

Accelerated Carbonation of High-Calcite Wollastonite Tailings


Abstract
1. Introduction
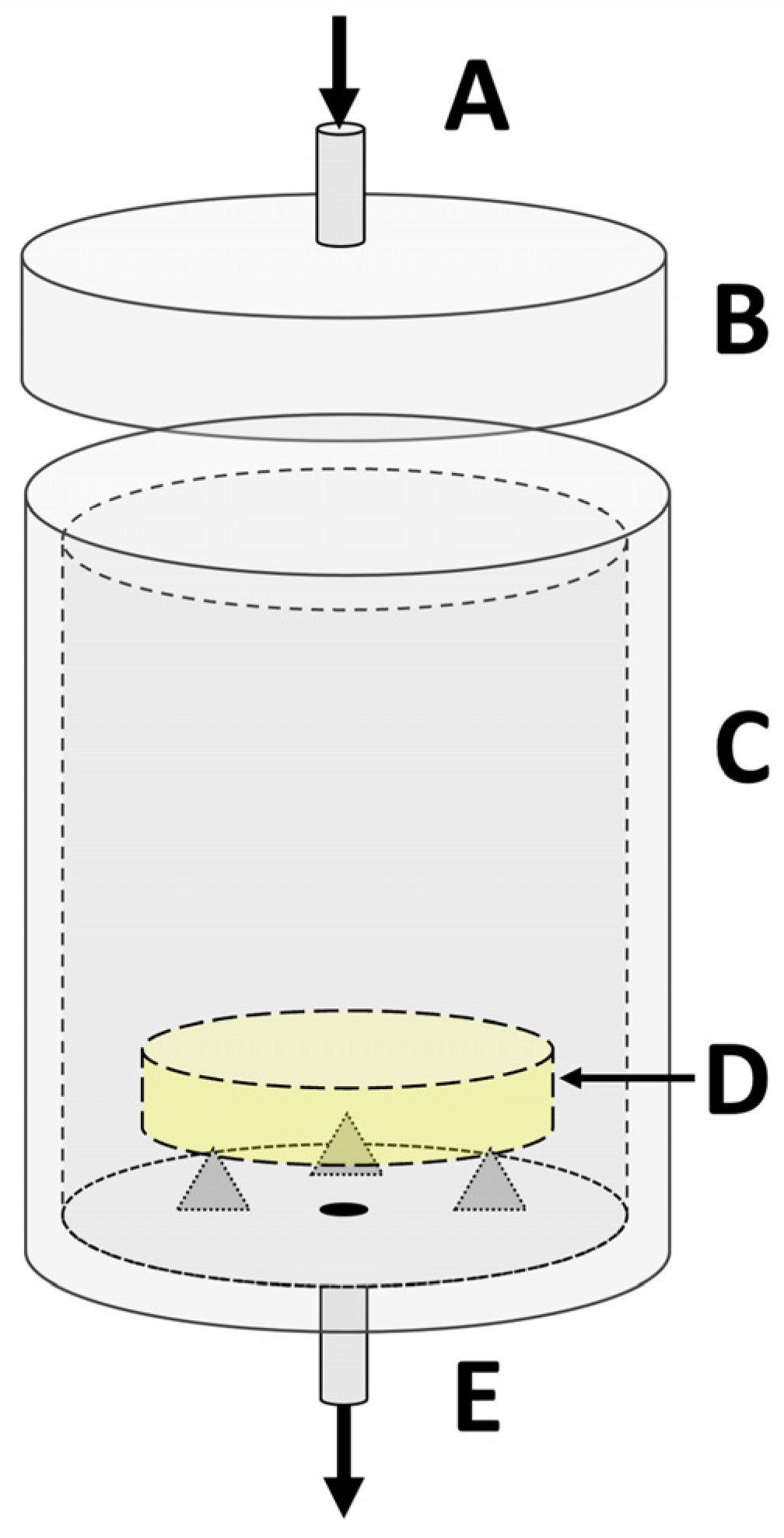
2. Materials and Methods
3. Results
3.1. Pre-Carbonation Characterization

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
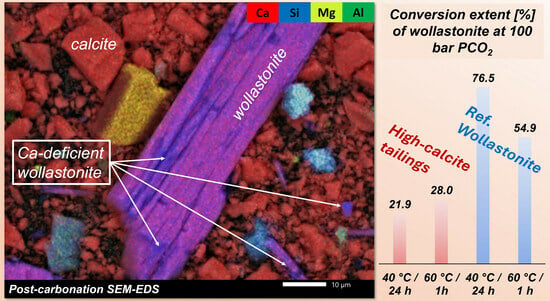
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Wollastonite Content [wt%] | Particle Size (D50) [µm] | SSA [m2/g] | LOI [%] |
