
In-Depth Analysis of Complex Multiphase Oxidative Transformations in Iron Sulfides (Pyrrhotite and Pyrite) Within Migmatitic Gneiss



Abstract
:1. Introduction
2. Geological Setting and Sampling
3. Methods
4. Results
4.1. Unaltered Pyrrhotite

4.2. Altered Pyrrhotite
4.2.1. First Stage of Alteration: IS Grains
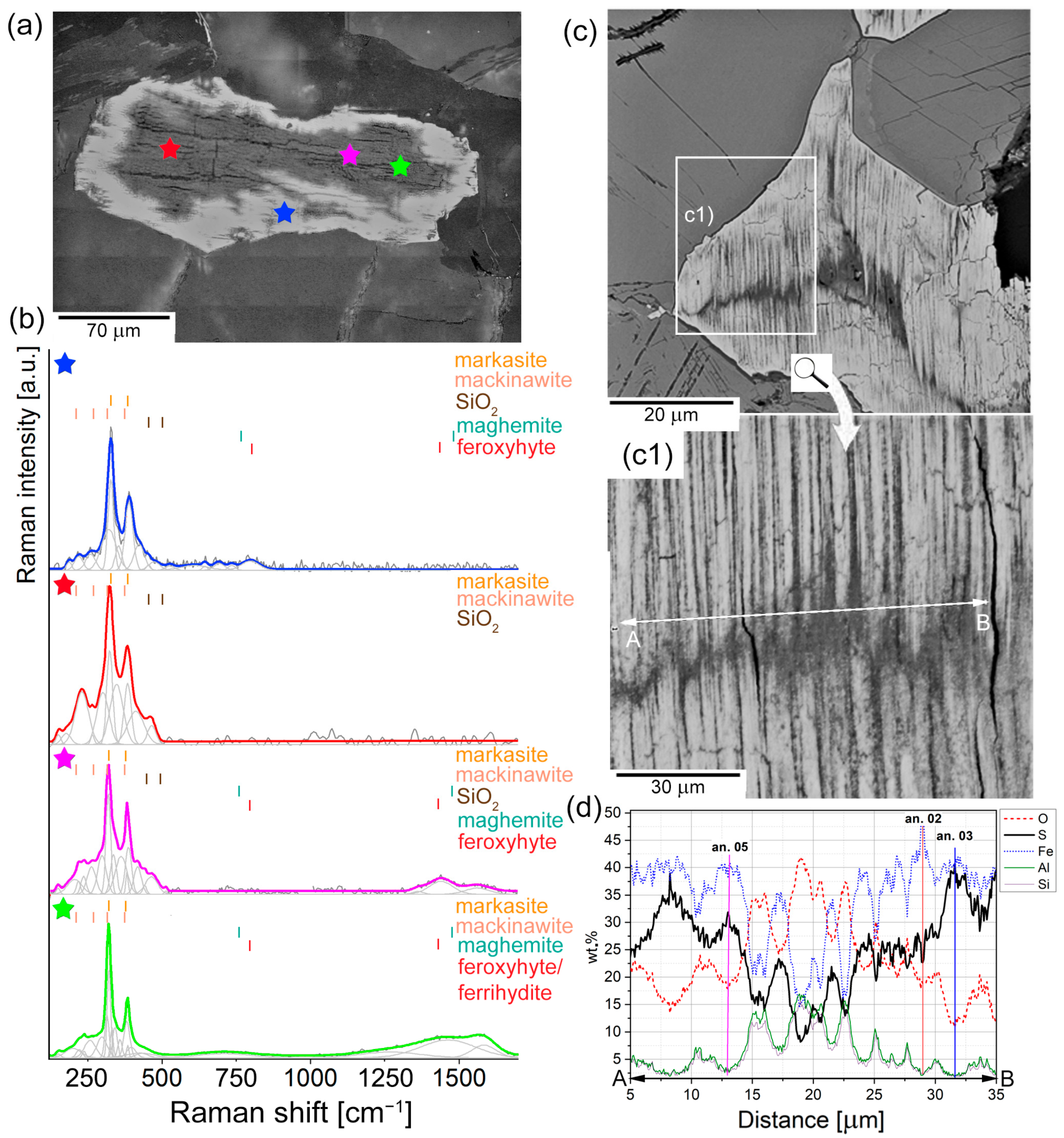

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Zn | 0.00 | 0.07 | 0.00 | 0.00 | 0.08 | 0.00 | 0.04 | 0.00 | 0.00 | |
| Co | 0.04 | 0.20 | 0.14 | 0.10 | 0.21 | 0.03 | 0.13 | 0.21 | 0.07 | |
| Ni | 0.16 | 0.42 | 0.12 | 0.22 | 0.24 | 0.12 | 0.19 | 0.31 | 0.11 | |
| Fe | 46.24 | 45.93 | 46.76 | 46.24 | 46.16 | 41.77 | 47.11 | 62.73 | 49.51 | 4.45 |
| S | 53.27 | 52.74 | 49.17 | 46.81 | 37.11 | 10.76 | 23.33 | 10.54 | 38.74 | 5.11 |
| Si | 0.06 | 0.30 | 0.05 | 0.83 | 0.86 | 3.67 | 3.60 | 0.41 | 0.88 | 19.46 |
| Al | 0.02 | 0.28 | 0.03 | 0.79 | 0.80 | 3.50 | 3.43 | 0.36 | 0.90 | 18.64 |
| Mg | 0.01 | 0.00 | 0.00 | 0.00 | 0.01 | 0.00 | 0.00 | 0.00 | 0.00 | |
| Ca | 0.05 | 0.04 | 0.29 | 0.33 | 0.14 | 0.08 | 0.19 | |||
| Na | 0.50 | 0.24 | ||||||||
| K | 0.39 | 0.33 | ||||||||
| P | 0.68 | 0.51 | ||||||||
| O | 0.75 | 1.80 | 4.40 | 13.06 | 37.26 | 20.70 | 24.13 | 9.20 | 49.80 | |
| Total | 99.79 | 100.74 | 98.07 | 99.53 | 98.82 | 99.01 | 98.67 | 98.77 | 100.68 | 97.45 |
