
Dissolution Mechanisms and Surface Charge of Clay Mineral Nanoparticles: Insights from Kinetic Monte Carlo Simulations


Abstract
:1. Introduction
2. Methods
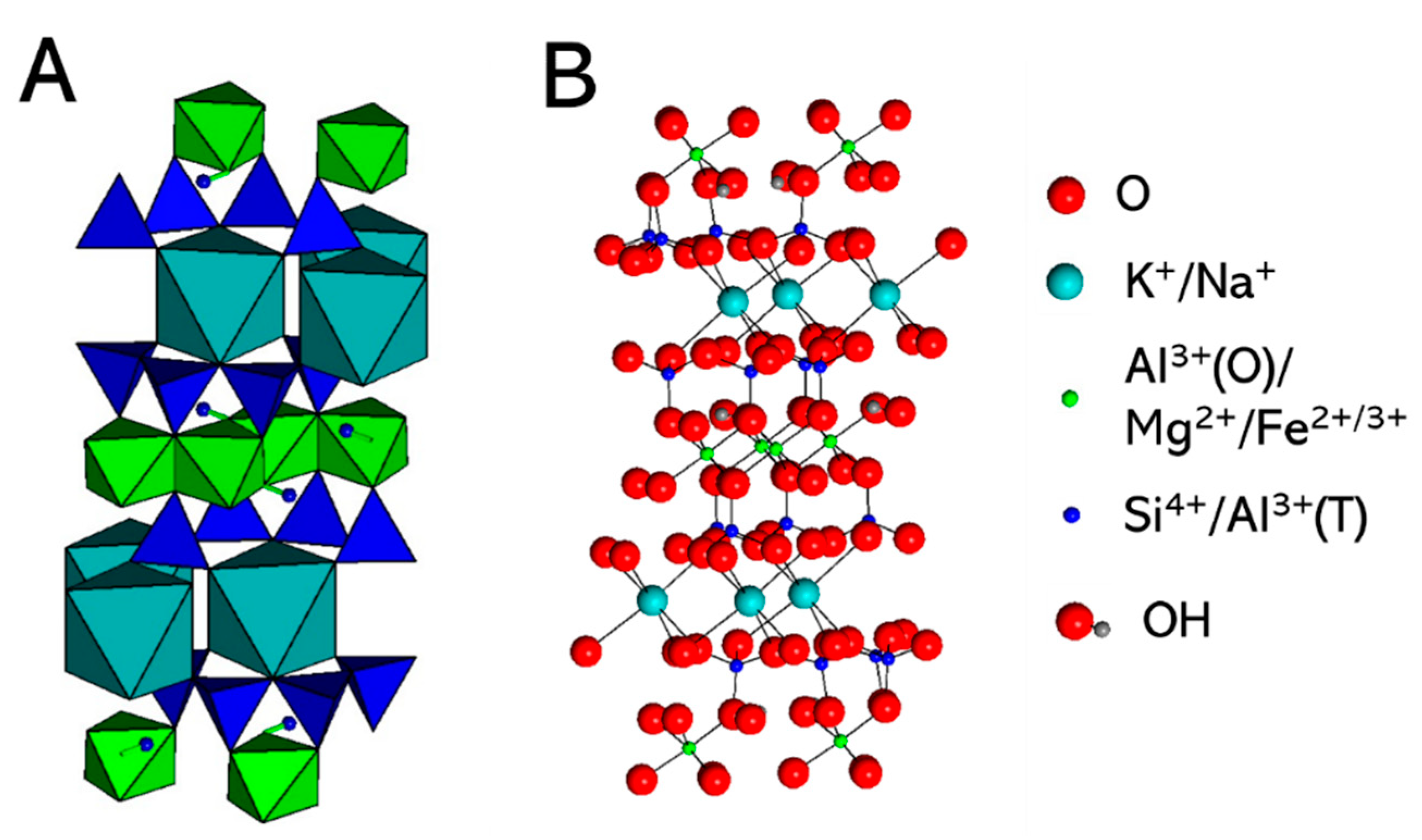
2.1. Simulated System
2.2. The Kinetic Monte Carlo Algorithm
2.3. Dissolution Rate Calculations
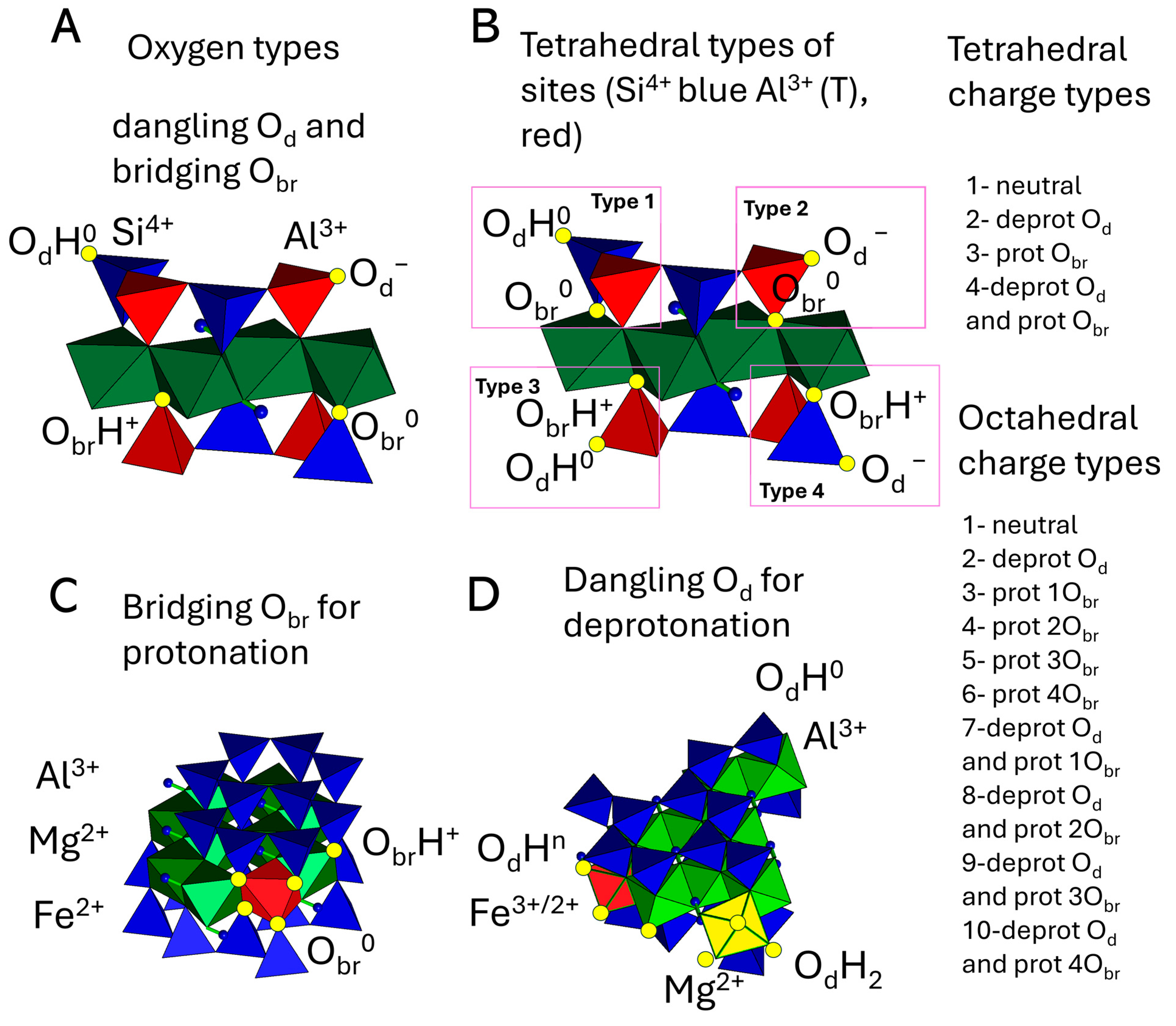
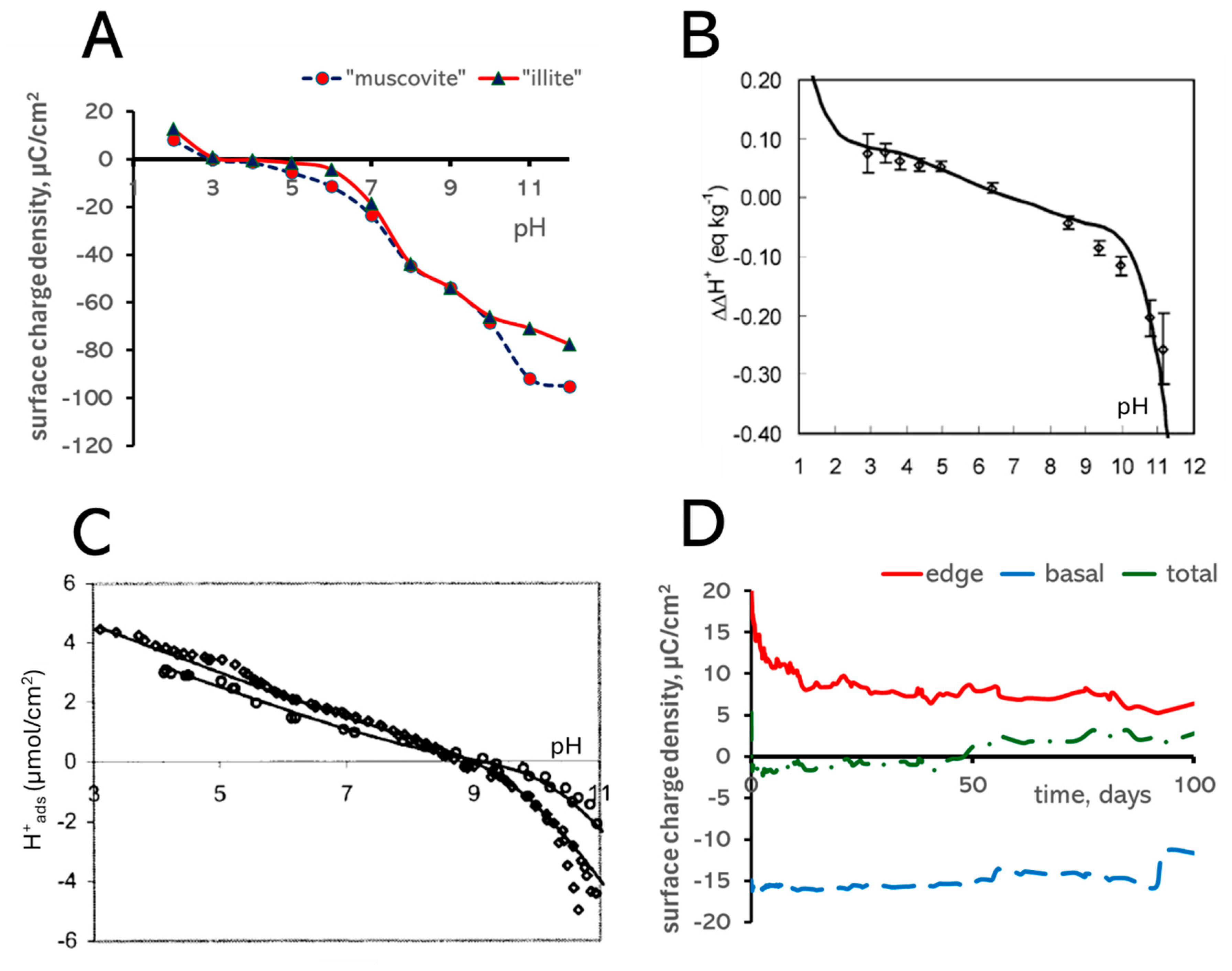
2.4. Acid–Base Property Modeling
3. Results and Discussion
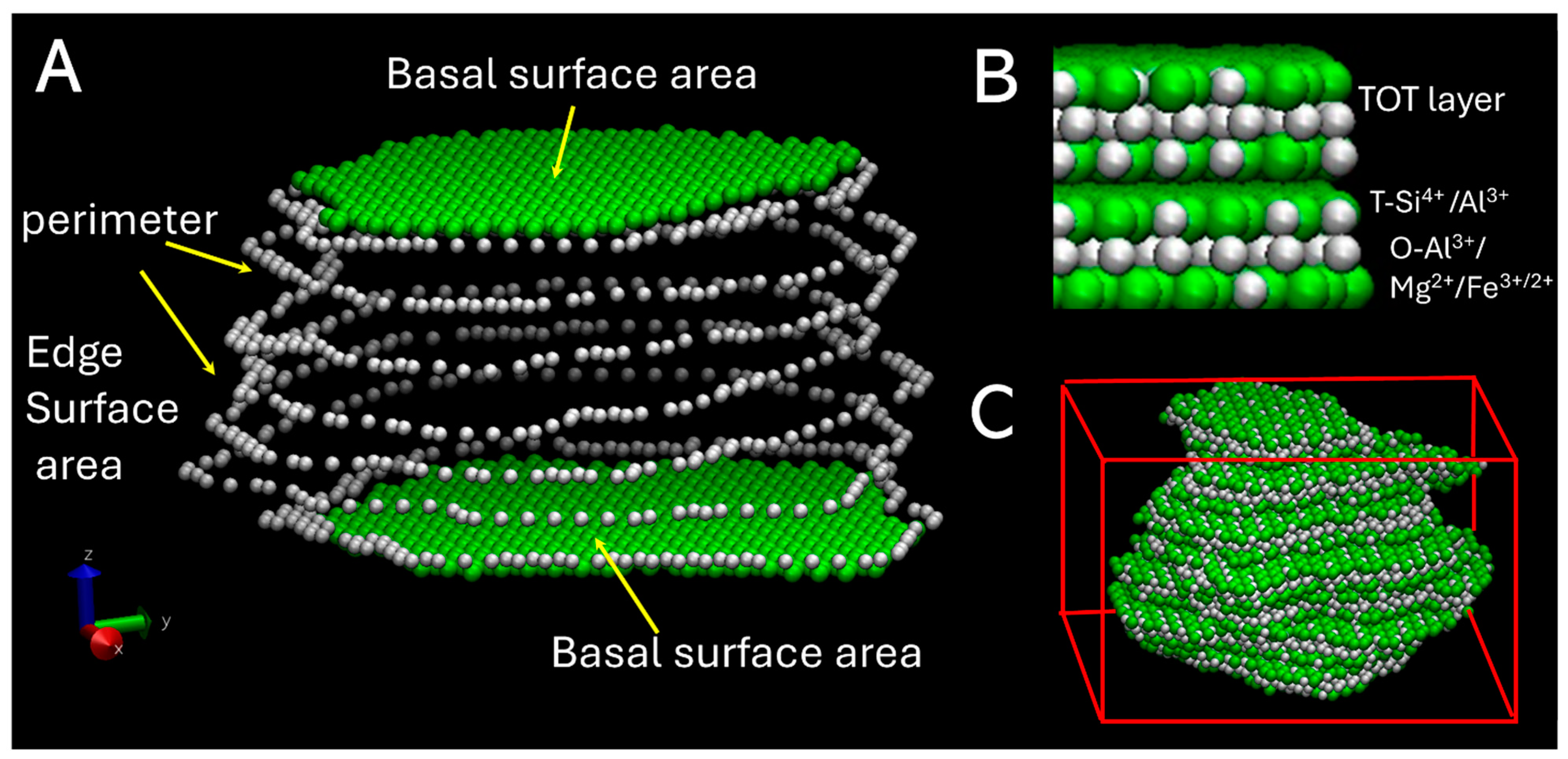
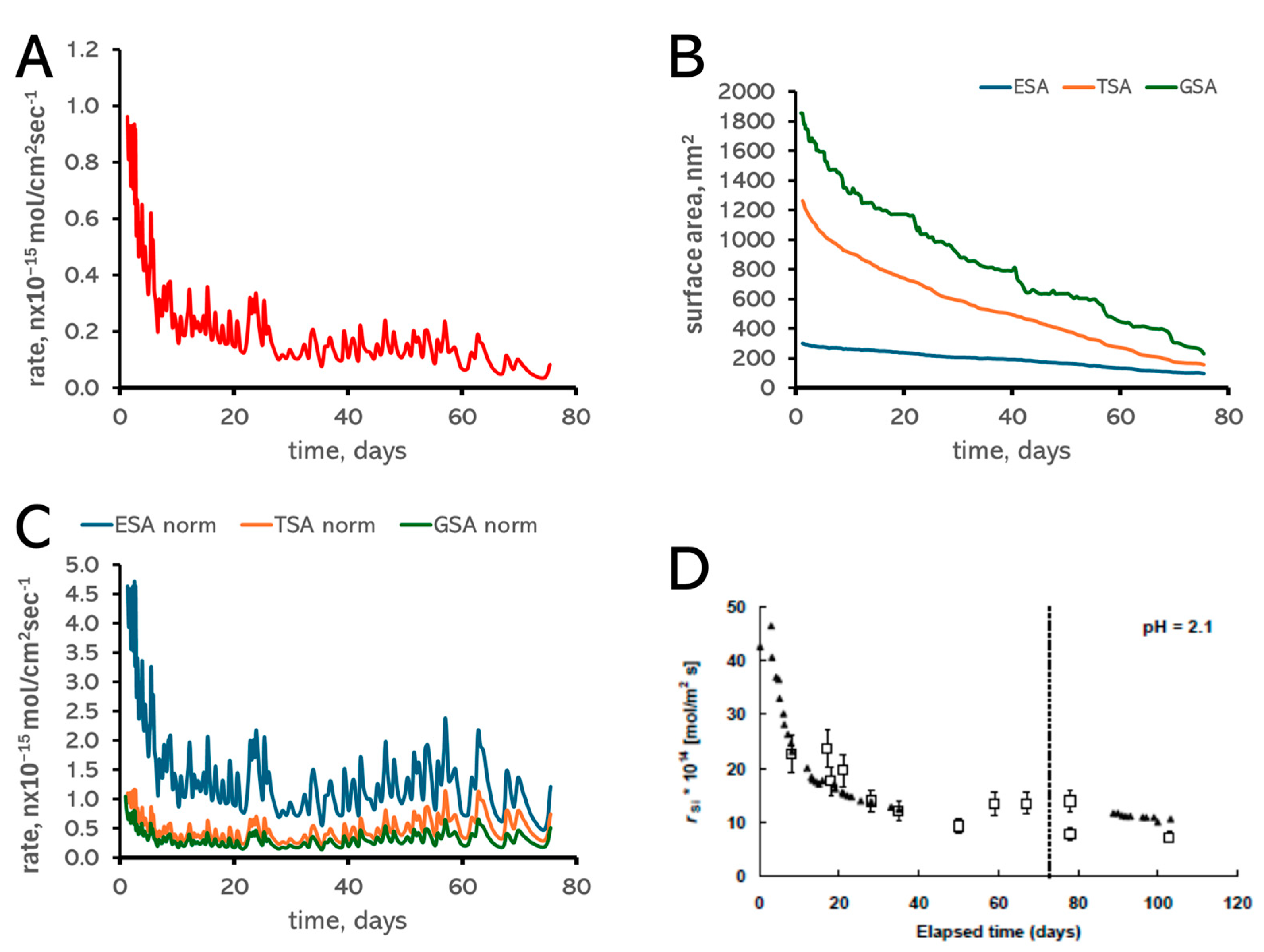
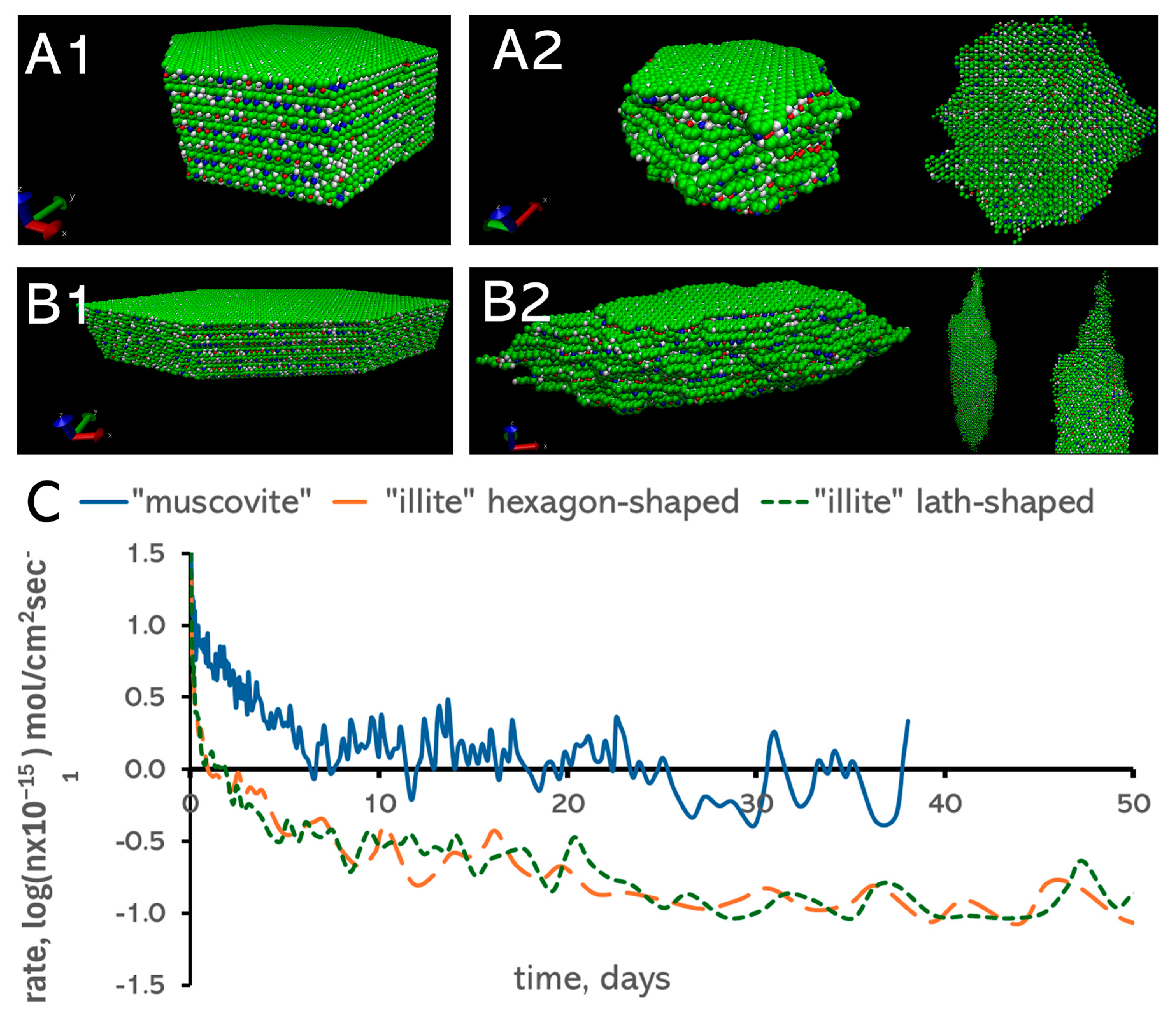
3.1. Surface Area Normalization
3.2. Morphology of Nanoparticles
3.3. Chemical Composition Effects
3.4. Surface Charge

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site | pKa | Mineral | Face | Formula(O)(T) | Method/Source |
|---|---|---|---|---|---|
| Si-OH | 7.0 ± 0.7 | montmorillonite | (010) | (Al3.5Mg0.5) (Si) | DFT-MD [82] |
| Al-(OH2)(OH) | 8.3 ± 1.0 | montmorillonite | (010) | (Al3.5Mg0.5) (Si) | DFT-MD [82] |
| Al-(OH2)2 | 3.1 ± 0.5 | montmorillonite | (010) | (Al3.5Mg0.5) (Si) | DFT-MD [82] |
| Mg-(OH2)2 | 13.2 ± 0.5 | montmorillonite | (010) | (Al3.5Mg0.5) (Si) | DFT-MD [82] |
| Fe(III)-(OH2)2 | 1.2 ± 0.7 | Nontronite | (010) | (Fe3Al1) (Si8) | DFT-MD [83] |
| Fe(III)-(OH2)(OH) | 4.4 ± 0.3 | Nontronite | (010) | (Fe3Al1) (Si8) | DFT-MD [83] |
| Si-OH | 8.7 ± 1.4 | Nontronite | (010) | (Fe3Al1) (Si8) | DFT-MD [83] |
| Fe(III)-(OH2)2 | 1.2 ± 0.5 | Fe-montmorillonite | (010) | (Fe0.5Al3) (Si8) | DFT-MD [83] |
| Fe(III)-(OH2)(OH) | 5.1 ± 1.0 | Fe-montmorillonite | (010) | (Fe0.5Al3) (Si8) | DFT-MD [83] |
| Si-OH | 8.6 ± 1.0 | Fe-montmorillonite | (010) | (Fe0.5Al3) (Si8) | DFT-MD [83] |
| ≡Si–OHn | 9.1 | montmorillonite | - | (Al,Fe,Mg) (Si,Al) | CD-MUSIC [66] |
| ≡Al–OHn | 10.5 | montmorillonite | - | (Al,Fe,Mg) (Si,Al) | CD-MUSIC [66] |
| ≡Mg–OHn | 12.7 | montmorillonite | - | (Al,Fe,Mg) (Si,Al) | CD-MUSIC [66] |
| ≡Fe(III)–OHn | 8.9 | montmorillonite | - | (Al,Fe,Mg) (Si,Al) | CD-MUSIC [66] |
| ≡SiAl–Ohn | 7.7 | montmorillonite | - | (Al,Fe,Mg) (Si,Al) | CD-MUSIC [66] |
| ≡SiMg–OHn | 9.8 | montmorillonite | - | (Al,Fe,Mg) (Si,Al) | CD-MUSIC [66] |
| ≡SiFe(III)–OHn | 6.1 | montmorillonite | - | (Al,Fe,Mg) (Si,Al) | CD-MUSIC [66] |
| ≡AlAl–OHn | 5.1 | montmorillonite | - | (Al,Fe,Mg) (Si,Al) | CD-MUSIC [66] |
| ≡AlMg–OHn | 7.3 | montmorillonite | - | (Al,Fe,Mg) (Si,Al) | CD-MUSIC [66] |
| ≡AlFe(III)–OHn | 3.5 | montmorillonite | - | (Al,Fe,Mg) (Si,Al) | CD-MUSIC [66] |
| Al-(OH2)2 | 9.5 ± 0.8 | Gibbsite | (100) | Al | DFT-MD [84] |
| Si-OH | 6.8 ± 0.4 | pyrophyllite | (010) | (Al)(Si) | DFT-MD [85] |
| Al-(OH2) | 7.6 ±1.3 | pyrophyllite | (010) | (Al)(Si) | DFT-MD [85] |
| Al(T)-OH | 15.1 | - | Si,Al | (Al) (Si,Al) | DFT-MD [86] |
| Mineral | Inflection Points | Background Electrolyte | Tetrahedral | Octahedral | Method |
|---|---|---|---|---|---|
| montmorillonite | 5/8 | 0.01 M NaCl | Si | Al,Mg | Exp [91] |
| montmorillonite | 5/8 | 0.02 M NaCl | Si3.98Al0.02 | Al1.55Fe(III)0.09Fe(II)0.08Mg0.28 | Exp [69] |
| illite | 6.3/10.6 | 0.001 M NaCl | Si6.89Al1.11 | Al2.82Fe0.57Mg0.44 | Exp [80] |
| illite | 7/10.7 | 0.001 M NaCl | Si7.70Al0.3 | Al0.28Fe2.71Mg0.86 | Exp [80] |
| hematite | 7 | 0.1 M NaClO4 | - | Fe | Exp [92] |
| pyrophillite | 3.8/9 | 0.1 mM 1-1 salt | Si | Al | GCMC [81] |
| montmorillonite | 5/9 | 0.1 mM 1-1 salt | Si | Al3.5Mg0.5 | GCMC [81] |
| Illite | 6.5/9.5 | 0.1 mM 1-1 salt | Si,Al | Al,Fe,Mg | GCMC [81] |