|---|---|---|---|---|
| A1 (coarse tailing) | 30.3 | 200 | 0.27 | 10.8 |
| B1 (fine tailing) | 21.1 | 5 | 2.25 | 33.7 |
| W325 | >87 * | 12.5 * | 1.35 * | 0.33 |
| W635 | >87 * | 3.5 * | 3.25 * | 3.26 |
3.2. Post-Carbonation Characterization
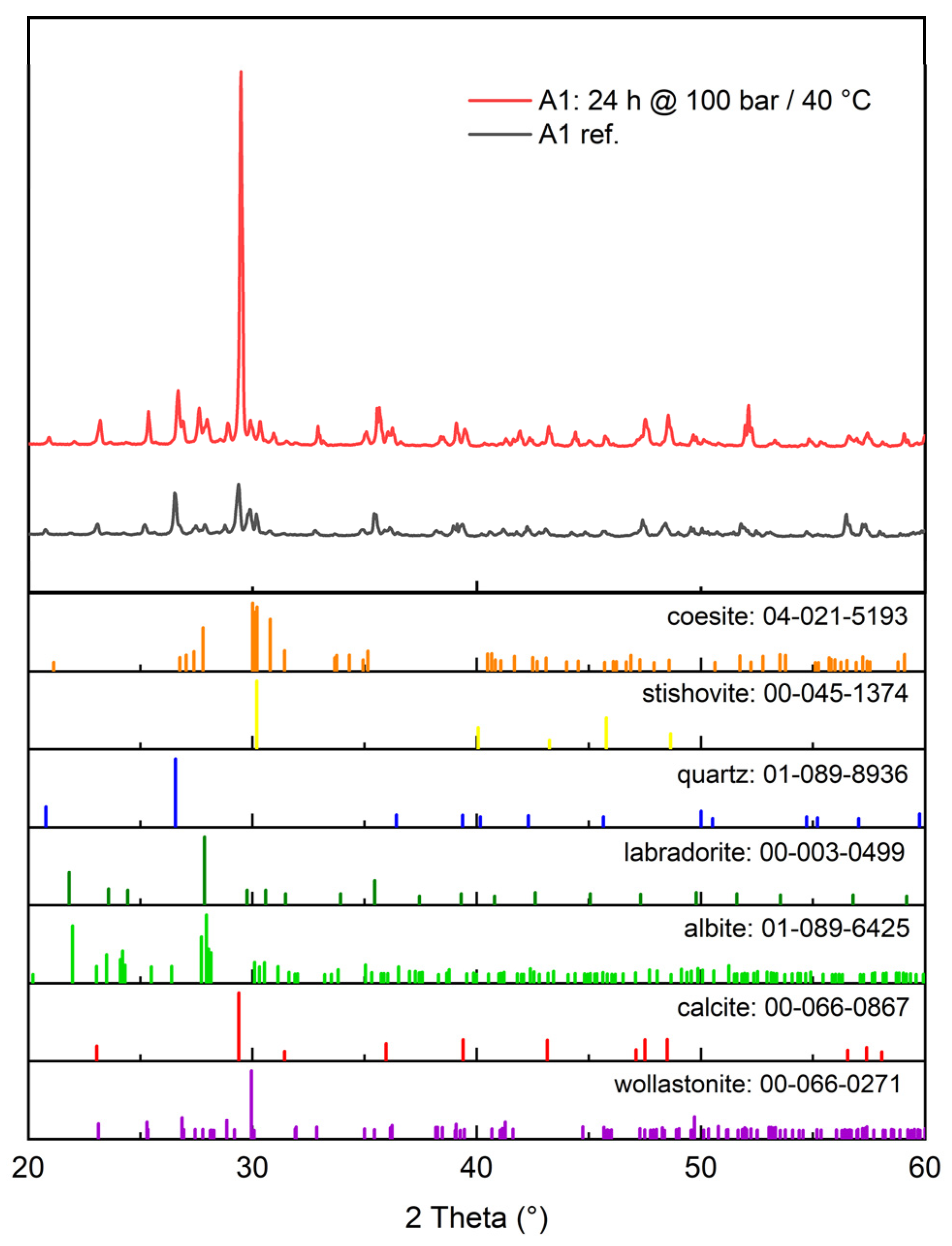
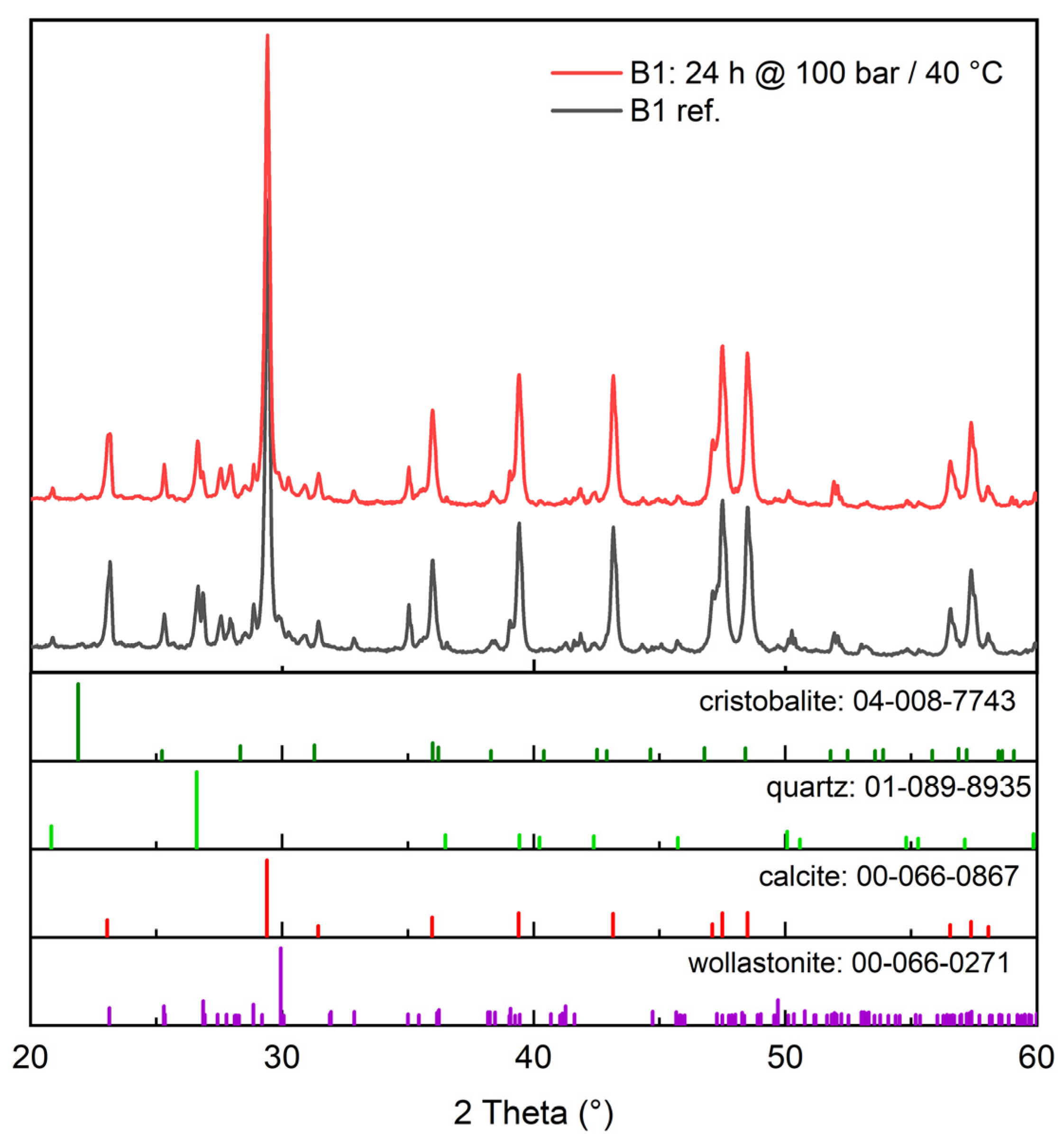
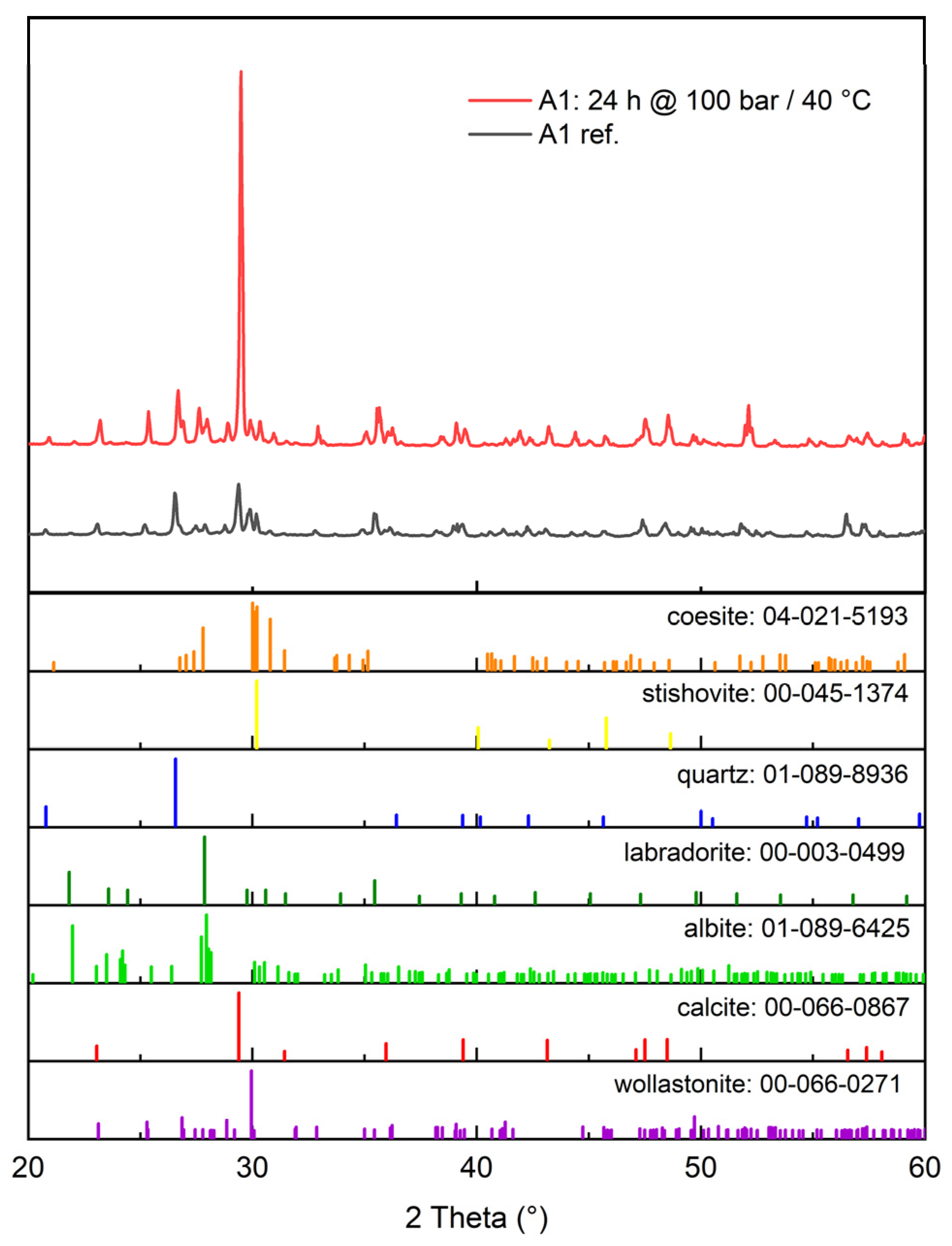
