vilagosh, Z.; Lajevardipour, A.; Appadoo, D.; Juodkazis, S.; Wood, A.W. Using attenuated total reflection (ATR) apparatus to investigate the temperature dependent dielectric properties of water, ice, and tissue-representative fats. Appl. Sci. 2021, 11, 2544. [Google Scholar] [CrossRef]
- Schramm, C. High temperature ATR-FTIR characterization of theinteraction of polycarboxylic acids and organotrialkoxysilanes with cellulosic material. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2020, 243, 118815. [Google Scholar] [CrossRef] [PubMed]
- Sobieski, B.; Chase, B.; Noda, I.; Rabolt, J. Artifact correction in temperature-dependent attenuated total reflection infrared (ATR-IR) spectra. Appl. Spectrosc. 2017, 71, 1868–1875. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; White, R.L. Variable temperature infrared spectroscopy with a button sample holder and thermoelectric heating/cooling. Anal. Chim. Acta 2022, 1198, 339558. [Google Scholar] [CrossRef]
- White, R.L. Thermal analysis by variable temperature infrared spectroscopy with a button sample holder and peltier heating/cooling. Talanta 2023, 258, 124474. [Google Scholar] [CrossRef]
- Kohutova, A.; Honcova, P.; Podzemna, V.; Bezdicka, P.; Vecernikova, E.; Louda, M.; Seidel, J. Thermal analysis of kidney stones and their characterization. J. Therm. Anal. Calorim. 2010, 101, 695–699. [Google Scholar] [CrossRef]
- Valido, I.H.; Rius-Barta, J.M.; Boada, R.; Resina-Gallego, M.; Valiente, M.; Lopez-Mesas, M. Characterization of calcium oxalate hydrates and the transformation process. ChemPhysChem 2020, 21, 2583–2593. [Google Scholar] [CrossRef]
- Conti, C.; Casati, M.; Colombo, C.; Possenti, E.; Realini, M.; Gatta, G.D.; Merlini, M.; Brambilla, L.; Zerbi, G. Synthesis of calcium oxalate trihydrate: New data by vibrational spectroscopy and synchrotron X-ray diffraction. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2015, 150, 721–730. [Google Scholar] [CrossRef]
- Fei, T.; Hao, X.; Bao-Lian, S. A simultaneous thermal study in quantitative analysis of calcium oxalate hydrates. Anal. Methods 2013, 5, 6900–6903. [Google Scholar] [CrossRef]
- Izatulina, A.R.; Gurzhiy, V.V.; Krzhizhanovskaya, M.G.; Kuzmina, M.A.; Leoni, M.; Frank-Kamenetskaya, O.V. Hydrated calcium oxalates: Crystal structures, thermal stability, and phase evolution. Cryst. Growth Des. 2018, 18, 5465–5478. [Google Scholar] [CrossRef]
- Zhao, W.; Sharma, N.; Jones, F.; Raiteri, P.; Gale, J.D.; Demichelis, R. Anhydrous calcium oxalate polymorphism: A combined computational and synchrotron X-ray diffraction study. Cryst. Growth Des. 2016, 16, 5954–5965. [Google Scholar] [CrossRef] [Green Version]
- Hochren, O.; Thomas, A.; Kniep, R. Revealing the crystal structure of anhydrous calcium oxalate, Ca[C2O4], by a combination of atomistic simulation and rietveld refinement. Z. Anorg. Allg. Chem. 2008, 634, 1826–1829. [Google Scholar] [CrossRef]
- Kolezynski, A.; Malecki, A. Theoretical analysis of electronic and structural properties of anhydrous calcium oxalate. J. Therm. Anal. Calorim. 2010, 99, 947–955. [Google Scholar] [CrossRef]
- Milosevic, M.; Berets, S.L. A review of FT-IR diffuse reflection sampling considerations. Appl. Spectrosc. Rev. 2002, 37, 347–364. [Google Scholar] [CrossRef]
- Maraoulaite, D.; White, R.L. Optical efficiency effects on infrared diffuse reflection spectroscopy signal-to-noise ratio. Am. Lab. 2018, 50, 14–17. [Google Scholar]
- White, R.L. Removal of baseline artifacts from variable-temperature diffuse reflectance infrared spectra. Anal. Chem. 1992, 64, 2010–2013. [Google Scholar] [CrossRef]
- Kubelka, P.; Munk, F. Ein Beitrag zur Optik der Farbanstriche. Z. Tech. Phys. 1931, 12, 593–601. [Google Scholar]
- Petrov, I.; Soptrajanov, B. Infrared spectrum of whewellite. Spectrochim. Acta 1975, 31, 309–316. [Google Scholar] [CrossRef]
- Shippey, T.A. Vibrational studies of calcium oxalate monohydrate (whewellite) and an anhydrous phase of calcium oxalate. J. Mol. Struct. 1980, 63, 157–166. [Google Scholar] [CrossRef]
- Guerra-Lopez, J.R.; Guida, J.A.; Vedova, C.O.D. Infrared and Raman studies on renal stones: The use of second derivative infrared spectra. Urol. Res. 2010, 38, 383–390. [Google Scholar] [CrossRef] [PubMed]
- White, R.L.; Ai, J. Variable-temperature diffuse reflectance infrared studies of calcium oxalate monohydrate. App. Spectrosc. 1992, 46, 93–99. [Google Scholar] [CrossRef]