| Percentage of minerals * | ||||||||||
| Mrc | 98.66 | 91.99 | 87.57 | 69.64 | 20.12 | 43.65 | 19.72 | 72.47 | 9.55 | |
| Gth | 24.48 * | |||||||||
| Fex | 62.10 * | |||||||||
| Mgh | 5.64 | 7.83 | 38.30 | 76.56 | 22.55 * | |||||
| Kln | 1.33 | 0.20 | 3.78 | 3.85 | 16.73 | 16.42 | 1.72 | 4.32 | 89.18 | |
| Total | 100.74 | 97.83 | 99.18 | 97.97 | 98.95 | 98.40 | 98.00 | 99.34 | 98.73 | |
4.2.2. The Second Stage of Alteration: ISO Grains

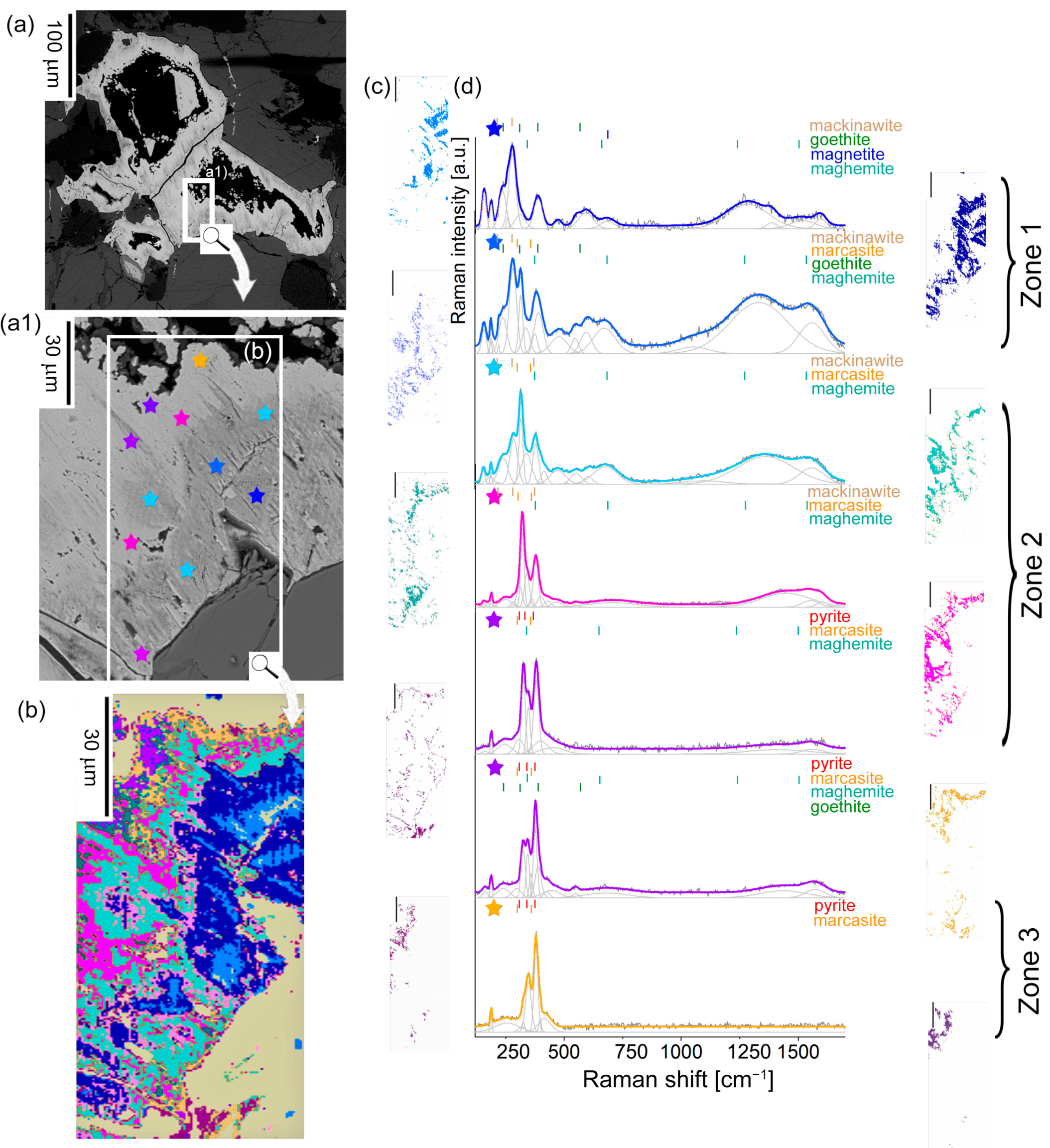
4.2.3. Stage Three: ISOS Grains
| Oxide | ISOS1 | ISOS2 | ||
|---|---|---|---|---|
| SiO2 | 17.68 | 44.32 | 42.17 | 40.48 |
| Al2O3 | 4.03 | 37.06 | 37.99 | 37.37 |
| Fe2O3 | 67.21 | 2.66 | 2.56 | 4.66 |
| MnO | 0.02 | 0.02 | 0.08 | 0.02 |
| MgO | 0.00 | 0.03 | 0.03 | 0.00 |
| CaO | 0.24 | 0.23 | 0.17 | 0.35 |
| K2O | 0.47 | 0.27 | 0.82 | 1.27 |
| Na2O | 0.00 | 0.00 | 0.09 | 0.10 |
| SO3 | 1.28 | 0.73 | 2.50 | 3.65 |
| P2O5 | 0.76 | 0.48 | 0.52 | 0.48 |
| Total | 91.70 | 85.80 | 86.92 | 87.23 |
| Percentage of minerals * | ||||
| Gth | 72.01 | 2.96 | 2.85 | 5.18 |
| Kln | 10.21 | 92.06 | 90.14 | 85.80 |
| Silica | 12.92 | 1.46 | 0.21 | 0.55 |
| Jrs | 5.69 | |||
| Alu | 1.89 | 6.47 | 9.44 | |
| Total | 100.83 | 98.37 | 99.67 | 100.97 |
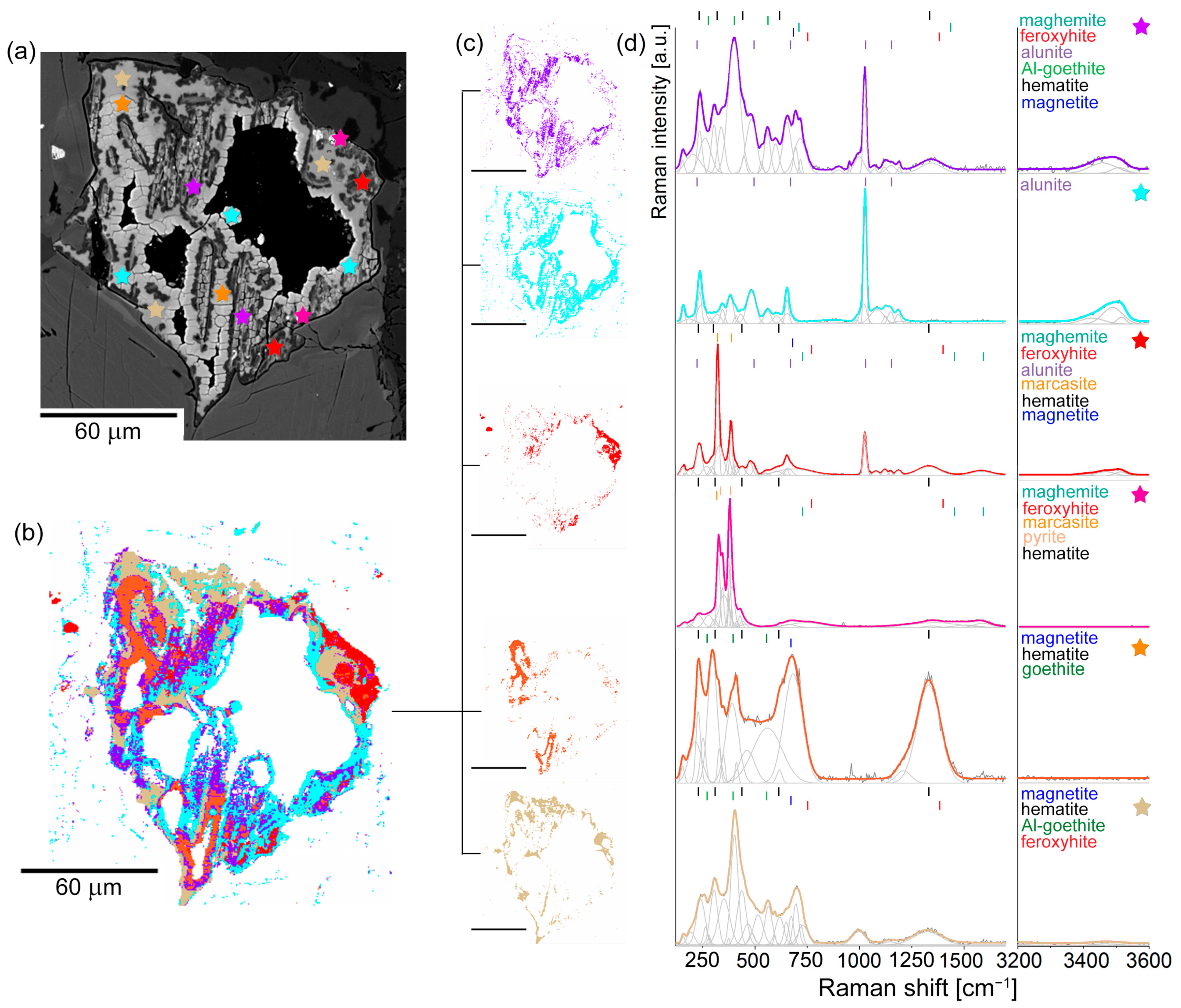
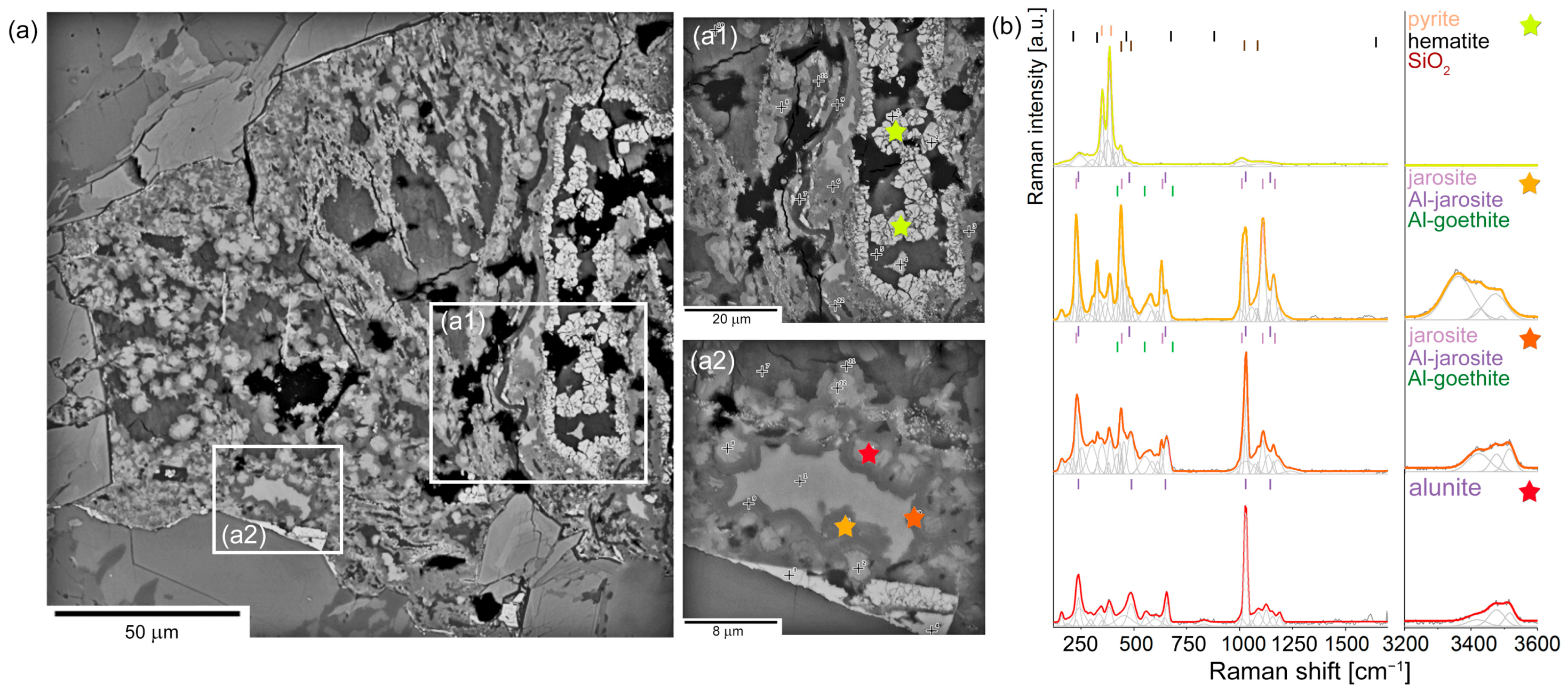
5. Pyrite Oxidation Products in Hydrothermal Veins