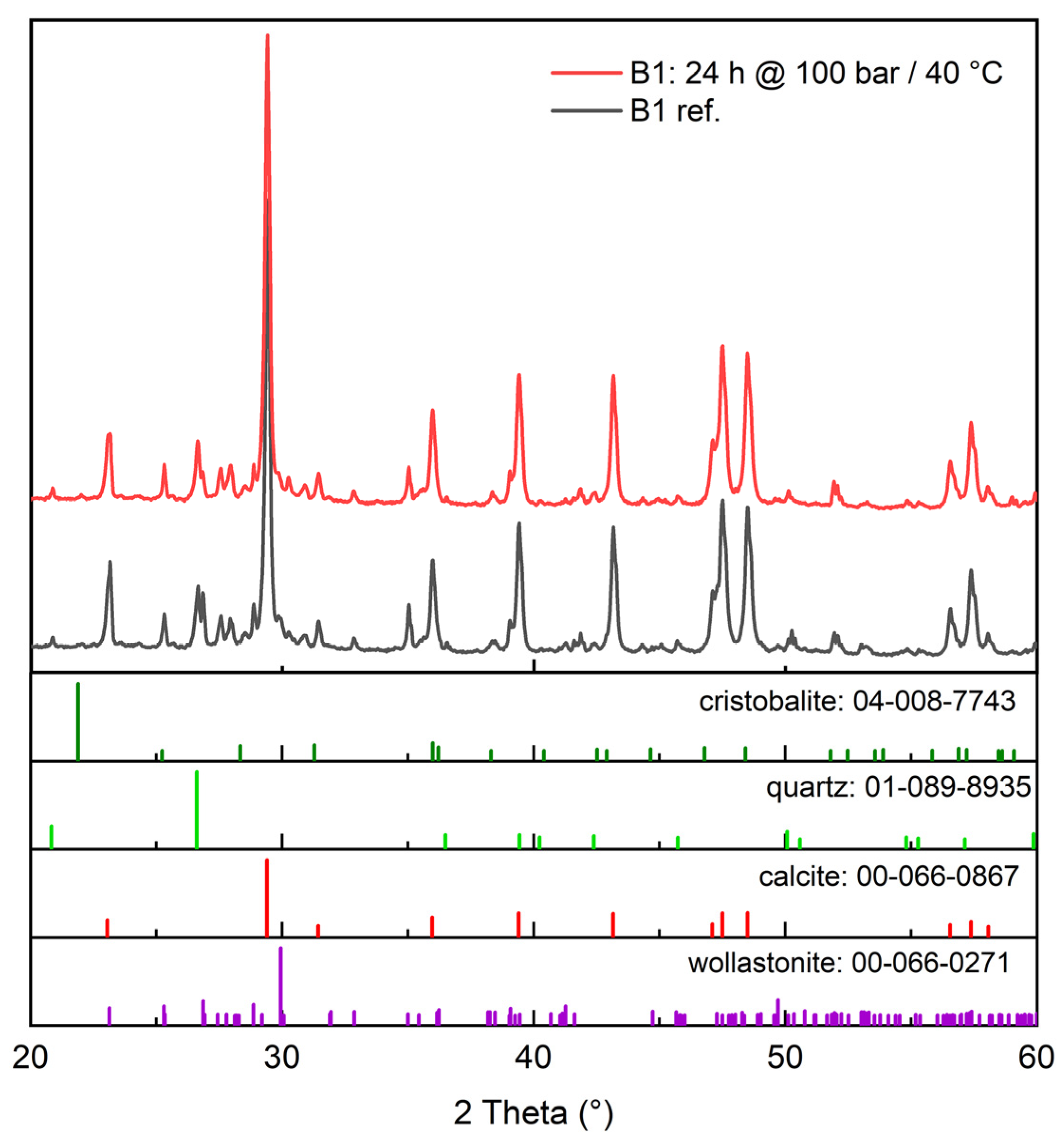
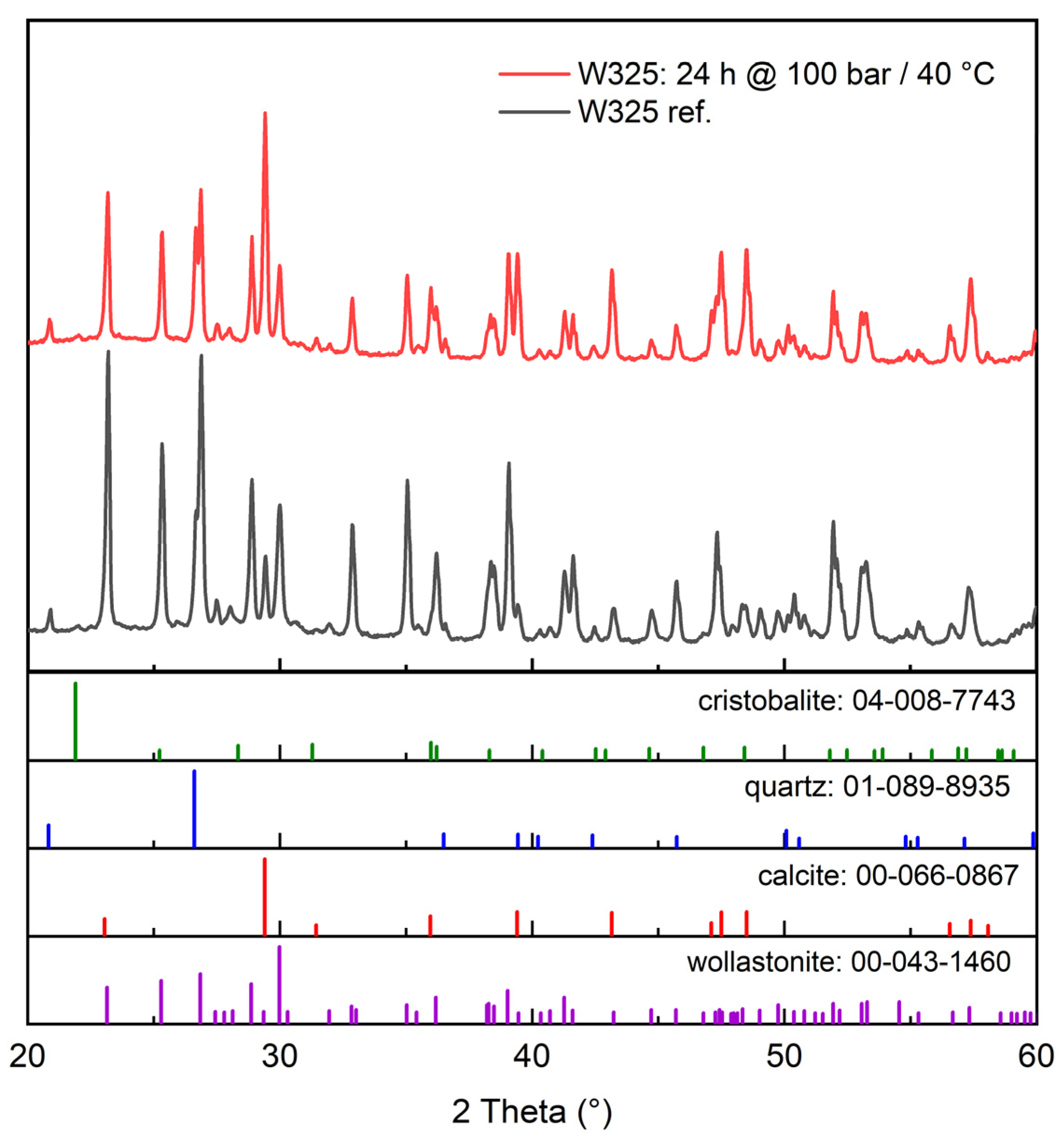
3.2.1. XRD (X-ray Diffraction)
3.2.2. TGA (Thermogravimetric Analysis)
3.2.3. SEM (Scanning Electron Microscopy)
4. Discussion and Conclusions
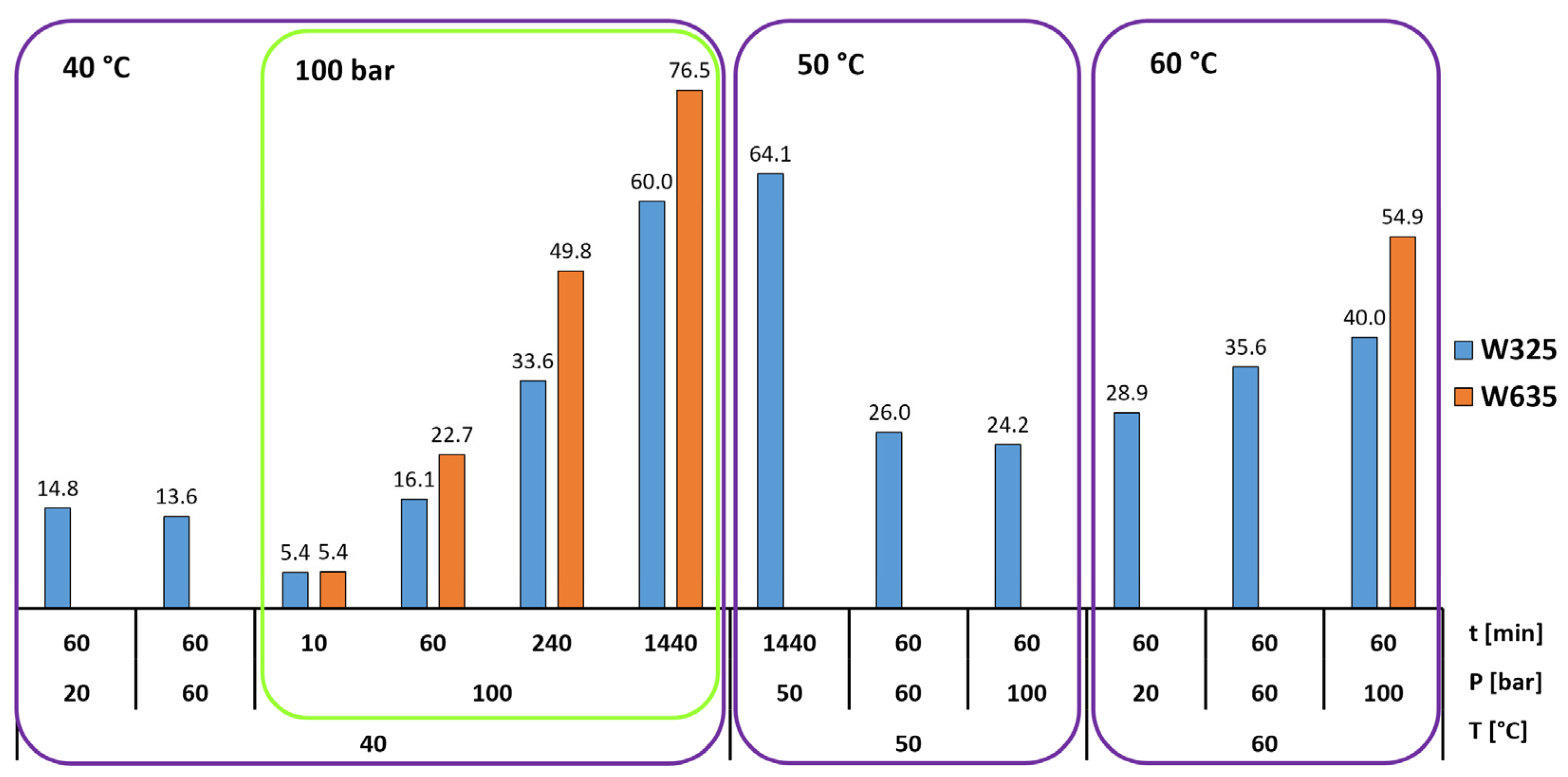
- Increasing the carbonation time at 40 °C and 100 bar increased the conversion extent, and it appears that in that case, 24 h was not enough time to reach the inevitable plateau dictated either by the formation of a passivating layer of calcite grains and residual silica or the simple depletion of the feedstock. This plateau was found by Min et al. [10] to occur at roughly 10 h under 100 bar pressure and 60 °C temperature, but at up to 10 days, according to Daval et al. [6], when using pCO2 = 25 MPa and 90 °C with several mild solutions.
