
A Novel Surface Passivation Method of Pyrite within Rocks in Underwater Environments to Mitigate Acid Mine Drainage at Its Source

Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Passivating Process
2.3. Electrochemical Measurements
2.4. Chemical Leaching Experiments
2.5. Characterization
3. Results and Discussion
3.1. Electrochemistry Results
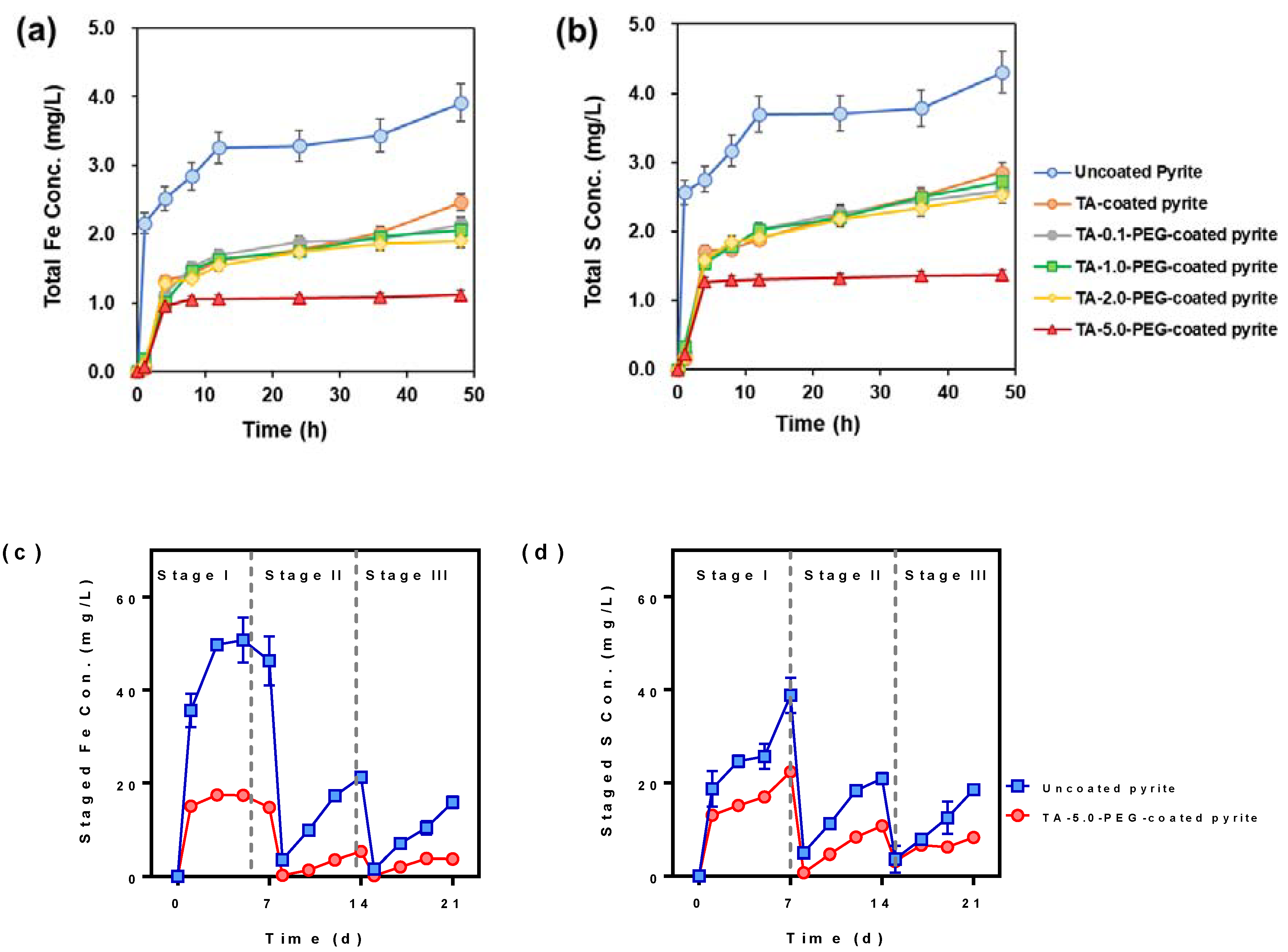
3.2. Chemical Leaching Experiments
3.3. Characterization of the Coated Pyrite
3.3.1. Surface Morphology of Coatings Using SEM
3.3.2. Wettability Properties of Coatings
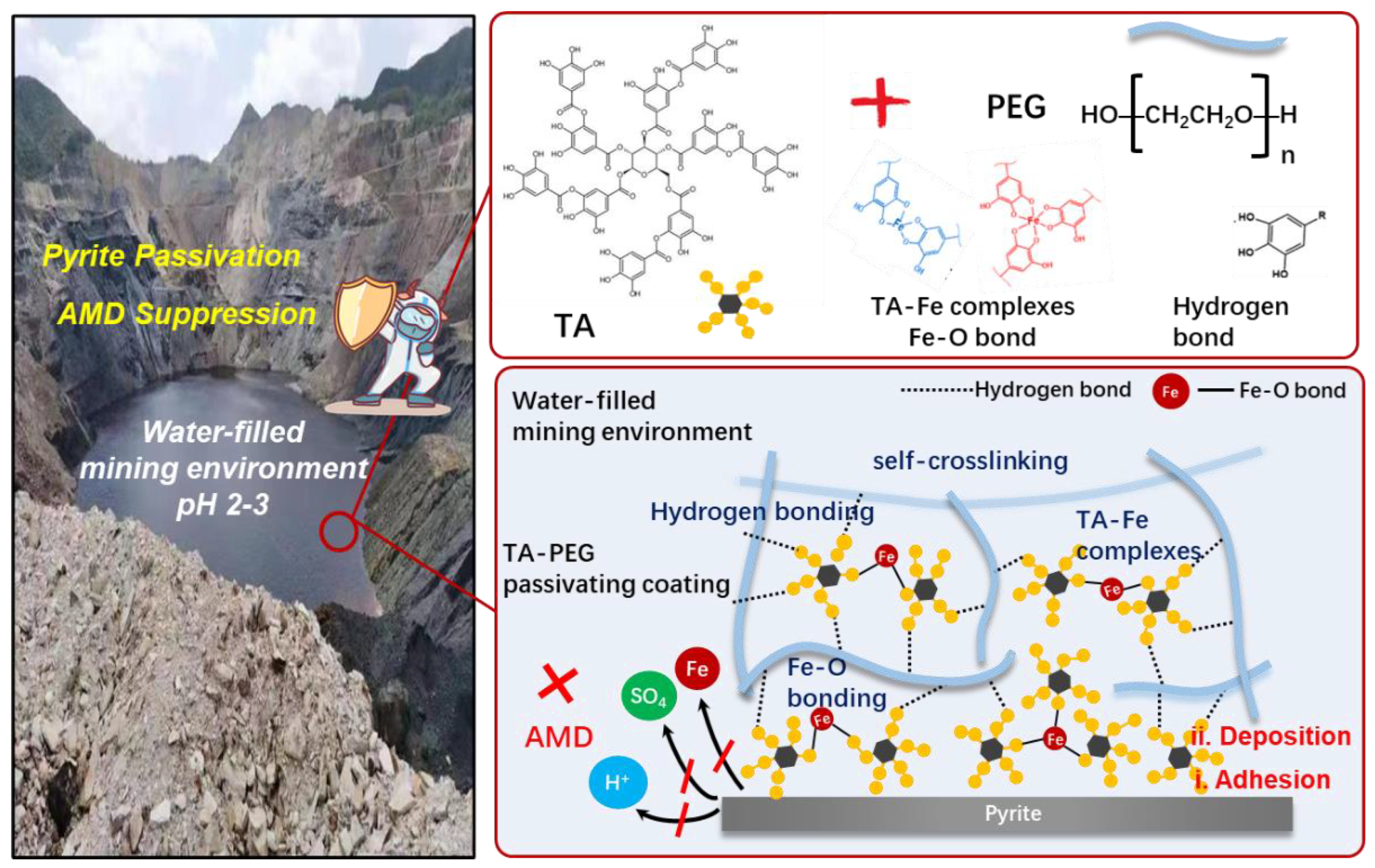
3.4. Mechanism of Passivation Coatings
3.4.1. FTIR Analysis
3.4.2. Raman Analysis
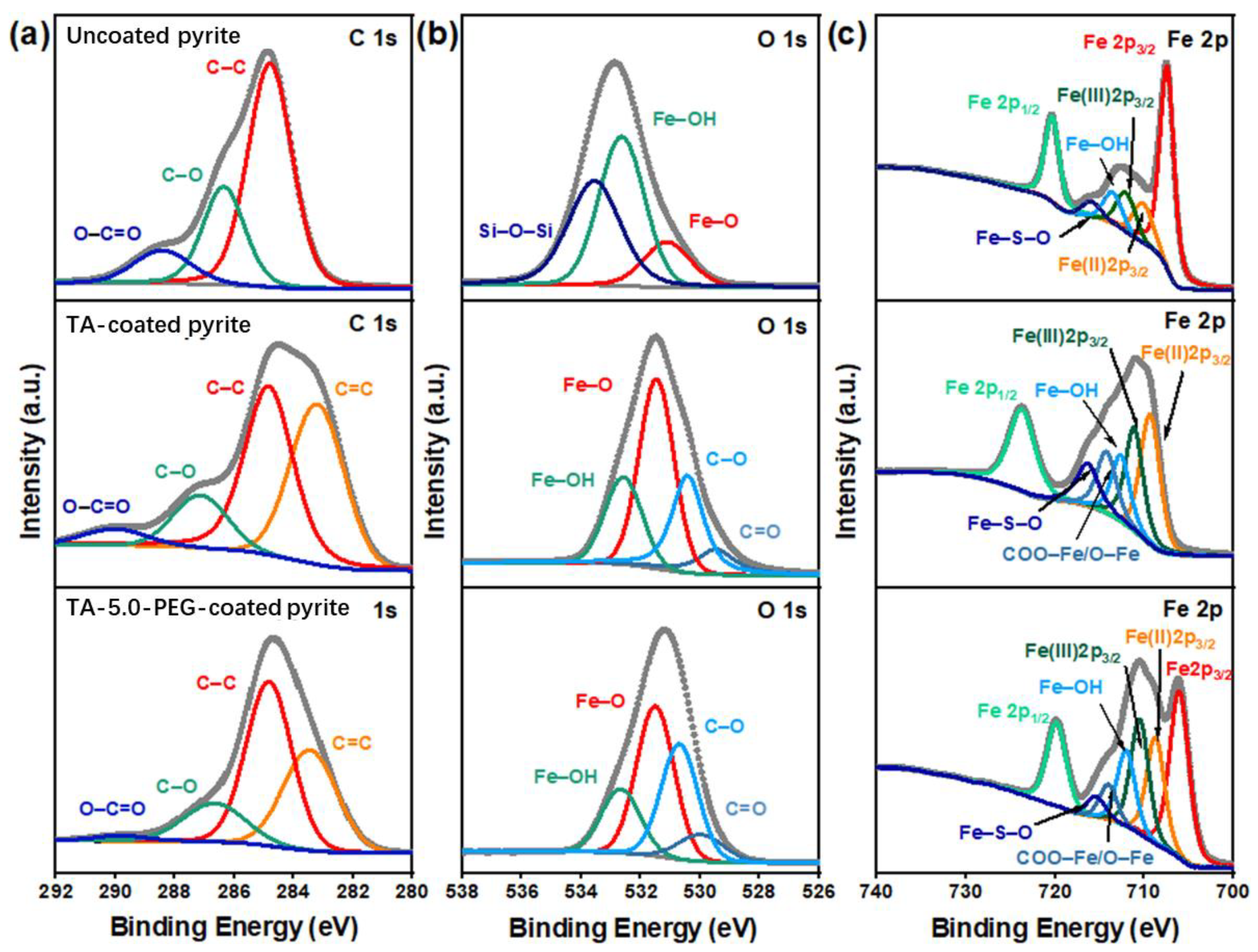
3.4.3. XPS Analysis
3.4.4. Summary of Passivating Mechanism on the Pyrite Surface
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hudson-Edwards, K. Tackling mine wastes. Science 2016, 352, 288–290. [Google Scholar] [CrossRef] [PubMed]
- Nordstrom, D.K. Mine waters: Acidic to circmneutral. Elements 2011, 7, 393–398. [Google Scholar] [CrossRef]
- Fan, L.; Zhao, F.; Liu, J.; Hudson-Edwards, K.A. Dissolution of realgar by Acidithiobacillus ferrooxidans in the presence and absence of zerovalent iron: Implications for remediation of iron-deficient realgar tailings. Chemosphere 2018, 209, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Burton, E.D.; Karimian, N.; Hamilton, J.L.; Frierdich, A.J. Iron Isotopes in Acid Mine Drainage: Extreme and Divergent Fractionation between Solid (Schwertmannite, Jarosite, and Ferric Arsenate) and Aqueous Species. Environ. Sci. Technol. 2022, 56, 18060–18068. [Google Scholar] [CrossRef] [PubMed]
- Hammond, C.M.; Root, R.A.; Maier, R.M.; Chorover, J. Arsenic and iron speciation and mobilization during phytostabilization of pyritic mine tailings. Geochim. Cosmochim. Acta 2020, 286, 306–323. [Google Scholar] [CrossRef]