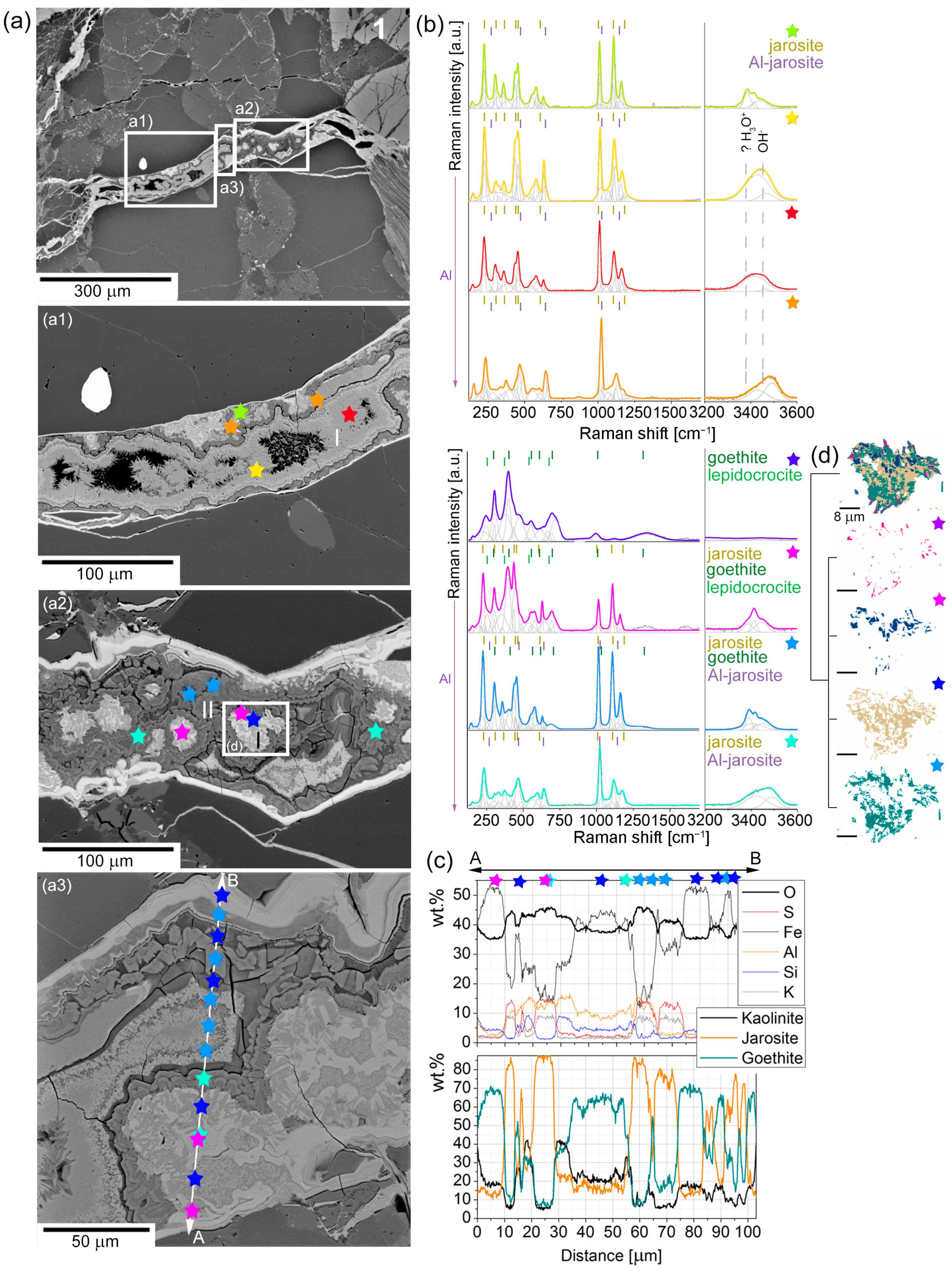
| Oxide | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
|---|---|---|---|---|---|---|---|---|---|
| SiO2 | 0.05 | 0.26 | 0.03 | 0.20 | 0.04 | 0.59 | 0.43 | 1.09 | 2.77 |
| Al2O3 | 0.52 | 2.52 | 6.10 | 26.43 | 0.46 | 3.05 | 7.38 | 24.98 | 30.65 |
| Fe2O3 | 46.02 | 43.26 | 37.43 | 13.52 | 46.96 | 45.15 | 39.64 | 20.40 | 20.27 |
| MgO | 0.04 | 0.00 | 0.03 | 0.01 | 0.05 | 0.00 | 0.02 | 0.00 | 0.01 |
| MnO | 0.07 | 0.00 | 0.00 | 0.00 | 0.12 | 0.00 | 0.03 | 0.01 | 0.05 |
| CaO | 0.05 | 0.01 | 0.04 | 0.16 | 0.08 | 0.24 | 0.08 | 0.11 | 0.41 |
| Na2O | 0.26 | 0.10 | 0.40 | 0.11 | 0.21 | 0.12 | 0.06 | 0.10 | 0.08 |
| K2O | 6.50 | 3.72 | 3.61 | 6.58 | 6.18 | 7.91 | 4.68 | 3.50 | 4.39 |
| SO3 | 32.20 | 31.62 | 30.35 | 36.82 | 29.77 | 30.25 | 29.61 | 35.43 | 35.96 |
| P2O5 | 0.00 | 0.16 | 1.07 | 0.40 | 0.00 | 1.59 | 0.29 | 0.85 | |
| Total | 85.71 | 81.65 | 79.06 | 84.22 | 83.87 | 88.91 | 81.93 | 85.91 | 95.42 |
| Number of ions calculated based on 2(XO4), where X = S and P | |||||||||
| Si | 0.004 | 0.022 | 0.003 | 0.014 | 0.004 | 0.051 | 0.039 | 0.082 | 0.202 |
| Al | 0.051 | 0.250 | 0.619 | 2.242 | 0.049 | 0.308 | 0.783 | 2.205 | 2.643 |
| Fe3+ | 2.866 | 2.736 | 2.425 | 0.732 | 3.163 | 2.907 | 2.685 | 1.149 | 1.116 |
| Mg | 0.005 | 0.000 | 0.004 | 0.001 | 0.007 | 0.000 | 0.003 | 0.000 | 0.001 |