- A finer particle size increased the carbonation extent in all tested cases. This is intuitively logical based on differences in surface area, and it is supported by the observations of Min et al. [10], where reacted layer thickness produced in certain conditions was proven to be independent of particle size. This of course assumes that the wollastonite component of the mineral feedstock is chemically the same, which is the case in our study, where both the tailings and the commercial wollastonites originated from the same quarry.
- An increase in temperature appears to be generally more beneficial for increasing the carbonation rate than an increase in pressure. This is in line with the findings of Gerdemann et al. [13], who observed that wollastonite carbonation had almost no sensitivity to pressure increase above ~40 atm, but exhibited almost linear scaling with a temperature increase between 25 °C and 100 °C. In our research, however, a pressure increase under elevated temperature (60 °C) quite clearly appears to increase the conversion extent. The positive effect of temperature is explained by the assumed increase in reaction and mass transfer rates [4], meaning the increased leaching of Ca-ions, which is almost universally agreed to be the rate-limiting step in wollastonite carbonation [4,6,17].
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

Appendix B

Appendix C

References
- Solismaa, S.; Ismailov, A.; Karhu, M.; Sreenivasan, H.; Lehtonen, M.; Kinnunen, P.; Illikainen, M.; Räisänen, M.L. Valorization of Finnish Mining Tailings for Use in the Ceramics Industry. Bull. Geol. Soc. Finl. 2018, 90, 33–54. [Google Scholar] [CrossRef]