3.5. pH Control of Dissolution Rates
4. Summary and Conclusions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, C.; Myshkin, V.F.; Khan, V.A.; Panamareva, A.N. A Review of the Migration of Radioactive Elements in Clay Minerals in the Context of Nuclear Waste Storage. J. Radioanal. Nucl. Chem. 2022, 331, 3401–3426. [Google Scholar] [CrossRef]
- Churakov, S.V.; Gimmi, T.; Unruh, T.; Van Loon, L.R.; Juranyi, F. Resolving Diffusion in Clay Minerals at Different Time Scales: Combination of Experimental and Modeling Approaches. Appl. Clay Sci. 2014, 96, 36–44. [Google Scholar] [CrossRef]
- Kalinichev, A.G.; Loganathan, N.; Wakou, B.F.N.; Chen, Z. Interaction of Ions with Hydrated Clay Surfaces: Computational Molecular Modeling for Nuclear Waste Disposal Applications. Procedia Earth Planet. Sci. 2017, 17, 566–569. [Google Scholar] [CrossRef]
- Murray, H.H. Overview—Clay Mineral Applications. Appl. Clay Sci. 1991, 5, 379–395. [Google Scholar] [CrossRef]
- Rehman, A.; Rukh, S.; Ayoubi, S.A.; Khattak, S.A.; Mehmood, A.; Ali, L.; Khan, A.; Malik, K.M.; Qayyum, A.; Salam, H. Natural Clay Minerals as Potential Arsenic Sorbents from Contaminated Groundwater: Equilibrium and Kinetic Studies. Int. J. Environ. Res. Public Health 2022, 19, 16292. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.K. A Review on the Adsorption of Heavy Metals by Clay Minerals, with Special Focus on the Past Decade. Chem. Eng. J. 2017, 308, 438–462. [Google Scholar] [CrossRef]
- Carretero, M.I. Clay Minerals and Their Beneficial Effects upon Human Health. A Review. Appl. Clay Sci. 2002, 21, 155–163. [Google Scholar] [CrossRef]
- Biswas, B.; Grathoff, G.; Naidu, R.; Warr, L.N. The Multidisciplinary Science of Applied Clay Research: A 2021–2023 Bibliographic Analysis. Appl. Clay Sci. 2024, 258, 107471. [Google Scholar] [CrossRef]
- Cygan, R.T.; Liang, J.-J.; Kalinichev, A.G. Molecular Models of Hydroxide, Oxyhydroxide, and Clay Phases and the Development of a General Force Field. J. Phys. Chem. B 2004, 108, 1255–1266. [Google Scholar] [CrossRef]
- Cygan, R.T.; Greathouse, J.A.; Kalinichev, A.G. Advances in Clayff Molecular Simulation of Layered and Nanoporous Materials and Their Aqueous Interfaces. J. Phys. Chem. C 2021, 125, 17573–17589. [Google Scholar] [CrossRef]
- Baston, G.M.N.; Berry, J.A.; Brownsword, M.; Cowper, M.M.; Heath, T.G.; Tweed, C.J. The Sorption of Uranium and Technetium on Bentonite, Tuff and Granodiorite. In Proceedings of the Symposium—Scientific Basis for Nuclear Waste Management XVIII, Kyoto, Japan, 23–27 October 1994; Volume 353. [Google Scholar]
- Churakov, S.V.; Hummel, W.; Fernandes, M.M. Fundamental Research on Radiochemistry of Geological Nuclear Waste Disposal. Chimia 2020, 74, 1000. [Google Scholar] [CrossRef] [PubMed]
- Zaoui, A.; Sekkal, W. Can Clays Ensure Nuclear Waste Repositories? Sci. Rep. 2015, 5, 8815. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Tank, R.W. Role of Clays in the Disposal of Nuclear Waste: A Review. Appl. Clay Sci. 1985, 1, 145–162. [Google Scholar] [CrossRef]
- Payne, T.E.; Brendler, V.; Ochs, M.; Baeyens, B.; Brown, P.L.; Davis, J.A.; Ekberg, C.; Kulik, D.A.; Lutzenkirchen, J.; Missana, T.; et al. Guidelines for Thermodynamic Sorption Modelling in the Context of Radioactive Waste Disposal. Environ. Model. Softw. 2013, 42, 143–156. [Google Scholar] [CrossRef]