- Von Gunten, K.; Bishop, B.; Plata Enriquez, I.; Alam, M.S.; Blanchard, P.; Robbins, L.J.; Feng, R.; Konhauser, K.O.; Alessi, D.S. Colloidal transport mechanisms and sequestration of U, Ni, and As in meromictic mine pit lakes. Geochim. Cosmochim. Acta 2019, 265, 292–312. [Google Scholar] [CrossRef]
- Fan, L.; Zhu, T.; Yang, Y.; Han, T.; Qiao, Z.; Huang, X.; Zhai, W.; Pan, X.; Zhang, D. Iron colloidal transport mechanisms and sequestration of As, Ni, and Cu along AMD-induced environmental gradients. Sci. Total. Environ. 2023, 898, 165513. [Google Scholar] [CrossRef] [PubMed]
- Park, I.; Tabelin, C.B.; Jeon, S.; Li, X.; Seno, K.; Ito, M.; Hiroyoshi, N. A review of recent strategies for acid mine drainage prevention and mine tailings recycling. Chemosphere 2019, 219, 588–606. [Google Scholar] [CrossRef]
- Ren, Y.; Cao, X.; Wu, P.; Li, L. Experimental insights into the formation of secondary minerals in acid mine drainage-polluted karst rivers and their effects on element migration. Sci. Total. Environ. 2023, 858, 160076. [Google Scholar] [CrossRef] [PubMed]
- Tu, Z.; Wu, Q.; He, H.; Zhou, S.; Liu, J.; He, H.; Liu, C.; Dang, Z.; Reinfelder, J.R. Reduction of acid mine drainage by passivation of pyrite surfaces: A review. Sci. Total. Environ. 2022, 832, 155116. [Google Scholar] [CrossRef]
- Anekwe, I.M.S.; Isa, Y.M. Bioremediation of acid mine drainage—Review. Alexandria Eng. J. 2023, 65, 1047–1075. [Google Scholar] [CrossRef]
- Qian, G.; Schumann, R.C.; Li, J.; Short, M.D.; Fan, R.; Li, Y.; Kawashima, N.; Zhou, Y.; Smart, R.S.C.; Gerson, A.R. Strategies for Reduced Acid and Metalliferous Drainage by Pyrite Surface Passivation. Minerals 2017, 7, 42. [Google Scholar] [CrossRef]
- Pabst, T.; Bussiere, B.; Aubertin, M.; Molson, J. Comparative performance of cover systems to prevent acid mine drainage from pre-oxidized tailings: A numerical hydro-geochemical assessment. J. Contam. Hydrol. 2018, 214, 39–53. [Google Scholar] [CrossRef]
- Beauchemin, S.; Clemente, J.S.; Thibault, Y.; Langley, S.; Gregorich, E.G.; Tisch, B. Geochemical stability of acid-generating pyrrhotite tailings 4 to 5 years after addition of oxygen-consuming organic covers. Sci. Total. Environ. 2018, 645, 1643–1655. [Google Scholar] [CrossRef] [PubMed]