- Lindfors, A.; Hakala, J. MEDIA RELEASE: Putting Nordkalk’s Circular Economy Strategy into Practice The Recovery of the Valuable Mineral Wollastonite Is Increasing in Lappeenranta. 2022. Available online: https://www.nordkalk.com/wp-content/uploads/2022/03/MEDIA-RELEASE_Recovery-of-wollastonite-increases.pdf (accessed on 10 April 2024).
- Yadav, S.; Mehra, A. A Review on Ex Situ Mineral Carbonation. Environ. Sci. Pollut. Res. 2021, 28, 12202–12231. [Google Scholar] [CrossRef] [PubMed]
- Huijgen, W.J.J.; Witkamp, G.J.; Comans, R.N.J. Mechanisms of Aqueous Wollastonite Carbonation as a Possible CO2 Sequestration Process. Chem. Eng. Sci. 2006, 61, 4242–4251. [Google Scholar] [CrossRef]
- Di Lorenzo, F.; Prieto, M. Dissolution-Recrystallization of (Mg,Fe)CO3 during Hydrothermal Cycles: FeII/FeIII Conundrums in the Carbonation of Ferromagnesian Minerals. Cryst. Growth Des. 2017, 17, 4170–4182. [Google Scholar] [CrossRef]
- Daval, D.; Martinez, I.; Corvisier, J.; Findling, N.; Goffé, B.; Guyot, F. Carbonation of Ca-Bearing Silicates, the Case of Wollastonite: Experimental Investigations and Kinetic Modeling. Chem. Geol. 2009, 265, 63–78. [Google Scholar] [CrossRef]
- Monasterio-Guillot, L.; Di Lorenzo, F.; Ruiz-Agudo, E.; Rodriguez-Navarro, C. Reaction of Pseudowollastonite with Carbonate-Bearing Fluids: Implications for CO2 Mineral Sequestration. Chem. Geol. 2019, 524, 158–173. [Google Scholar] [CrossRef]
- Wu, J.C.S.; Sheen, J.D.; Chen, S.Y.; Fan, Y.C. Feasibility of CO2 Fixation via Artificial Rock Weathering. Ind. Eng. Chem. Res. 2001, 40, 3902–3905. [Google Scholar] [CrossRef]
- Saarimaa, V.; Kaleva, A.; Ismailov, A.; Laihinen, T.; Virtanen, M.; Levänen, E.; Väisänen, P. Corrosion Product Formation on Zinc-Coated Steel in Wet Supercritical Carbon Dioxide. Arab. J. Chem. 2022, 15, 103636. [Google Scholar] [CrossRef]
- Min, Y.; Li, Q.; Voltolini, M.; Kneafsey, T.; Jun, Y.S. Wollastonite Carbonation in Water-Bearing Supercritical CO2: Effects of Particle Size. Environ. Sci. Technol. 2017, 51, 13044–13053. [Google Scholar] [CrossRef] [PubMed]