- Sinitsyn, V.A.; Aja, S.U.; Kulik, D.A.; Wood, S.A. Acid–Base Surface Chemistry and Sorption of Some Lanthanides on K+-Saturated Marblehead Illite: I. Results of an Experimental Investigation. Geochim. Cosmochim. Acta 2000, 64, 185–194. [Google Scholar] [CrossRef]
- Kulik, D.A.; Aja, S.U.; Sinitsyn, V.A.; Wood, S.A. Acid–Base Surface Chemistry and Sorption of Some Lanthanides on K+-Saturated Marblehead Illite: II. a Multisite–Surface Complexation Modeling. Geochim. Cosmochim. Acta 2000, 64, 195–213. [Google Scholar] [CrossRef]
- Li, C.; Adeniyi, E.O.; Zarzycki, P. Machine Learning Surrogates for Surface Complexation Model of Uranium Sorption to Oxides. Sci. Rep. 2024, 14, 6603. [Google Scholar] [CrossRef]
- Okumura, M.; Kerisit, S.; Bourg, I.C.; Lammers, L.N.; Ikeda, T.; Sassi, M.; Rosso, K.M.; Machida, M. Radiocesium Interaction with Clay Minerals: Theory and Simulation Advances Post–Fukushima. J. Environ. Radioact. 2018, 189, 135–145. [Google Scholar] [CrossRef]
- Zhang, J.; Mallants, D.; Brady, P.V. Molecular Dynamics Study of Uranyl Adsorption from Aqueous Solution to Smectite. Appl. Clay Sci. 2022, 218, 106361. [Google Scholar] [CrossRef]
- Brigatti, M.F.; Medici, L.; Poppi, L. Sepiolite and Industrial Waste-Water Purification: Removal of Zn2+ and Pb2+ from Aqueous Solutions. Appl. Clay Sci. 1996, 11, 43–54. [Google Scholar] [CrossRef]
- Manning, B.A.; Goldberg, S. Adsorption and Stability of Arsenic(III) at the Clay Mineral−Water Interface. Environ. Sci. Technol. 1997, 31, 2005–2011. [Google Scholar] [CrossRef]
- Khan, S.; Ajmal, S.; Hussain, T.; Rahman, M.U. Clay-Based Materials for Enhanced Water Treatment: Adsorption Mechanisms, Challenges, and Future Directions. J. Umm Al-Qura Univ. Appll. Sci. 2023. [Google Scholar] [CrossRef]
- Lefticariu, L.; Sutton, S.R.; Lanzirotti, A.; Flynn, T.M. Enhanced Immobilization of Arsenic from Acid Mine Drainage by Detrital Clay Minerals. ACS Earth Space Chem. 2019, 3, 2525–2538. [Google Scholar] [CrossRef]
- Bibi, I.; Singh, B.; Silvester, E. Dissolution Kinetics of Soil Clays in Sulfuric Acid Solutions: Ionic Strength and Temperature Effects. Appl. Geochem. 2014, 51, 170–183. [Google Scholar] [CrossRef]
- Auboiroux, M.; Baillif, P.; Touray, J.C.; Bergaya, F. Fixation of Zn2+ and Pb2+ by a Ca-Montmorillonite in Brines and Dilute Solutions: Preliminary Results. Appl. Clay Sci. 1996, 11, 117–126. [Google Scholar] [CrossRef]
- Lazarević, S.; Janković-Častvan, I.; Jovanović, D.; Milonjić, S.; Janaćković, D.; Petrović, R. Adsorption of Pb2+, Cd2+ and Sr2+ Ions onto Natural and Acid-Activated Sepiolites. Appl. Clay Sci. 2007, 37, 47–57. [Google Scholar] [CrossRef]
- Brigatti, M.F.; Colonna, S.; Malferrari, D.; Medici, L.; Poppi, L. Mercury Adsorption by Montmorillonite and Vermiculite: A Combined XRD, TG-MS, and EXAFS Study. Appl. Clay Sci. 2005, 28, 1–8. [Google Scholar] [CrossRef]
- Celis, R.; HermosÍn, M.C.; Cornejo, J. Heavy Metal Adsorption by Functionalized Clays. Environ. Sci. Technol. 2000, 34, 4593–4599. [Google Scholar] [CrossRef]
- Churakov, S.V.; Dähn, R. Zinc Adsorption on Clays Inferred from Atomistic Simulations and EXAFS Spectroscopy. Environ. Sci. Technol. 2012, 46, 5713–5719. [Google Scholar] [CrossRef]
- Škorňa, P.; Jankovič, L.; Scholtzová, E.; Tunega, D. Hexavalent Chromium Adsorption by Tetrahexylphosphonium Modified Beidellite Clay. Appl. Clay Sci. 2022, 228, 106623. [Google Scholar] [CrossRef]