- Pierre Louis, A.M.; Yu, H.; Shumlas, S.L.; Van Aken, B.; Schoonen, M.A.; Strongin, D.R. Effect of Phospholipid on Pyrite Oxidation and Microbial Communities under Simulated Acid Mine Drainage (AMD) Conditions. Environ. Sci. Technol. 2015, 49, 7701–7708. [Google Scholar] [CrossRef]
- Acharya, B.S.; Kharel, G. Acid mine drainage from coal mining in the United States—An overview. J. Hydrol. 2020, 588, 125061. [Google Scholar] [CrossRef]
- Park, I.; Higuchi, K.; Tabelin, C.B.; Jeon, S.; Ito, M.; Hiroyoshi, N. Suppression of arsenopyrite oxidation by microencapsulation using ferric-catecholate complexes and phosphate. Chemosphere 2021, 269, 129413. [Google Scholar] [CrossRef]
- Li, X.; Park, I.; Tabelin, C.B.; Naruwa, K.; Goda, T.; Harada, C.; Jeon, S.; Ito, M.; Hiroyoshi, N. Enhanced pyrite passivation by carrier-microencapsulation using Fe-catechol and Ti-catechol complexes. J. Hazard. Mater. 2021, 416, 126089. [Google Scholar] [CrossRef]
- Park, I.; Tabelin, C.B.; Seno, K.; Jeon, S.; Ito, M.; Hiroyoshi, N. Simultaneous suppression of acid mine drainage formation and arsenic release by Carrier-microencapsulation using aluminum-catecholate complexes. Chemosphere 2018, 205, 414–425. [Google Scholar] [CrossRef]
- Yu, M.; Feng, J.; Yang, Q.; Dang, Z.; Zhang, L. Inhibition of organosilane/ATP@HQ self-healing passivator for pyrite oxidation. Chemosphere 2022, 287, 132342. [Google Scholar] [CrossRef]
- Ouyang, Y.; Liu, Y.; Zhu, R.; Ge, F.; Xu, T.; Luo, Z.; Liang, L. Pyrite oxidation inhibition by organosilane coatings for acid mine drainage control. Miner. Eng. 2015, 72, 57–64. [Google Scholar] [CrossRef]
- Dong, Y.; Zeng, W.; Lin, H.; He, Y. Preparation of a novel water-soluble organosilane coating and its performance for inhibition of pyrite oxidation to control acid mine drainage at the source. Appl. Surf. Sci. 2020, 531, 147328. [Google Scholar] [CrossRef]