| Mn | 0.005 | 0.000 | 0.000 | 0.000 | 0.009 | 0.000 | 0.002 | 0.001 | 0.003 |
| Ca | 0.004 | 0.001 | 0.004 | 0.012 | 0.008 | 0.022 | 0.008 | 0.009 | 0.032 |
| Na | 0.042 | 0.016 | 0.067 | 0.016 | 0.036 | 0.020 | 0.010 | 0.015 | 0.011 |
| K | 0.686 | 0.399 | 0.396 | 0.604 | 0.706 | 0.864 | 0.537 | 0.334 | 0.409 |
| S | 2.000 | 1.994 | 1.961 | 1.988 | 2.000 | 1.942 | 2.000 | 1.991 | 1.974 |
| P | 0.000 | 0.006 | 0.039 | 0.012 | 0.000 | 0.058 | 0.000 | 0.018 | 0.026 |
| H3O+ * | 0.272 | 0.585 | 0.537 | 0.380 | 0.258 | 0.117 | 0.452 | 0.651 | 0.579 |
| Total | 5.935 | 6.008 | 6.054 | 6.002 | 6.239 | 6.288 | 6.519 | 6.455 | 6.997 |
| ΣB | 2.931 | 2.986 | 3.052 | 2.988 | 3.235 | 3.237 | 3.481 | 3.364 | 3.794 |

6. Discussion
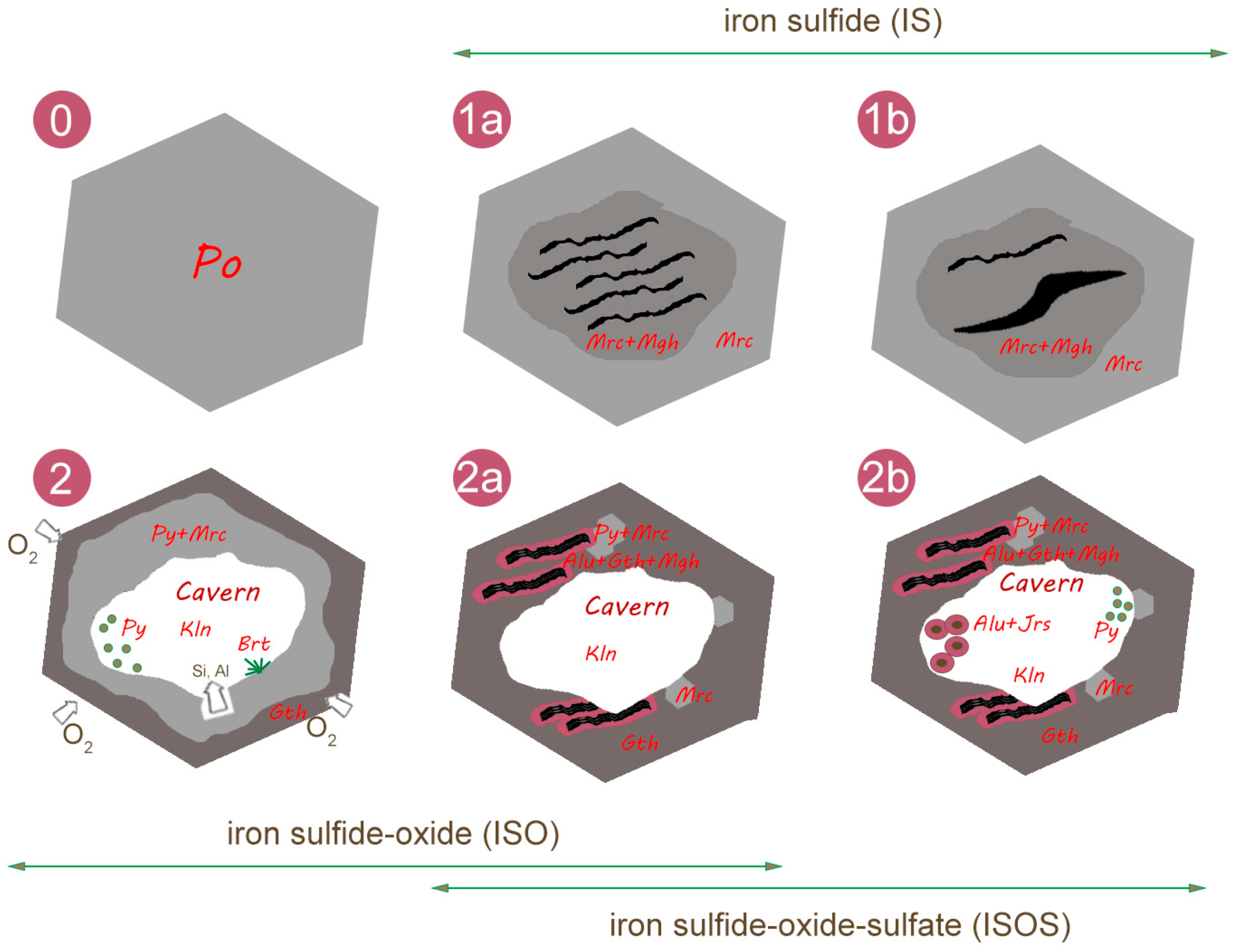
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dold, B. Acid rock drainage prediction: A critical review. J. Geochem. Explor. 2017, 172, 120–132. [Google Scholar] [CrossRef]
- Blowes, D.W.; Ptacek, C.J.; Jambor, J.L.; Weisener, C.G. 9.05—The Geochemistry of Acid Mine Drainage. In Treatise on Geochemistry; Holland, H.D., Turekian, K.K., Eds.; Pergamon: Oxford, UK, 2003; pp. 149–204. ISBN 978-0-08-043751-4. [Google Scholar]
- Moncur, M.C.; Jambor, J.L.; Ptacek, C.J.; Blowes, D.W. Mine drainage from the weathering of sulfide minerals and magnetite. Appl. Geochem. 2009, 24, 2362–2373. [Google Scholar] [CrossRef]