- Melo, T.M.; Schauerte, M.; Bluhm, A.; Slaný, M.; Paller, M.; Bolan, N.; Bosch, J.; Fritzsche, A.; Rinklebe, J. Ecotoxicological Effects of Per- and Polyfluoroalkyl Substances (PFAS) and of a New PFAS Adsorbing Organoclay to Immobilize PFAS in Soils on Earthworms and Plants. J. Hazard. Mater. 2022, 433, 128771. [Google Scholar] [CrossRef] [PubMed]
- Ewis, D.; Ba-Abbad, M.M.; Benamor, A.; El-Naas, M.H. Adsorption of Organic Water Pollutants by Clays and Clay Minerals Composites: A Comprehensive Review. Appl. Clay Sci. 2022, 229, 106686. [Google Scholar] [CrossRef]
- Preocanin, T.; Abdelmonem, A.; Montavon, G.; Lützenkirchen, J. Charging Behavior of Clays and Clay Minerals in Aqueous Electrolyte Solutions—Experimental Methods for Measuring the Charge and Interpreting the Results. In Clays, Clay Minerals and Ceramic Materials Based on Clay Minerals; Do Nascimento, G.M., Ed.; InTech: London, UK, 2016; Volume 51. [Google Scholar] [CrossRef]
- Developments in Clay Science; Tournassat, C.; Steefel, C.I.; Bourg, I.C.; Bergaya, F. (Eds.) Natural and Engineered Clay Barriers; Elsevier: Amsterdam, The Netherlands, 2015; Volume 6, ISBN 978-0-08-100050-2. [Google Scholar]
- Kuwahara, Y. Surface Microtopography of Lath-Shaped Hydrothermal Illite by Tapping-ModeTM and Contact-Mode AFM. Clays Clay Miner. 1998, 46, 574–582. [Google Scholar] [CrossRef]
- Kuwahara, Y.; Uehara, S.; Aoki, Y. Atomic Force Microscopy Study of Hydrothermal Illite in Izumiyama Pottery Stone from Arita, Saga Prefecture, Japan. Clays Clay Miner. 2001, 49, 300–309. [Google Scholar] [CrossRef]
- Kurganskaya, I.; Luttge, A. A Comprehensive Stochastic Model of Phyllosilicate Dissolution: Structure and Kinematics of Etch Pits Formed on Muscovite Basal Face. Geochim. Cosmochim. Acta 2013, 120, 545–560. [Google Scholar] [CrossRef]
- Lowe, B.M.; Skylaris, C.-K.; Green, N.G. Acid-Base Dissociation Mechanisms and Energetics at the Silica–Water Interface: An Activationless Process. J. Colloid Interface Sci. 2015, 451, 231–244. [Google Scholar] [CrossRef]
- Schliemann, R.; Churakov, S.V. Atomic Scale Mechanism of Clay Minerals Dissolution Revealed by Ab Initio Simulations. Geochim. Cosmochim. Acta 2021, 293, 438–460. [Google Scholar] [CrossRef]
- Criscenti, L.J.; Kubicki, J.D.; Brantley, S.L. Silicate Glass and Mineral Dissolution: Calculated Reaction Paths and Activation Energies for Hydrolysis of a Q3 Si by H3O+ Using Ab Initio Methods. J. Phys. Chem. A 2006, 110, 198–206. [Google Scholar] [CrossRef]
- Morrow, C.P.; Nangia, S.; Garrison, B.J. Ab Initio Investigation of Dissolution Mechanisms in Aluminosilicate Minerals. J. Phys. Chem. A 2009, 113, 1343–1352. [Google Scholar] [CrossRef]
- Pelmenschikov, A.; Leszczynski, J.; Pettersson, L.G.M. Mechanism of Dissolution of Neutral Silica Surfaces: Including Effect of Self-Healing. J. Phys. Chem. A 2001, 105, 9528–9532. [Google Scholar] [CrossRef]
- Pelmenschikov, A.; Strandh, H.; Pettersson, L.G.M.; Leszczynski, J. Lattice Resistance to Hydrolysis of Si−O−Si Bonds of Silicate Minerals: Ab Initio Calculations of a Single Water Attack onto the (001) and (111) β-Cristobalite Surfaces. J. Phys. Chem. B 2000, 104, 5779–5783. [Google Scholar] [CrossRef]
- Gin, S.; Delaye, J.-M.; Angeli, F.; Schuller, S. Aqueous Alteration of Silicate Glass: State of Knowledge and Perspectives. npj Mater. Degrad. 2021, 5, 42. [Google Scholar] [CrossRef]
- Kurganskaya, I.; Churakov, S.V. Carbonate Dissolution Mechanisms in the Presence of Electrolytes Revealed by Grand Canonical and Kinetic Monte Carlo Modeling. J. Phys. Chem. C 2018, 122, 29285–29297. [Google Scholar] [CrossRef]