- Huijgen, W.J.J. Carbon Dioxide Sequestration by Mineral Carbonation—Feasibility of Enhanced Natural Weathering as a CO2 Emission Reduction Technology; Wageningen University and Research: Wageningen, The Netherlands, 2007; ISBN 9085045738. [Google Scholar]
- O’Connor, W.K.; Dahlin, D.C.; Rush, G.E.; Dahlin, C.L.; Collins, W.K. Carbon Dioxide Sequestration by Direct Mineral Carbonation: Process Mineralogy of Feed and Products. Miner. Metall. Process. 2002, 19, 95–101. [Google Scholar] [CrossRef]
- Gerdemann, S.J.; O’Connor, W.K.; Dahlin, D.C.; Penner, L.R.; Rush, H. Ex Situ Aqueous Mineral Carbonation. Environ. Sci. Technol. 2007, 41, 2587–2593. [Google Scholar] [CrossRef] [PubMed]
- Ewell, R.H.; Bunting, E.N.; Geller, R.F. Thermal Decomposition of Talc. J. Res. Natl. Bur. Stand. 1935, 15, 551–556. [Google Scholar] [CrossRef]
- Kojima, T.; Nagamine, A.; Ueno, N.; Uemiya, S. Absorption and Fixation of Carbon Dioxide by Rock Weathering. Energy Convers. Manag. 1997, 38, S461–S466. [Google Scholar] [CrossRef]
- Penner, L.; O’Connor, W.K.; Dahlin, D.C.; Gerdemann, S.; Rush, G.E. Mineral Carbonation: Energy Costs of Pretreatment Options and Insights Gained from Flow Loop Reaction Studies. In Proceedings of the 3rd Annual Conference on Carbon Capture & Sequestration, Alexandria, VA, USA, 3–6 May 2004; Volume 5, pp. 1–18. [Google Scholar]
- Di Lorenzo, F.; Ruiz-Agudo, C.; Ibañez-Velasco, A.; Gil-San Millán, R.; Navarro, J.A.R.; Ruiz-Agudo, E.; Rodriguez-Navarro, C. The Carbonation of Wollastonite: A Model Reaction to Test Natural and Biomimetic Catalysts for Enhanced CO2 Sequestration. Minerals 2018, 8, 209. [Google Scholar] [CrossRef]