- Christy, A.A.; Nodland, E.; Burnham, A.K.; Kvalheim, O.M.; Dahl, B. Determination of kinetic paramters for the dehydration of calcium oxalate monohydrate by diffuse reflectance FT-IR spectroscopy. Appl. Spectrosc. 1994, 48, 561–568. [Google Scholar] [CrossRef]
- Conti, C.; Casati, M.; Colombo, C.; Realini, M.; Brambilla, L.; Zerbi, G. Phase transformation of calcium oxalate dihydrate-monohydrate: Effects of relative humidity and new spectroscopic data. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2014, 128, 413–419. [Google Scholar] [CrossRef]
- Gulley-Stahl, H.J.; Haas, J.A.; Schmidt, K.A.; Evan, A.P.; Sommer, A.J. Attenuated total internal reflection Fourier transform infrared spectroscopy: A quantitative approach for kidney stone analysis. Appl. Spectrosc. 2009, 63, 759–766. [Google Scholar] [CrossRef] [Green Version]
- Liavitskaya, T.; Vyazovkin, S. Delving into the kinetics of reversible thermal decomposition of solids measured on heating and cooling. J. Phys. Chem. C 2017, 121, 15392–15401. [Google Scholar] [CrossRef]
- Garn, P.D. Thermal analysis: A critique. Anal. Chem. 1961, 33, 1247–1251. [Google Scholar] [CrossRef]
- Nair, C.G.R.; Ninan, K.N. Thermal decomposition studies: Part X. Thermal decomposition kinetics of calcium oxalate monohydrate-correlations with heating rate and sample mass. Thermochim. Acta 1978, 23, 161–169. [Google Scholar] [CrossRef]
- Ninan, K.N. Thermal decomposition kinetics. Part XIV. Kinetics and mechanism of isothermal decomposition of calcium oxalate monohydrate and correlation with sample mass. Thermochim. Acta 1986, 98, 221–236. [Google Scholar] [CrossRef]
- Blazejowski, J.; Zadykowicz, B. Computational prediction of the pattern of thermal gravimetry data for the thermal decomposition of calcium oxalate monohydrate. J. Therm. Anal. Calorim. 2013, 113, 1497–1503. [Google Scholar] [CrossRef] [Green Version]
- Tazzoli, V.; Domeneghetti, C. The crystal structures of whewellite and weddellite: Re-examination and comparison. Am. Miner. 1980, 65, 327–334. [Google Scholar]
- Knoll, C.; Muller, D.; Artner, W.; Welch, J.M.; Werner, A.; Harasek, M.; Weinberger, P. Probing cycle stability and reversibility in thermochemical energy storage-CaC2O4•H2O as perfect match? Appl. Energy 2017, 187, 1–9. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Calcium Oxalate Monohydrate | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Water Vibrations | Oxalate Vibrations | Method | Ref. | ||||||
| ν(OH)2 | ν(OH)1 | ν(OH)1 | 2δ(OH) | ν(OH)2 | νa | νs | νδ | ||
| 3486 | 3433 | 3338 | 3257 | 3063 | 1631 | 1321 | 782 | Button | Figure 2 |
| 3488 | 3428 | 3338 | 3258 | 3058 | 1627 | 1320 | 782 | DRIFTS | [33] |
| 3486 | 3434 | 3334 | 3254 | 3060 | 1627 | 1319 | 781 | DRIFTS | [32] |
| 3485 | 3428 | 3339 | 3259 | 3059 | - | 1328 | 781 | Microscope | [34] |
| 3483 | 3429 | 3336 | 3258 | 3058 | 1624–1622 | 1320–1316 | 783–781 | Microscope | [19] |
| 3496 | 3429 | 3341 | 3269 | 3065 | 1619 | 1318 | 780 | KBr Pellet | [31] |
| 3495 | 3440 | 3340 | 3250 | 3060 | 1620 | 1316 | 782 | KBr Pellet | [29] |
| 3488 | 3430 | 3338 | 3258 | 3057 | 1622 | 1317 | 781 | Mull | [30] |
| 3481 | 3427 | 3336 | 3255 | 3050 | 1620 * | 1313 | 778 | ATR | [18] |
| - | - | - | - | - | 1620 | 1315 | 780 | ATR | [35] |
| Anhydrous Calcium Oxalate | |||||||||
| 1643 | 1323 | 787 | Button | Figure 2 | |||||
| 1636 | 1320 | 787 | Mull | [30] | |||||
| 1640–1638 | 1330–1327 | 784–782 | Microscope | [19] | |||||
| 1638 | 1335 | 780 | Microscope | [34] | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
White, R.L. Infrared Spectroscopic Investigations of Calcium Oxalate Monohydrate (Whewellite) Dehydration/Rehydration. Minerals 2023, 13, 783. https://doi.org/10.3390/min13060783
White RL. Infrared Spectroscopic Investigations of Calcium Oxalate Monohydrate (Whewellite) Dehydration/Rehydration. Minerals. 2023; 13(6):783. https://doi.org/10.3390/min13060783
Chicago/Turabian StyleWhite, Robert L. 2023. "Infrared Spectroscopic Investigations of Calcium Oxalate Monohydrate (Whewellite) Dehydration/Rehydration" Minerals 13, no. 6: 783. https://doi.org/10.3390/min13060783
APA StyleWhite, R. L. (2023). Infrared Spectroscopic Investigations of Calcium Oxalate Monohydrate (Whewellite) Dehydration/Rehydration. Minerals, 13(6), 783. https://doi.org/10.3390/min13060783