- Cabri, L.J.; Vaughan, D. Modern Approaches to Ore and Environmental Mineralogy; Short Course Series (Mineralogical Association of Canada); Mineralogical Association of Canada: Ottawa, ON, Canada, 1998; Volume 27, ISBN 978-0-921294-00-9. [Google Scholar]
- Jambor, J.L.; Blowes, D.W.; Ritchie, A.I.M. (Eds.) Environmental Aspects of Mine Wastes; Short Course Series; Mineralogical Association of Canada: Ottawa, ON, Canada, 2003; ISBN 978-0-921294-31-3. [Google Scholar]
- Belzile, N.; Chen, Y.-W.; Cai, M.-F.; Li, Y. A review on pyrrhotite oxidation. J. Geochem. Explor. 2004, 84, 65–76. [Google Scholar] [CrossRef]
- Jamieson, H.E. Geochemistry and Mineralogy of Solid Mine Waste: Essential Knowledge for Predicting Environmental Impact. Elements 2011, 7, 381–386. [Google Scholar] [CrossRef]
- Ss En 12620 2008 Specification For Aggregates For Concrete PDF Free | PDF. Available online: https://www.scribd.com/document/587261300/pdfcoffee-com-ss-en-12620-2008-specification-for-aggregates-for-concrete-pdf-free (accessed on 30 December 2024).
- EN 12620:2002+A1:2008; Aggregates for Concrete. European Committee for Standardization: Brussels, Belgium, 2008. Available online: https://standards.iteh.ai/catalog/standards/cen/27c8e34e-993f-4b1a-989f-8a3263dbe9fb/en-12620-2002a1-2008 (accessed on 30 December 2024).