- Li, D.; Gong, B.; Liu, Y.; Dang, Z. Self-healing coatings based on PropS-SH and pH-responsive HNT-BTA nanoparticles for inhibition of pyrite oxidation to control acid mine drainage. Chem. Eng. J. 2021, 415, 128993. [Google Scholar] [CrossRef]
- Liu, Y.; Hu, X.; Xu, Y. PropS-SH/SiO2 nanocomposite coatings for pyrite oxidation inhibition to control acid mine drainage at the source. J. Hazard. Mater. 2017, 338, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Zhou, C.; Yang, Q.; Dang, Z.; Zhang, L. Performance and mechanisms of PropS-SH/Ce(dbp)(3) coatings in the inhibition of pyrite oxidationtion for acid mine drainage control. Environ. Pollut. 2023, 322, 121162. [Google Scholar] [CrossRef] [PubMed]
- Messersmith, R.E.; Bartlett, M.E.; Rose, D.J.; Smith, D.A.; Patchan, M.W.; Benkoski, J.J.; Trexler, M.M.; Hoffman, C.M. Rapid Underwater Adhesive Utilizing Crosslinker and Amine Catalyst-Filled Microcapsules. ACS Appl. Polym. Mater. 2021, 3, 996–1002. [Google Scholar] [CrossRef]
- Lee, D.; Hwang, H.; Kim, J.S.; Park, J.; Youn, D.; Kim, D.; Hahn, J.; Seo, M.; Lee, H. VATA: A Poly(vinyl alcohol)- and Tannic Acid-Based Nontoxic Underwater Adhesive. ACS Appl. Mater. Interfaces 2020, 12, 20933–20941. [Google Scholar] [CrossRef]
- Guo, Z.; Xie, W.; Lu, J.; Guo, X.; Zhao, L. Tannic Acid-based Metal Phenolic Networks for Bio-applications: A Review. J. Mater. Chem. B 2021, 9, 4098–4110. [Google Scholar] [CrossRef]
- Pucci, C.; Martinelli, C.; De Pasquale, D.; Battaglini, M.; di Leo, N.; Degl’Innocenti, A.; Belenli Gumus, M.; Drago, F.; Ciofani, G. Tannic Acid-Iron Complex-Based Nanoparticles as a Novel Tool against Oxidative Stress. ACS Appl. Mater. Interfaces 2022, 14, 15927–15941. [Google Scholar] [CrossRef] [PubMed]
- Gülçin, İ.; Huyut, Z.; Elmastaş, M.; Aboul-Enein, H.Y. Radical scavenging and antioxidant activity of tannic acid. Arabian J. Chem. 2010, 3, 43–53. [Google Scholar] [CrossRef]
- Kaghazchi, L.; Naderi, R.; Ramezanzadeh, B. Construction of a high-performance anti-corrosion film based on the green tannic acid molecules and zinc cations on steel: Electrochemical/Surface investigations. Constr. Build. Mater. 2020, 262, 120861. [Google Scholar] [CrossRef]