- Downs, B.; Bartelmehs, K.; Sinnaswamy, K. Database of XtalDraw 1.0 Software. XtalDraw 2004. Available online: https://www.geo.arizona.edu/xtal/group/software.htm#xtaldraw (accessed on 27 August 2024).
- Köhler, S.J.; Dufaud, F.; Oelkers, E.H. An Experimental Study of Illite Dissolution Kinetics as a Function of Ph from 1.4 to 12.4 and Temperature from 5 to 50 °C. Geochim. Cosmochim. Acta 2003, 67, 3583–3594. [Google Scholar] [CrossRef]
- Voter, A.F. Introduction to the Kinetic Monte Carlo Method. In Radiation Effects in Solids; Sickafus, K.E., Kotomin, E.A., Uberuaga, B.P., Eds.; Springer: Dordrecht, The Netherlands, 2007; pp. 1–23. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD-Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. Available online: http://www.ks.uiuc.edu/Research/vmd/ (accessed on 27 August 2024). [CrossRef]
- Köhler, S.J.; Bosbach, D.; Oelkers, E.H. Do Clay Mineral Dissolution Rates Reach Steady State? Geochim. Cosmochim. Acta 2005, 69, 1997–2006. [Google Scholar] [CrossRef]
- Kuwahara, Y. In Situ Observations of Muscovite Dissolution under Alkaline Conditions at 25–50 °C by AFM with an Air/Fluid Heater System. Am. Mineral. 2008, 93, 1028–1033. [Google Scholar] [CrossRef]
- Kuwahara, Y. In-Situ AFM Study of Smectite Dissolution under Alkaline Conditions at Room Temperature. Am. Mineral. 2006, 91, 1142–1149. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of Gases in Multimolecular Layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Naderi, M. Chapter Fourteen—Surface Area: Brunauer–Emmett–Teller (BET). In Progress in Filtration and Separation; Tarleton, S., Ed.; Academic Press: Oxford, UK, 2015; pp. 585–608. ISBN 978-0-12-384746-1. [Google Scholar]
- Sing, K.S.W. Reporting Physisorption Data for Gas/Solid Systems with Special Reference to the Determination of Surface Area and Porosity (Recommendations 1984). Pure Appl. Chem. 1985, 57, 603–619. [Google Scholar] [CrossRef]
- Shimizu, S.; Matubayasi, N. Surface Area Estimation: Replacing the Brunauer–Emmett–Teller Model with the Statistical Thermodynamic Fluctuation Theory. Langmuir 2022, 38, 7989–8002. [Google Scholar] [CrossRef] [PubMed]
- Cama, J.; Ganor, J.; Ayora, C.; Lasaga, C.A. Smectite Dissolution Kinetics at 80 °C and pH 8.8. Geochimica et Cosmochimica Acta 2000, 64, 2701–2717. [Google Scholar] [CrossRef]
- Bibi, I.; Singh, B.; Silvester, E. Dissolution of Illite in Saline–Acidic Solutions at 25 °C. Geochimica et Cosmochimica Acta 2011, 75, 3237–3249. [Google Scholar] [CrossRef]
- Kurganskaya, I.; Niazi, N.K.; Luttge, A. A Modeling Approach for Unveiling Adsorption of Toxic Ions on Iron Oxide Nanocrystals. J. Hazard. Mater. 2021, 417, 126005. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.J.; Wilson, L.; Patey, I. The Influence of Individual Clay Minerals on Formation Damage of Reservoir Sandstones: A Critical Review with Some New Insights. Clay Miner. 2014, 49, 147–164. [Google Scholar] [CrossRef]
- Peltz, M.; Jacob, A.; Grathoff, G.H.; Enzmann, F.; Kersten, M.; Warr, L.N. A FIB-SEM Study of Illite Morphology in Aeolian Rotliegend Sandstones: Implications for Understanding the Petrophysical Properties of Reservoir Rocks. Clays Clay Miner. 2022, 70, 84–105. [Google Scholar] [CrossRef]
- Kurganskaya, I.; Arvidson, R.S.; Fischer, C.; Luttge, A. Does the Stepwave Model Predict Mica Dissolution Kinetics? Geochim. Cosmochim. Acta 2012, 97, 120–130. [Google Scholar] [CrossRef]