- Marcelino, A.; Calixto, J.; Gumieri, A.; Ferreira, M.; Caldeira, C.; Silva, M.; Costa, A. Evaluation of pyrite and pyrrhotite in concretes. Rev. Ibracon Estrut. E Mater. 2016, 9, 484–493. [Google Scholar] [CrossRef]
- Courtin-Nomade, A.; Grosbois, C.; Marcus, M.A.; Fakra, S.C.; Beny, J.-M.; Foster, A.L. The weathering of a sulfide orebody: Speciation and fate of some potential contaminants. Can. Mineral. 2009, 47, 493–508. [Google Scholar] [CrossRef]
- Hayes, S.M.; Root, R.A.; Perdrial, N.; Maier, R.; Chorover, J. Surficial weathering of iron sulfide mine tailings under semi-arid climate. Geochim. Et Cosmochim. Acta 2014, 141, 240–257. [Google Scholar] [CrossRef]
- Lara, R.H.; Monroy, M.G.; Mallet, M.; Dossot, M.; González, M.A.; Cruz, R. An experimental study of iron sulfides weathering under simulated calcareous soil conditions. Environ. Earth Sci. 2015, 73, 1849–1869. [Google Scholar] [CrossRef]
- Gu, X.; Heaney, P.J.; Reis, F.D.A.A.; Brantley, S.L. Deep abiotic weathering of pyrite. Science 2020, 370, eabb8092. [Google Scholar] [CrossRef]
- Nasdala, L.; Smith, D.; Kaindl, R.; Ziemann, M. Raman spectroscopy: Analytical perspectives in mineralogical research. In EMU Notes in Mineralogy; Eötvös University Press: Budapest, Hungary, 2004; Volume 12, pp. 281–343. ISBN 978-963-463-662-5. [Google Scholar]
- Mazur, S.; Aleksandrowski, P.; Kryza, R.; Oberc-Dziedzic, T. The Variscan Orogen in Poland. Geol. Q. 2006, 50, 89–118. [Google Scholar]
- Kryza, R. Migmatization in gneisses of the northern part of the Sowie Góry, Sudetes. Geol. Sudet. 1981, 16, 7–100. [Google Scholar]
- Ilnicki, S.; Nejbert, K.; Pieczka, A.; Szełęg, E.; Turniak, K.; Szuszkiewicz, A.; Łodziński, M.; Banach, M.; Michałowski, P.; Rożniak, R. Metamorphic record of retrogressed eclogites from the Piława Górna (Sowie Góry block, SW Poland). Mineral. Pol.—Spec. Pap. 2011, 38, 103–104. [Google Scholar]
- Muszer, A.; Szuszkiewicz, A.; Łobos, K. New Occurrence of Clausthalite (PbSe) in the Sudetes (SW Poland). Mineralogia 2006, 37, 123–132. [Google Scholar] [CrossRef]
- Pieczka, A.; Łodziński, M.; Szełęg, E.; Ilnicki, S.; Nejbert, K.; Szuszkiewicz, A.; Turniak, K.; Banach, M.; Michałowski, P.; Różniak, R. The Sowie Mts. pegmatites (Lower Silesia, SW Poland): A current knowledge. Acta Mineral.–Petrogr. 2012, 7, 105–106. [Google Scholar]
- Piestrzyński, A.; Kowalik, K. Argentopentlandite from barite vein in Zagórze Śląskie, Lower Silesia; a first occurrence in Poland. Mineral. Pol. 2015, 45, 13–25. [Google Scholar] [CrossRef]
- Wala, A.; Wolska, A.; Jeleń, P. Stary Julianów Fault in Sowie Góry gneiss complex and geological situation in the place of its occurrence. (Wałbrzych Foothills, Central Sudetes, Southern Poland. Biul. Panstw. Inst. Geol. 2015, 463, 77–116. [Google Scholar] [CrossRef]
- Pieczka, A.; Szuszkiewicz, A.; Szełęg, E.; Janeczek, J.; Nejbert, K. Granitic pegmatites of the Polish part of the Sudetes (NE Bohemian massif, SW Poland). In Fieldtrip Guidebook, Proceedings of the 7th International Symposium on Granitic Pegmatites, Książ, Poland, 17–19 June 2015; PEG: Książ, Poland, 2015; pp. 73–103. [Google Scholar]
- Turniak, K.; Pieczka, A.; Kennedy, A.K.; Szełęg, E.; Ilnicki, S.; Nejbert, K.; Szuszkiewicz, A. Crystallisation age of the Julianna pegmatite system (Góry Sowie Block, NE margin of the Bohemian massif): Evidence from U-Th-Pb SHRIMP monazite and CHIME uraninite studies. In Fieldtrip Guidebook, Proceedings of the 7th International Symposium on Granitic Pegmatites, Książ, Poland, 17–19 June 2015; PEG: Książ, Poland, 2015; pp. 111–112. [Google Scholar]
- Muller, A.; Szuszkiewicz, A.; Ilnicki, S.; Nejbert, K. Quartz chemistry of the Julianna pegmatites and their wall rocks, Piława Górna, Poland: Implications for the origin of pegmatite melts. In Abstracts, Proceedings of the 8th International Symposium on Granitic Pegmatites, PEG2017, Kristiansand, Norway, 13–15 June 2017; Norsk Geologisk Forening: Trondheim, Norway, 2017; pp. 84–88. [Google Scholar]
- Nejbert, K.; Ilnicki, S.; Anczkiewicz, R.; Pieczka, A.; Szełęg, E.; Szuszkiewicz, A.; Turniak, K. Geochronological constraints on metamorphism of eclogites from Piława Górna (Góry Sowie block, Sudetes). Mineral. Spec. Pap. 2013, 41, 66. [Google Scholar]
- Szuszkiewicz, A.; Szełęg, E.; Pieczka, A.; Ilnicki, S.; Nejbert, K.; Turniak, K.; Banach, M.; Łodziński, M.; Różniak, R.; Michałowski, P. The Julianna pegmatite vein system at the Piława Górna Mine, Góry Sowie Block, SW Poland—Preliminary data on geology and descriptive mineralogy. Geol. Q. 2013, 57, 467–484. [Google Scholar] [CrossRef]
- Xi, S.; Zhang, X.; Luan, Z.; Du, Z.; Li, L.; Liang, Z.; Lian, C.; Yan, J. Micro-Raman Study of Thermal Transformations of Sulfide and Oxysalt Minerals Based on the Heat Induced by Laser. Minerals 2019, 9, 751. [Google Scholar] [CrossRef]
- Weber, I.; Böttger, U.; Pavlov, S.G.; Hübers, H.-W.; Hiesinger, H.; Jessberger, E.K. Laser alteration on iron sulfides under various environmental conditions. J. Raman Spectrosc. 2017, 48, 1509–1517. [Google Scholar] [CrossRef]
- Mendili, Y.E.; Abdelouas, A.; Hajj, H.E.; Bardeau, J.-F. Phase transitions of iron sulphides formed by steel microbial corrosion. RSC Adv. 2013, 3, 26343–26351. [Google Scholar] [CrossRef]
- Arnold, R.G. Range in composition and structure of 82 natural terrestrial pyrrhotite. Can. Mineral. 1967, 31–50. [Google Scholar]
- Qian, G.; Xia, F.; Brugger, J.; Skinner, W.M.; Bei, J.; Chen, G.; Pring, A. Replacement of pyrrhotite by pyrite and marcasite under hydrothermal conditions up to 220 °C: An experimental study of reaction textures and mechanisms. Am. Mineral. 2011, 96, 1878–1893. [Google Scholar] [CrossRef]