- Magdalena, K. Chelation of Cu(II), Zn(II), and Fe(II) by tannin constituents of selected edible nuts. Int. J. Mol. Sci. 2009, 10, 5485–5497. [Google Scholar] [CrossRef] [PubMed]
- Ejima, H.; Richardson, J.J.; Liang, K.; Best, J.P.; Koeverden, M.P.V.; Such, G.K.; Cui, J.; Caruso, F. One-Step Assembly of Coordination Complexes for Versatile Film and Particle Engineering. Science 2013, 341, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Geng, X.W.; Pan, Y.H.; Ma, Y.N.; Ma, Y.X.; Gao, S.Z.; Huang, X.J. Synthesis and characterization of tannic acid-PEG hydrogel via Mitsunobu polymerization. RSC Adv. 2020, 10, 1724–1732. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, S.; Zhao, S.; Kang, H.; Wang, Z.; Xia, C.; Yu, Y.; Li, J. Facile biomimetic self-coacervation of tannic acid and polycation: Tough and wide pH range of underwater adhesives. Chem. Eng. J. 2021, 404, 127069. [Google Scholar] [CrossRef]
- Espina, A.; Canamares, M.V.; Jurasekova, Z.; Sanchez-Cortes, S. Analysis of Iron Complexes of Tannic Acid and Other Related Polyphenols as Revealed by Spectroscopic Techniques: Implications in the Identification and Characterization of Iron Gall Inks in Historical Manuscripts. ACS Omega 2022, 7, 27937–27949. [Google Scholar] [CrossRef]
- Li, D.; Chen, X.; Liu, C.; Tian, J.; Li, F.; Liu, Y. Suppression of pyrite oxidation by co-depositing bio-inspired PropS-SH-tannic acid coatings for the source control acid mine drainage. Sci. Total. Environ. 2023, 862, 160857. [Google Scholar] [CrossRef]
- Fan, H.; Wang, J.; Jin, Z. Tough, Swelling-Resistant, Self-Healing, and Adhesive Dual-Cross-Linked Hydrogels Based on Polymer–Tannic Acid Multiple Hydrogen Bonds. Macromolecules 2018, 51, 1696–1705. [Google Scholar] [CrossRef]
- Sun, D.; Li, M.; Zhang, M.; Cui, R.; Yang, Z.; Yu, L.; Wang, D.; Yao, W. Utilization and Mechanisms of Tannic Acid as a Depressant for Chalcopyrite and Pyrite Separation. ACS Omega 2023, 8, 30474–30482. [Google Scholar] [CrossRef]