- Labbez, C.; Jönsson, B.; Pochard, I.; Nonat, A.; Cabane, B. Surface Charge Density and Electrokinetic Potential of Highly Charged Minerals: Experiments and Monte Carlo Simulations on Calcium Silicate Hydrate. J. Phys. Chem. B 2006, 110, 9219–9230. [Google Scholar] [CrossRef]
- Churakov, S.V.; Labbez, C.; Pegado, L.; Sulpizi, M. Intrinsic Acidity of Surface Sites in Calcium Silicate Hydrates and Its Implication to Their Electrokinetic Properties. J. Phys. Chem. C 2014, 118, 11752–11762. [Google Scholar] [CrossRef]
- Bourg, I.C.; Sposito, G.; Bourg, A.C.M. Modeling the Acid–Base Surface Chemistry of Montmorillonite. J. Colloid Interface Sci. 2007, 312, 297–310. [Google Scholar] [CrossRef]
- Tournassat, C.; Davis, J.A.; Chiaberge, C.; Grangeon, S.; Bourg, I.C. Modeling the Acid–Base Properties of Montmorillonite Edge Surfaces. Environ. Sci. Technol. 2016, 50, 13436–13445. [Google Scholar] [CrossRef]
- Tournassat, C.; Bourg, I.C.; Steefel, C.I.; Bergaya, F. Chapter 1—Surface Properties of Clay Minerals. In Developments in Clay Science; Tournassat, C., Steefel, C.I., Bourg, I.C., Bergaya, F., Eds.; Natural and Engineered Clay Barriers; Elsevier: Amsterdam, The Netherlands, 2015; Volume 6, pp. 5–31. [Google Scholar]
- Tournassat, C.; Ferrage, E.; Poinsignon, C.; Charlet, L. The Titration of Clay Minerals: II. Structure-Based Model and Implications for Clay Reactivity. J. Colloid Interface Sci. 2004, 273, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Rosenqvist, J.; Persson, P.; Sjöberg, S. Protonation and Charging of Nanosized Gibbsite (α-Al(OH)3) Particles in Aqueous Suspension. Langmuir 2002, 18, 4598–4604. [Google Scholar] [CrossRef]
- Gao, P.; Liu, X.; Guo, Z.; Tournassat, C. Acid–Base Properties of Cis-Vacant Montmorillonite Edge Surfaces: A Combined First-Principles Molecular Dynamics and Surface Complexation Modeling Approach. Environ. Sci. Technol. 2023, 57, 1342–1352. [Google Scholar] [CrossRef] [PubMed]
- Hiemstra, T.; Venema, P.; Riemsdijk, W.H.V. Intrinsic Proton Affinity of Reactive Surface Groups of Metal (Hydr)Oxides: The Bond Valence Principle. J. Colloid Interface Sci. 1996, 184, 680–692. [Google Scholar] [CrossRef]
- Pauling, L. The principles determining the structure of complex ionic crystals. J. Am. Chem. Soc. 1929, 51, 1010–1026. [Google Scholar] [CrossRef]
- Brown, I.D. 14—The Bond-Valence Method: An Empirical Approach to Chemical Structure and Bonding. In Industrial Chemistry Library; O’Keeffe, M., Navrotsky, A., Eds.; Structure and Bonding in Crystals; Elsevier: Amsterdam, The Netherlands, 1981; Volume 2, pp. 1–30. [Google Scholar]
- Brown, I.D. Recent Developments in the Methods and Applications of the Bond Valence Model. Chem. Rev. 2009, 109, 6858–6919. [Google Scholar] [CrossRef]
- Bickmore, B.R.; Tadanier, C.J.; Rosso, K.M.; Monn, W.D.; Eggett, D.L. Bond-Valence Methods for pKa Prediction: Critical Reanalysis and a New Approach1. Geochim. Cosmochim. Acta 2004, 68, 2025–2042. [Google Scholar] [CrossRef]
- Bickmore, B.R.; Rosso, K.M.; Tadanier, C.J.; Bylaska, E.J.; Doud, D. Bond-Valence Methods for pKa Prediction. II. Bond-Valence, Electrostatic, Molecular Geometry, and Solvation Effects. Geochim. Cosmochim. Acta 2006, 70, 4057–4071. [Google Scholar] [CrossRef]
- Sulpizi, M.; Sprik, M. Acidity Constants from DFT-Based Molecular Dynamics Simulations. J. Phys. Condens. Matter 2010, 22, 284116. [Google Scholar] [CrossRef]