- Daval, D.; Bernard, S.; Rémusat, L.; Wild, B.; Guyot, F.; Micha, J.S.; Rieutord, F.; Magnin, V.; Fernandez-Martinez, A. Dynamics of Altered Surface Layer Formation on Dissolving Silicates. Geochim. Cosmochim. Acta 2017, 209, 51–69. [Google Scholar] [CrossRef]


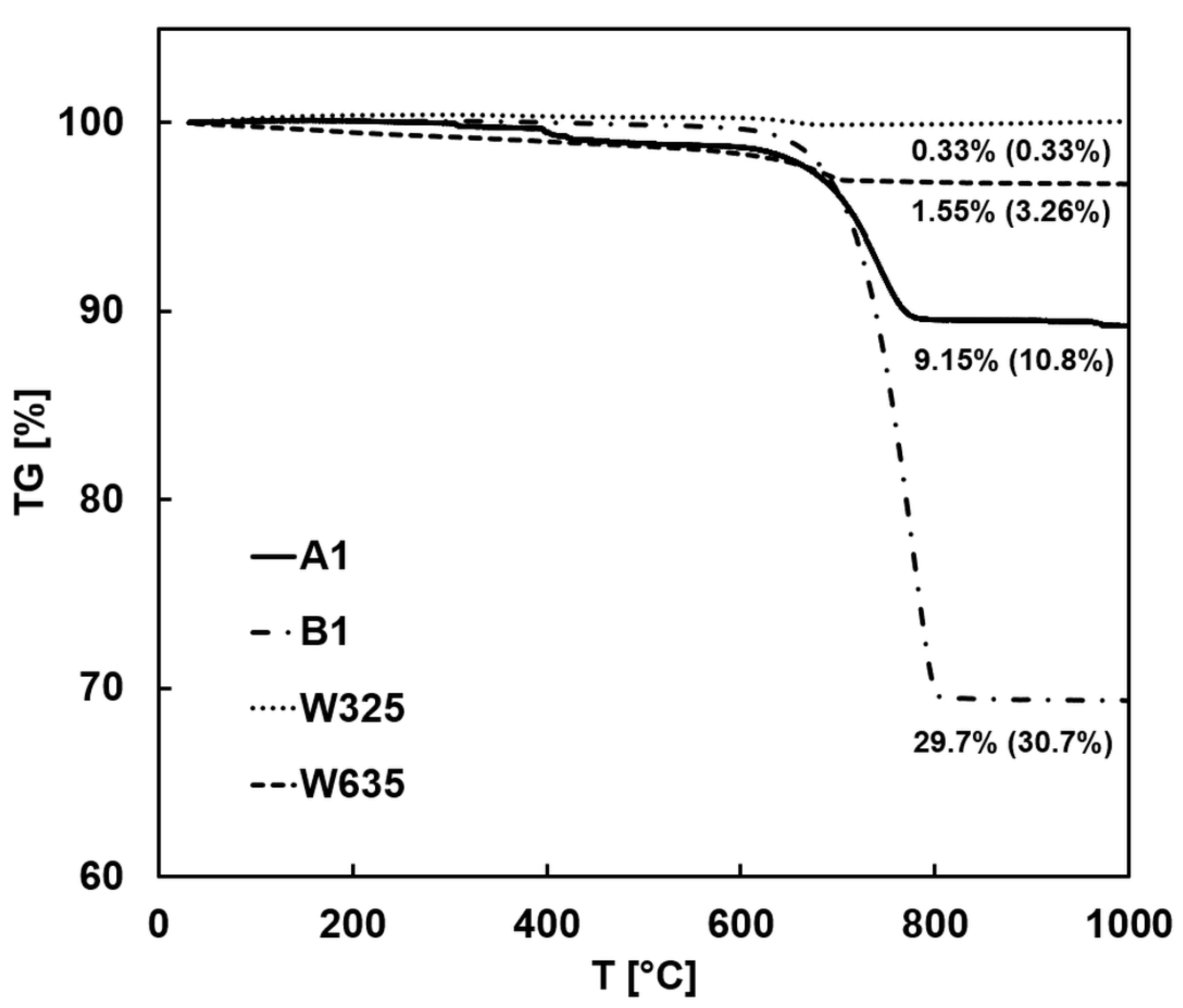

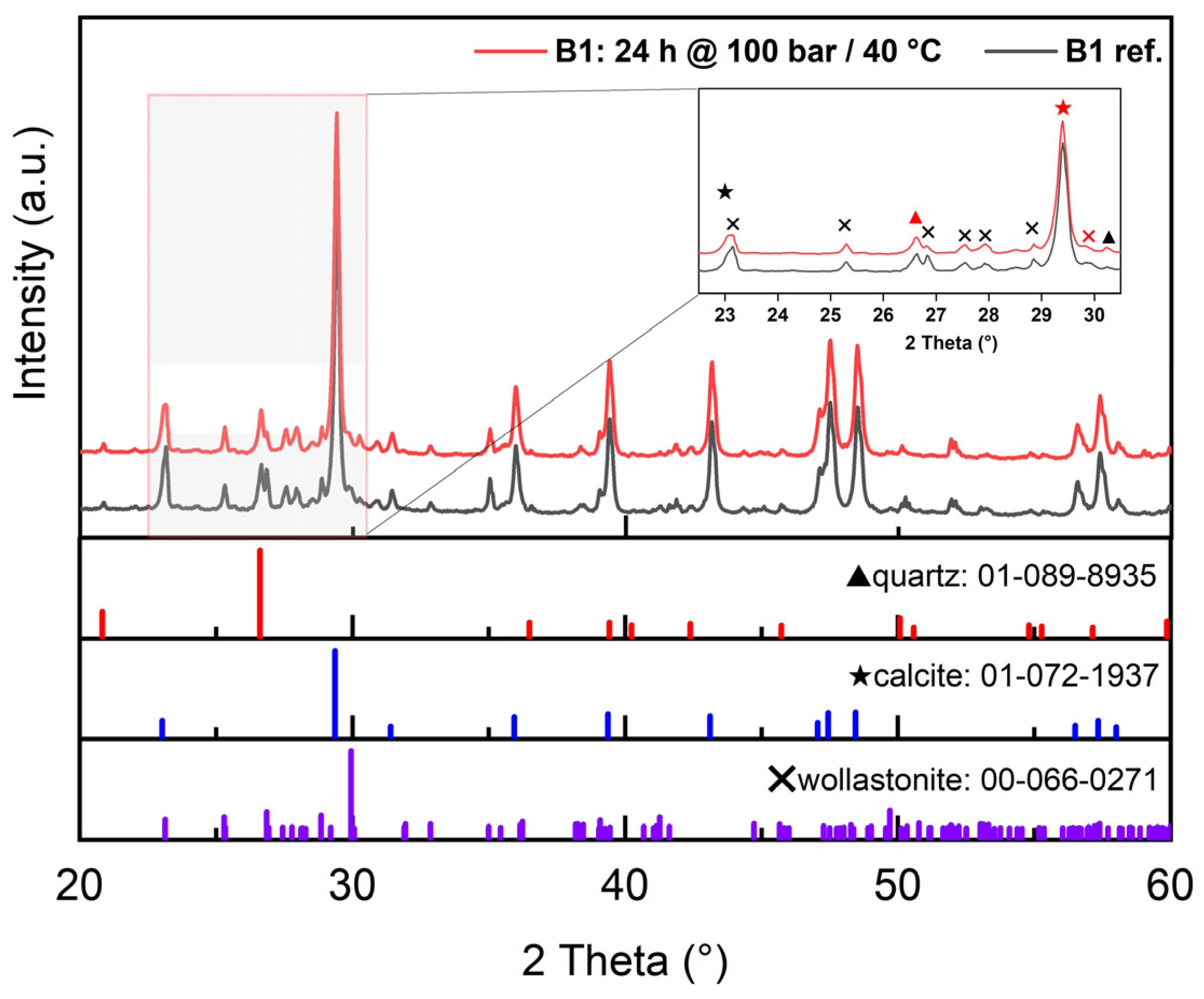
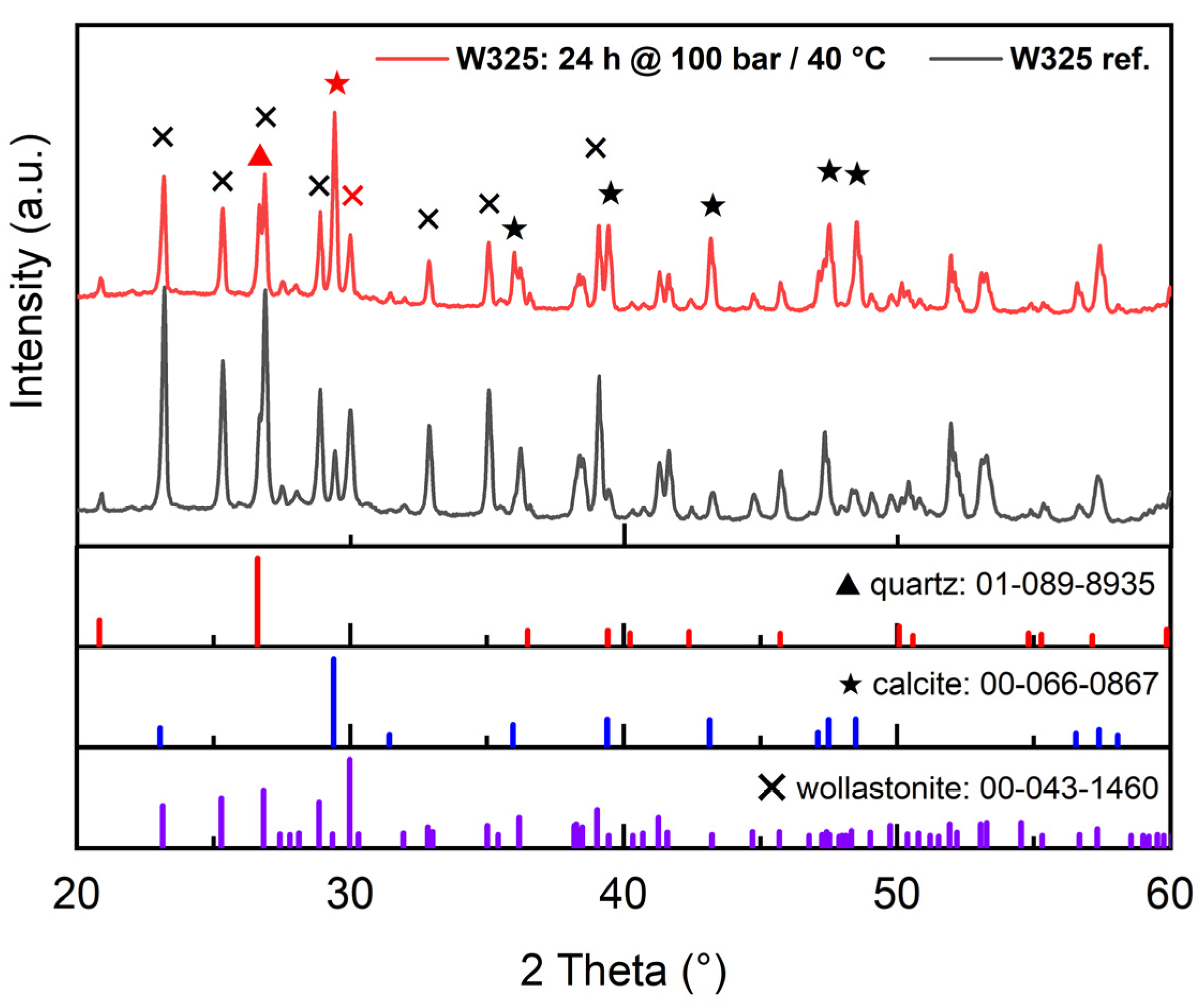
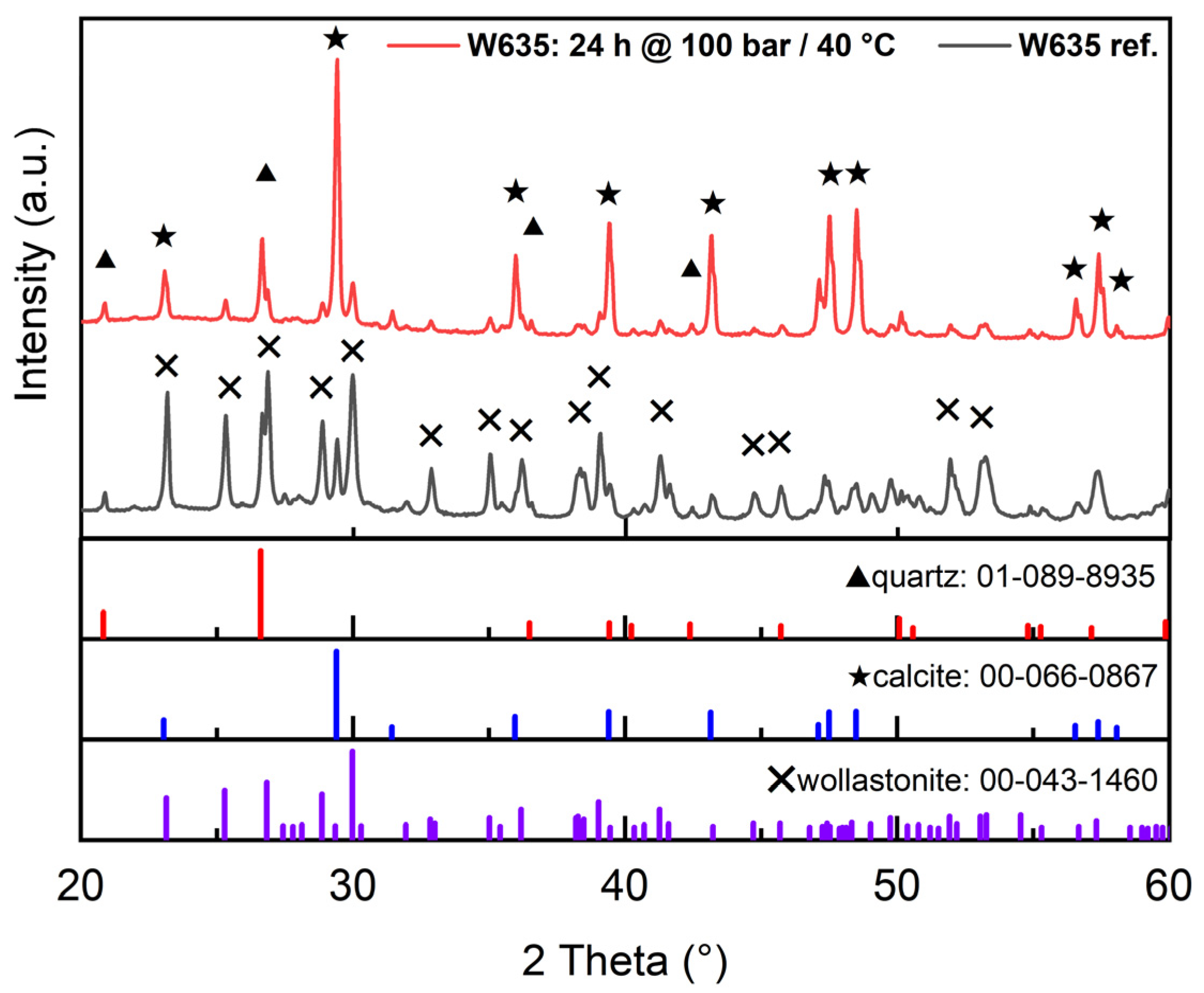





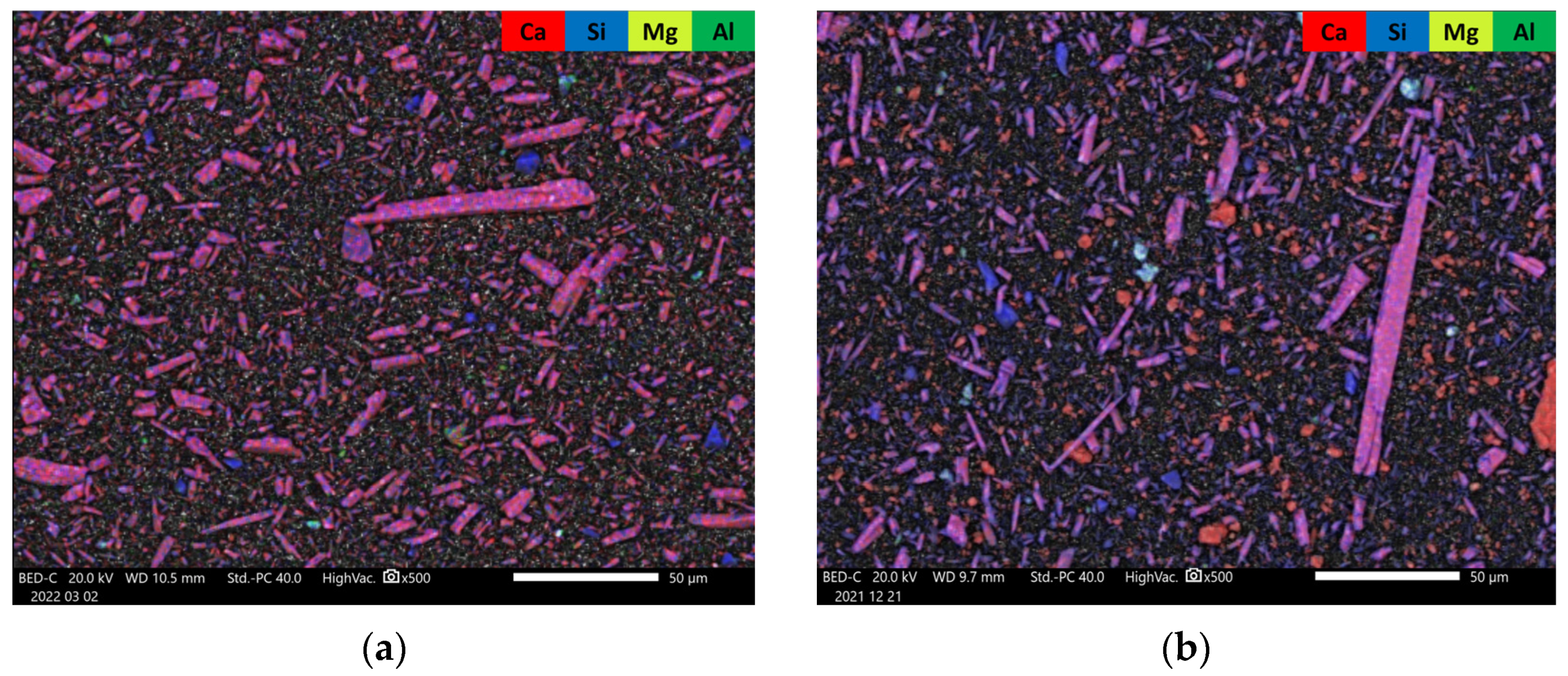

| Tailing Fractions | Wollastonite | Other Ca-Silicates * | Carbonate Minerals | Plagioclase, Feldspar, Quartz, and Other |
|---|---|---|---|---|
| A1 (coarse) | 30.3 | 33.8 | 17.1 | 20.7 |
| B1 (fine) | 21.1 | 7.1 | 54.0 | 17.8 |
| Sample | O | Na | Mg | Al | Si | Ca | Fe | S | Total |
|---|---|---|---|---|---|---|---|---|---|
| A1 | 66.4 | 0.5 | 4.7 | 1.1 | 15.3 | 11.9 | - | - | 100 |
| B1 | 75.2 | - | 1.0 | 0.7 | 4.7 | 18.2 | 0.2 | 0.2 | 100 |
| Slurry | pH: Initial | pH: 1 h | pH: Δ |
|---|---|---|---|
| W325 | 9.37 | 9.28 | −0.09 |
| W635 | 9.66 | 10.21 | +0.55 |
| A1 | 9.18 | 9.02 | −0.16 |
| B1 | 9.86 | 9.59 | −0.27 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ismailov, A.; Merilaita, N.; Levänen, E. Accelerated Carbonation of High-Calcite Wollastonite Tailings. Minerals 2024, 14, 415. https://doi.org/10.3390/min14040415
Ismailov A, Merilaita N, Levänen E. Accelerated Carbonation of High-Calcite Wollastonite Tailings. Minerals. 2024; 14(4):415. https://doi.org/10.3390/min14040415
Chicago/Turabian StyleIsmailov, Arnold, Niina Merilaita, and Erkki Levänen. 2024. "Accelerated Carbonation of High-Calcite Wollastonite Tailings" Minerals 14, no. 4: 415. https://doi.org/10.3390/min14040415
APA StyleIsmailov, A., Merilaita, N., & Levänen, E. (2024). Accelerated Carbonation of High-Calcite Wollastonite Tailings. Minerals, 14(4), 415. https://doi.org/10.3390/min14040415