- You, G.-X.; Yu, C.-C.; Lu, Y.; Dang, Z. Evaluation of the protective effect of polysiloxane coating on pyrite with electrochemical techniques. Electrochim. Acta 2013, 93, 65–71. [Google Scholar] [CrossRef]
- Cai, Y.; Pan, Y.; Xue, J.; Sun, Q.; Su, G.; Li, X. Comparative XPS study between experimentally and naturally weathered pyrites. Appl. Surf. Sci. 2009, 255, 8750–8760. [Google Scholar] [CrossRef]
- Feng, X.; Chen, Z.; Wang, S.; Cen, L.; Ni, B.J.; Liu, Q. Insights into the weathering behavior of pyrite in alkaline soil through electrochemical characterizations: Actual hazards or potentially benefits? J. Hazard. Mater. 2023, 451, 131145. [Google Scholar] [CrossRef]
- Solmaz, R.; Karda, G.; Yazc, B.; Erbil, M. Adsorption and corrosion inhibitive properties of 2-amino-5-mercapto-1,3,4-thiadiazole on mild steel in hydrochloric acid media. Colloids Surf. A 2008, 312, 7–17. [Google Scholar] [CrossRef]
- Zheng, K.; Li, H.; Xu, L.; Li, S.; Wang, L.; Wen, X.; Liu, Q. The influence of humic acids on the weathering of pyrite: Electrochemical mechanism and environmental implications. Environ. Pollut. 2019, 251, 738–745. [Google Scholar] [CrossRef]
- Mukarrom, F.; Pranoto; Karsidi, R.; Gravitiani, E.; Astuti, F.; Maharditya, W. The assessment of claystone, quartz and coconut shell charcoal for adsorbing heavy metals ions in acid mine drainage. IOP Conf. Ser. Mater. Sci. Eng. 2020, 858, 012040. [Google Scholar] [CrossRef]
- Shafiq, M.; Yasin, T.; Saeed, S. Synthesis and characterization of linear low-density polyethylene/sepiolite nanocomposites. J. Appl. Polym. Sci. 2011, 123, 1718–1723. [Google Scholar] [CrossRef]
- Justin Dhanaraj, C.; Sivasankaran Nair, M. Synthesis, characterization, and antimicrobial studies of some Schiff-base metal (II) complexes. J. Coord. Chem. 2009, 62, 4018–4028. [Google Scholar] [CrossRef]
- Kaghazchi, L.; Naderi, R.; Ramezanzadeh, B. Synergistic mild steel corrosion mitigation in sodium chloride-containing solution utilizing various mixtures of phytic acid molecules and Zn2+ ions. J. Mol. Liq. 2021, 323, 114589. [Google Scholar] [CrossRef]