- Meites, L. Theory of Titration Curves. VIII. A New Interpretation of Acid-Base Titration Curves for Polymeric Acids. The Significance of the Kern-Equation Parameters pKa and n. J. Polym. Sci. Part A-1 Polym. Chem. 1972, 10, 779–790. [Google Scholar] [CrossRef]
- Kriaa, A.; Hamdi, N.; Srasra, E. Surface Properties and Modeling Potentiometric Titration of Aqueous Illite Suspensions. Surf. Engin. Appl. Electrochem. 2008, 44, 217–229. [Google Scholar] [CrossRef]
- Delhorme, M.; Labbez, C.; Caillet, C.; Thomas, F. Acid−Base Properties of 2:1 Clays. I. Modeling the Role of Electrostatics. Langmuir 2010, 26, 9240–9249. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Lu, X.; Sprik, M.; Cheng, J.; Meijer, E.J.; Wang, R. Acidity of Edge Surface Sites of Montmorillonite and Kaolinite. Geochim. Cosmochim. Acta 2013, 117, 180–190. [Google Scholar] [CrossRef]
- Liu, X.; Cheng, J.; Sprik, M.; Lu, X.; Wang, R. Interfacial Structures and Acidity of Edge Surfaces of Ferruginous Smectites. Geochim. Cosmochim. Acta 2015, 168, 293–301. [Google Scholar] [CrossRef]
- Liu, X.; Cheng, J.; Sprik, M.; Lu, X.; Wang, R. Understanding Surface Acidity of Gibbsite with First Principles Molecular Dynamics Simulations. Geochim. Cosmochim. Acta 2013, 120, 487–495. [Google Scholar] [CrossRef]
- Tazi, S.; Rotenberg, B.; Salanne, M.; Sprik, M.; Sulpizi, M. Absolute Acidity of Clay Edge Sites from Ab-Initio Simulations. Geochim. Cosmochim. Acta 2012, 94, 1–11. [Google Scholar] [CrossRef]
- Liu, X.; Cheng, J.; Sprik, M.; Lu, X.; Wang, R. Surface Acidity of 2:1-Type Dioctahedral Clay Minerals from First Principles Molecular Dynamics Simulations. Geochim. Cosmochim. Acta 2014, 140, 410–417. [Google Scholar] [CrossRef]
- Tournassat, C.; Greneche, J.-M.; Tisserand, D.; Charlet, L. The Titration of Clay Minerals: I. Discontinuous Backtitration Technique Combined with CEC Measurements. J. Colloid Interface Sci. 2004, 273, 224–233. [Google Scholar] [CrossRef]
- Guo, Y.; Yu, X.; Guo, Y.; Yu, X. Characterizing the Surface Charge of Clay Minerals with Atomic Force Microscope (AFM). Aimsmates 2017, 4, 582–593. [Google Scholar] [CrossRef]
- Labbez, C.; Jönsson, B. A New Monte Carlo Method for the Titration of Molecules and Minerals. In Applied Parallel Computing. State of the Art in Scientific Computing; Springer: Berlin/Heidelberg, Germany, 18 June 2006; pp. 66–72. [Google Scholar]
- Labbez, C.; Jonsson, B.; Skarba, M.; Borkovec, M. Ion-Ion Correlation and Charge Reversal at Titrating Solid Interfaces. arXiv 2008, arXiv:0812.0300. [Google Scholar] [CrossRef] [PubMed]
- Tombácz, E.; Nyilas, T.; Libor, Z.; Csanaki, C. Surface Charge Heterogeneity and Aggregation of Clay Lamellae in Aqueous Suspensions. In From Colloids to Nanotechnology; Zrínyi, M., Hórvölgyi, Z.D., Eds.; Springer: Berlin/Heidelberg, Germany, 2004; pp. 206–215. [Google Scholar]
- Zebardast, H.R.; Pawlik, M.; Rogak, S.; Asselin, E. Potentiometric Titration of Hematite and Magnetite at Elevated Temperatures Using a ZrO2-Based pH Probe. Colloids Surf. A Physicochem. Eng. Asp. 2014, 444, 144–152. [Google Scholar] [CrossRef]
- Marty, N.C.M.; Claret, F.; Lassin, A.; Tremosa, J.; Blanc, P.; Madé, B.; Giffaut, E.; Cochepin, B.; Tournassat, C. A Database of Dissolution and Precipitation Rates for Clay-Rocks Minerals. Appl. Geochem. 2015, 55, 108–118. [Google Scholar] [CrossRef]
- Xiao, Y.; Lasaga, A.C. Ab Initio Quantum Mechanical Studies of the Kinetics and Mechanisms of Quartz Dissolution: OH− Catalysis. Geochim. Cosmochim. Acta 1996, 60, 2283–2295. [Google Scholar] [CrossRef]