- Kontny, A.; Friedrich, G.; Behr, H.J.; Wall, H.; Horn, E.E.; Möller, P.; Zulauf, G. Formation of ore minerals in metamorphic rocks of the German continental deep drilling site (KTB). J. Geophys. Res. Solid Earth 1997, 102, 18323–18336. [Google Scholar] [CrossRef]
- Kontny, A.; de Wall, H.; Sharp, T.G.; Pósfai, M. Mineralogy and magnetic behavior of pyrrhotite from a 260 °C section at the KTB drilling site, Germany. Am. Mineral. 2000, 85, 1416–1427. [Google Scholar] [CrossRef]
- Yuan, B.; Luan, W.; Tu, S. One-step synthesis of cubic FeS2 and flower-like FeSe2 particles by a solvothermal reduction process. Dalton Trans. 2011, 41, 772–776. [Google Scholar] [CrossRef]
- Teo, M.Y.C.; Kulinich, S.A.; Plaksin, O.A.; Zhu, A.L. Photoinduced Structural Conversions of Transition Metal Chalcogenide Materials. J. Phys. Chem. A 2010, 114, 4174–4180. [Google Scholar] [CrossRef]
- Bourdoiseau, J.-A.; Jeannin, M.; Sabot, R.; Rémazeilles, C.; Refait, P. Characterisation of mackinawite by Raman spectroscopy: Effects of crystallisation, drying and oxidation. Corros. Sci. 2008, 50, 3247–3255. [Google Scholar] [CrossRef]
- Zhang, W.; Young, D.; Brown, B.; Shafer, C.; Lu, F.; Anyanwu, E.; Singer, M. An in-situ Raman study on the oxidation of mackinawite as a corrosion product layer formed on mild steel in marginally sour environments. Corros. Sci. 2021, 188, 109516. [Google Scholar] [CrossRef]
- Hansson, E.B.; Odziemkowski, M.S.; Gillham, R.W. Formation of poorly crystalline iron monosulfides: Surface redox reactions on high purity iron, spectroelectrochemical studies. Corros. Sci. 2006, 48, 3767–3783. [Google Scholar] [CrossRef]
- Pineau, S.; Sabot, R.; Quillet, L.; Jeannin, M.; Caplat, C.; Dupont-Morral, I.; Refait, P. Formation of the Fe(II-III) hydroxysulphate green rust during marine corrosion of steel associated to molecular detection of dissimilatory sulphite-reductase. Corros. Sci. 2008, 50, 1099–1111. [Google Scholar] [CrossRef]
- Mendili, Y.E.; Minisini, B.; Abdelouas, A.; Bardeau, J.-F. Assignment of Raman-active vibrational modes of tetragonal mackinawite: Raman investigations and ab initio calculations. RSC Adv. 2014, 4, 25827–25834. [Google Scholar] [CrossRef]
- Sweeney, R.E.; Kaplan, I.R. Pyrite Framboid Formation; Laboratory Synthesis and Marine Sediments. Econ. Geol. 1973, 68, 618–634. [Google Scholar] [CrossRef]
- Rickard, D.T. The chemistry of iron sulphide formation at low temperatures. Stockh. Contrib. Geol. 1969, 20, 67–95. [Google Scholar]
- Mendili, Y.E.; Abdelouas, A.; Bardeau, J.-F. Insight into the mechanism of carbon steel corrosion under aerobic and anaerobic conditions. Phys. Chem. Chem. Phys. 2013, 15, 9197–9204. [Google Scholar] [CrossRef]
- Kergourlay, F.; Rémazeilles, C.; Neff, D.; Foy, E.; Conforto, E.; Guilminot, E.; Reguer, S.; Dillmann, P.; Nicot, F.; Mielcarek, F.; et al. Mechanisms of the dechlorination of iron archaeological artefacts extracted from seawater. Corros. Sci. 2011, 53, 2474–2483. [Google Scholar] [CrossRef]
- Müller, K.; Ciminelli, V.S.T.; Dantas, M.S.S.; Willscher, S. A comparative study of As(III) and As(V) in aqueous solutions and adsorbed on iron oxy-hydroxides by Raman spectroscopy. Water Res. 2010, 44, 5660–5672. [Google Scholar] [CrossRef]
- Ferguson, G.A.; Hass, M. Magnetic structure and vacancy distribution in g-Fe2O3 by neutron diffraction. Phys. Rev. 1958, 112, 1130–1131. [Google Scholar] [CrossRef]
- White, W.B.; DeAngelis, B.A. Interpretation of the vibrational spectra of spinels. Spectrochim. Acta Part A Mol. Spectrosc. 1967, 23, 985–995. [Google Scholar] [CrossRef]
- Verble, J.L. Temperature-dependent light-scattering studies of the Verwey transition and electronic disorder in magnetite. Phys. Rev. B 1974, 9, 5236–5248. [Google Scholar] [CrossRef]
- Berner, R.A. Iron Sulfides Formed from Aqueous Solution at Low Temperatures and Atmospheric Pressure. J. Geol. 1964, 72, 293–306. [Google Scholar] [CrossRef]
- Benning, L.G.; Wilkin, R.T.; Barnes, H.L. Reaction pathways in the Fe–S system below 100 °C. Chem. Geol. 2000, 167, 25–51. [Google Scholar] [CrossRef]
- Bourdoiseau, J.-A.; Jeannin, M.; Rémazeilles, C.; Sabot, R.; Refait, P. The transformation of mackinawite into greigite studied by Raman spectroscopy. J. Raman Spectrosc. 2011, 42, 496–504. [Google Scholar] [CrossRef]
- Maubec, N.; Lahfid, A.; Lerouge, C.; Wille, G.; Michel, K. Characterization of alunite supergroup minerals by Raman spectroscopy. Spectrochim. Acta Part A: Mol. Biomol. Spectrosc. 2012, 96, 925–939. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, W.W.; Mason, R. Study of Aqueous Al2(SO4)3 Solution under Hydrothermal Conditions: Sulfate Ion Pairing, Hydrolysis, and Formation of Hydronium Alunite. J. Solut. Chem. 2001, 30, 527–548. [Google Scholar] [CrossRef]
- Frost, R.L.; Wills, R.-A.; Weier, M.L.; Martens, W.; Theo Kloprogge, J. A Raman spectroscopic study of alunites. J. Mol. Struct. 2006, 785, 123–132. [Google Scholar] [CrossRef]
- Toumi, M.; Tlili, A. Rietveld refinement and vibrational spectroscopic study of alunite from el Gnater, central Tunisia. Russ. J. Inorg. Chem. 2008, 53, 1845–1853. [Google Scholar] [CrossRef]
- Chamritski, I.; Burns, G. Infrared- and Raman-Active Phonons of Magnetite, Maghemite, and Hematite: A Computer Simulation and Spectroscopic Study. J. Phys. Chem. B 2005, 109, 4965–4968. [Google Scholar] [CrossRef]
- Bersani, D.; Lottici, P.P.; Montenero, A. Micro-Raman investigation of iron oxide films and powders produced by sol–gel syntheses. J. Raman Spectrosc. 1999, 30, 355–360. [Google Scholar] [CrossRef]