- Berkh, K.; Rammlmair, D. The effect of Co substitution on the Raman spectra of pyrite: Potential as an assaying tool. Eur. J. Mineral. 2022, 34, 259–274. [Google Scholar] [CrossRef]
- Chan-Rosado, G.; Pech-Canul, M.A. Influence of native oxide film age on the passivation of carbon steel in neutral aqueous solutions with a dicarboxylic acid. Corros. Sci. 2019, 153, 19–31. [Google Scholar] [CrossRef]
- Mei, L.; Liao, L.; Wang, Z.; Xu, C. Interactions between Phosphoric/Tannic Acid and Different Forms of FeOOH. Adv. Mater. Sci. Eng. 2015, 2015, 1–10. [Google Scholar] [CrossRef]
- Wang, B.; Moon, J.R.; Ryu, S.; Park, K.D.; Kim, J. Antibacterial 3D graphene composite gel with polyaspartamide and tannic acid containing in situ generated Ag nanoparticle. Polym. Compos. 2020, 41, 2578–2587. [Google Scholar] [CrossRef]
- Geißler, S.; Barrantes, A.; Tengvall, P.; Messersmith, P.B.; Tiainen, H.J.L. Deposition kinetics of bioinspired phenolic coatings on titanium surfaces. Langmuir 2016, 32, 8050–8060. [Google Scholar] [CrossRef] [PubMed]
- Primo, E.N.; Bracamonte, M.V.; Luque, G.L.; Bercoff, P.G.; Leiva, E.P.M.; Barraco, D.E. Mechanochemically synthesized pyrite and its electrochemical behavior as cathode for lithium batteries. J. Solid State Electrochem. 2019, 23, 1929–1938. [Google Scholar] [CrossRef]
- Zhu, J.; Xian, H.; Lin, X.; Tang, H.; Du, R.; Yang, Y.; Zhu, R.; Liang, X.; Wei, J.; Teng, H.H.; et al. Surface structure-dependent pyrite oxidation in relatively dry and moist air: Implications for the reaction mechanism and sulfur evolution. Geochim. Cosmochim. Acta 2018, 228, 259–274. [Google Scholar] [CrossRef]
- Herbert, F.W.; Krishnamoorthy, A.; Ma, W.; Van Vliet, K.J.; Yildiz, B. Dynamics of point defect formation, clustering and pit initiation on the pyrite surface. Electrochim. Acta 2014, 127, 416–426. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrodes | Rs/Ω·cm2 | Q1 | Rct/104 Ω·cm2 | Q2 | Rf/104 Ω·cm2 | ||