- Xiao, Y.; Lasaga, A.C. Ab Initio Quantum Mechanical Studies of the Kinetics and Mechanisms of Silicate Dissolution: H+(H3O+) Catalysis. Geochim. Cosmochim. Acta 1994, 58, 5379–5400. [Google Scholar] [CrossRef]
- Fenter, P.; Zapol, P.; He, H.; Sturchio, N.C. On the Variation of Dissolution Rates at the Orthoclase (0 0 1) Surface with pH and Temperature. Geochim. Cosmochim. Acta 2014, 141, 598–611. [Google Scholar] [CrossRef]
- Lasaga, A.C.; Soler, J.M.; Ganor, J.; Burch, T.E.; Nagy, K.L. Chemical Weathering Rate Laws and Global Geochemical Cycles. Geochim. Cosmochim. Acta 1994, 58, 2361–2386. [Google Scholar] [CrossRef]
- Ganor, J.; Mogollón, J.L.; Lasaga, A.C. The Effect of pH on Kaolinite Dissolution Rates and on Activation Energy. Geochim. Et Cosmochim. Acta 1995, 59, 1037–1052. [Google Scholar] [CrossRef]
- Lüttge, A.; Arvidson, R.S.; Fischer, C. A Stochastic Treatment of Crystal Dissolution Kinetics. Elements 2013, 9, 183–188. [Google Scholar] [CrossRef]
- Arvidson, R.S.; Ertan, I.E.; Amonette, J.E.; Luttge, A. Variation in Calcite Dissolution Rates:: A Fundamental Problem? Geochim. Cosmochim. Acta 2003, 67, 1623–1634. [Google Scholar] [CrossRef]
- Fischer, C.; Arvidson, R.S.; Lüttge, A. How Predictable Are Dissolution Rates of Crystalline Material? Geochim. Cosmochim. Acta 2012, 98, 177–185. [Google Scholar] [CrossRef]
- Kurganskaya, I.; Luttge, A. Mineral Dissolution Kinetics: Pathways to Equilibrium. ACS Earth Space Chem. 2021, 5, 1657–1673. [Google Scholar] [CrossRef]
- Schliemann, R.; Churakov, S.V. Pyrophyllite Dissolution at Elevated Pressure Conditions: An Ab Initio Study. Geochim. Cosmochim. Acta 2021, 307, 42–55. [Google Scholar] [CrossRef]


| Type of Bond, i | ∆Ei, kT Units | Type of Bond, i | ∆Ei, kT Units |
|---|---|---|---|
| Si-Si | 8.0 | Si-Mg(O) | 9.0 |
| Si-Al(T) | 6.0 | Si-Fe(O) | 10.0 |
| Si-Al(O) | 5.0 | Al(T)-Mg(O) | 7.0 |
| Al(T)-Al(O) | 5.0 | Al(T)-Fe(O) | 8.0 |
| Al(O)-Al(O) | 5.0 | Al(O)-Mg(O) | 6.0 |
| OH-steric factor | 1.0 | Al(O)-Fe(O) | 7.0 |
| 2nd order T-T | 4.0 | Mg(O)-Fe(O) | 7.5 |
| 2nd order O-O | 2.5 | Mg(O)-Mg(O) | 7.0 |
| 2nd order T-O | 2.5 | Fe(O)-Fe(O) | 8.0 |
| 2nd order O-T | 2.0 |
| Site | pKa | Deprotonated/Protonated Site | Activation Energy Correction, kT Units |
|---|---|---|---|
| Si-OH0 | 7 | Si-O− | −10 |
| Al(T)-OH2 | 15 | Al(T)-OH− | −7 |
| Al(O)-(OH2)(OH)0 | 10 | Al(O)-(OH)(OH)− | −10 |
| Fe(III)-(OH2)(OH)0 | 9 | Fe(III)-(OH)(OH)− | −10 |
| Mg-(OH2)20 | 13 | Mg-(OH2)(OH)− | −10 |
| Si-O(br)-Al(O) | 2 | Si-O(br)H+-Al(O) | −15 |
| Al(T)-O(br)-Al(O) | 5 | Al(T)-O(br)H+-Al(O) | −10 |
| Si-O(br)-Mg(O) | 2 | Si-O(br)H+-Mg(O) | −10 |
| Al(T)-O(br)-Mg(O) | 5 | Al(T)-O(br)H+-Mg(O) | −10 |
| Si-O(br)-Fe(O) | 2 | Si-O(br)H+-Fe(O) | −10 |
| Al(T)-O(br)-Fe(O) | 5 | Al(T)-O(br)H+-Fe(O) | −10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurganskaya, I. Dissolution Mechanisms and Surface Charge of Clay Mineral Nanoparticles: Insights from Kinetic Monte Carlo Simulations. Minerals 2024, 14, 900. https://doi.org/10.3390/min14090900
Kurganskaya I. Dissolution Mechanisms and Surface Charge of Clay Mineral Nanoparticles: Insights from Kinetic Monte Carlo Simulations. Minerals. 2024; 14(9):900. https://doi.org/10.3390/min14090900
Chicago/Turabian StyleKurganskaya, Inna. 2024. "Dissolution Mechanisms and Surface Charge of Clay Mineral Nanoparticles: Insights from Kinetic Monte Carlo Simulations" Minerals 14, no. 9: 900. https://doi.org/10.3390/min14090900