|---|---|---|---|---|---|---|---|
| Y1/10−4 S/sn cm−2 n | Y2/10−4 S/sn cm−2 n | ||||||
| Uncoated pyrite | 30.9 | 4.1 | 0.9 | 1.8 | 37.9 | 0.8 | 0.8 |
| TA-coated pyrite | 35.7 | 3.6 | 0.8 | 3.6 | 32.5 | 0.8 | 1.9 |
| TA-0.1–PEG-coated pyrite | 38.3 | 3.1 | 0.8 | 5.9 | 25.4 | 0.8 | 2.8 |
| TA-1.0–PEG-coated pyrite | 38.9 | 3.1 | 0.9 | 10.7 | 17.8 | 0.6 | 19.8 |
| TA-2.0–PEG-coated pyrite | 39.2 | 2.9 | 0.9 | 11.2 | 19.7 | 0.9 | 27.1 |
| TA-5.0–PEG-coated pyrite | 34.1 | 1.7 | 0.9 | 15.6 | 16.4 | 0.6 | 39.7 |
| Electrodes | Ecorr (mV/SCE) | icorr (μA/cm2) |
|---|---|---|
| Uncoated pyrite | 284 | 6.71 |
| TA-coated pyrite | 312 | 4.09 |
| TA-0.1–PEG-coated pyrite | 371 | 3.12 |
| TA-1.0–PEG-coated pyrite | 384 | 2.51 |
| TA-2.0–PEG-coated pyrite | 389 | 2.35 |
| TA-5.0–PEG-coated pyrite | 411 | 1.89 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, L.; Han, T.; Huang, X.; Yang, Y.; Zhu, T.; Zhai, W.; Zhang, D.; Pan, X. A Novel Surface Passivation Method of Pyrite within Rocks in Underwater Environments to Mitigate Acid Mine Drainage at Its Source. Minerals 2024, 14, 973. https://doi.org/10.3390/min14100973
Fan L, Han T, Huang X, Yang Y, Zhu T, Zhai W, Zhang D, Pan X. A Novel Surface Passivation Method of Pyrite within Rocks in Underwater Environments to Mitigate Acid Mine Drainage at Its Source. Minerals. 2024; 14(10):973. https://doi.org/10.3390/min14100973
Chicago/Turabian StyleFan, Lijun, Tiancheng Han, Xianxing Huang, Yixuan Yang, Tao Zhu, Weiwei Zhai, Daoyong Zhang, and Xiangliang Pan. 2024. "A Novel Surface Passivation Method of Pyrite within Rocks in Underwater Environments to Mitigate Acid Mine Drainage at Its Source" Minerals 14, no. 10: 973. https://doi.org/10.3390/min14100973
APA StyleFan, L., Han, T., Huang, X., Yang, Y., Zhu, T., Zhai, W., Zhang, D., & Pan, X. (2024). A Novel Surface Passivation Method of Pyrite within Rocks in Underwater Environments to Mitigate Acid Mine Drainage at Its Source. Minerals, 14(10), 973. https://doi.org/10.3390/min14100973