- Gasparov, L.V.; Tanner, D.B.; Romero, D.B.; Berger, H.; Margaritondo, G.; Forró, L. Infrared and Raman studies of the Verwey transition in magnetite. Phys. Rev. B 2000, 62, 7939–7944. [Google Scholar] [CrossRef]
- Jubb, A.M.; Allen, H.C. Vibrational Spectroscopic Characterization of Hematite, Maghemite, and Magnetite Thin Films Produced by Vapor Deposition. ACS Appl. Mater. Interfaces 2010, 10, 2804–2812. [Google Scholar] [CrossRef]
- de Faria, D.L.A.; Venâncio Silva, S.; de Oliveira, M.T. Raman microspectroscopy of some iron oxides and oxyhydroxides. J. Raman Spectrosc. 1998, 28, 873–878. [Google Scholar] [CrossRef]
- Das, S.; Hendry, M.J. Application of Raman spectroscopy to identify iron minerals commonly found in mine wastes. Chem. Geol. 2011, 3–4, 101–108. [Google Scholar] [CrossRef]
- Oh, S.J.; Cook, D.C.; Townsend, H.E. Characterization of Iron Oxides Commonly Formed as Corrosion Products on Steel. Hyperfine Interact. 1998, 112, 59–66. [Google Scholar] [CrossRef]
- Dünnwald, J.; Otto, A. An investigation of phase transitions in rust layers using Raman spectroscopy. Corros. Sci. 1989, 29, 1167–1176. [Google Scholar] [CrossRef]
- Hanesch, M. Raman spectroscopy of iron oxides and (oxy)hydroxides at low laser power and possible applications in environmental magnetic studies. Geophys. J. Int. 2009, 177, 941–948. [Google Scholar] [CrossRef]
- Ohtsuka, T.; Kubo, K.; Sato, N. Raman Spectroscopy of Thin Corrosion Films on Iron at 100 to 150 C in Air. Corrosion 1986, 42, 476–481. [Google Scholar] [CrossRef]
- Liu, H.; Chen, T.; Zou, X.; Qing, C.; Frost Ray, L. Effect of Al content on the structure of Al-substituted goethite: A micro-Raman spectroscopic study. J. Raman Spectrosc. 2013, 44, 1609–1614. [Google Scholar] [CrossRef]
- van der Weerd, J.; Rehren, T.; Firth, S.; Clark, R.J.H. Identification of iron oxide impurities in earliest industrial-scale processed platinum. Mater. Charact. 2004, 53, 63–70. [Google Scholar] [CrossRef]
- Nie, X.; Li, X.; Du, C.; Huang, Y.; He, D. Characterization of corrosion products formed on the surface of carbon steel by Raman spectroscopy. J. Raman Spectrosc. 2009, 40, 76–79. [Google Scholar] [CrossRef]
- Frost Ray, L.; Rachael-Anne, W.; Weier Matt, L. Martens Wayde Comparison of the Raman spectra of natural and synthetic K- and Na-jarosites at 298 and 77 K. J. Raman Spectrosc. 2005, 36, 435–444. [Google Scholar] [CrossRef]
- Sasaki, K.; Tanaike, O.; Konno, H. Distinction of jarosite-group compounds by Raman spectroscopy. Can. Mineral. 1998, 36, 1225–1235. [Google Scholar]
- Stoffregen, R.E.; Alpers, C.N.; Jambor, J.L. Alunite-Jarosite Crystallography, Thermodynamics, and Geochronology. Rev. Mineral. Geochem. 2000, 40, 453–479. [Google Scholar] [CrossRef]
- Frost, R.L.; Wills, R.-A.; Weier, M.L.; Martens, W.; Mills, S. A Raman spectroscopic study of selected natural jarosites. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2006, 63, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Härtig, C.; Brand, P.; Bohmhammel, K. Fe-Al-Isomorphie and Strukturwasser in Kristallen vom Jarosit-Alunit-Typ. Z. Anorg. Allg. Chem. 1984, 508, 159–164. [Google Scholar] [CrossRef]
- Kubisz, J. Studies on synthetic alkali–hydronium jarosites. I. Synthesis of jarosite and natrojarosite. Mineral. Pol. 1970, 1, 47–57. [Google Scholar]
- Murphy, P.J.; Smith, A.M.L.; Hudson-Edwards, K.A.; Dubbin, W.E.; Wright, K. Raman and IR spectroscopic studies of alunite-supergroup compounds containing Al, Cr3+, Fe3+ and V3+ at the B site. Can. Mineral. 2009, 47, 663–681. [Google Scholar] [CrossRef]
- Li, S.; Hihara, L.H. A Micro-Raman Spectroscopic Study of Marine Atmospheric Corrosion of Carbon Steel: The Effect of Akaganeite. J. Electrochem. Soc. 2015, 162, C495. [Google Scholar] [CrossRef]
- Murowchick, J.B. Marcasite inversion and the petrographic determination of pyrite ancestry. Econ. Geol. 1992, 87, 1141–1152. [Google Scholar] [CrossRef]
- Qian, G.; Brugger, J.; Skinner, W.; Chen, G.; Pring, A. An experimental study of the mechanism of the replacement of magnetite by pyrite up to 300 degrees C. Geochim. Cosmochim. Acta 2010, 74, 5610–5630. [Google Scholar] [CrossRef]
- Rottier, B.; Kouzmanov, K.; Wälle, M.; Bendezú, R.; Fontboté, L. Sulfide Replacement Processes Revealed by Textural and LA-ICP-MS Trace Element Analyses: Example from the Early Mineralization Stages at Cerro de Pasco, Peru. Econ. Geol. 2016, 111, 1347–1367. [Google Scholar] [CrossRef]
- Drobner, E.; Huber, H.; Wächtershäuser, G.; Rose, D.; Stetter, K.O. Pyrite formation linked with hydrogen evolution under anaerobic conditions. Nature 1990, 346, 742–744. [Google Scholar] [CrossRef]
- Lennie, A.; Vaughan, D.J. Spectroscopic studies of iron sulfide formation and phase relations at low temperatures. Geochem. Soc. Spec. Publ. 1996, 5, 117–131. [Google Scholar]
- Jolivet, J.-P.; Tronc, E.; Chanéac, C. Iron oxides: From molecular clusters to solid. A nice example of chemical versatility. Comptes Rendus Geosci. 2006, 338, 488–497. [Google Scholar] [CrossRef]
- Jolivet, J.-P.; Chanéac, C.; Tronc, E. Iron oxide chemistry. From molecular clusters to extended solid networks. Chem. Commun. 2004, 481–483. [Google Scholar] [CrossRef]
- Sobanska, S.; Deneele, D.; Barbillat, J.; Ledésert, B. Natural weathering of slags from primary Pb–Zn smelting as evidenced by Raman microspectroscopy. Appl. Geochem. 2016, 64, 107–117. [Google Scholar] [CrossRef]
- Andrés, M.P.A.; López, R.P.; Ross, G.R.; Illera, V.; Cama, J.; Cotte, M.; Tucoulou, R. Analysis of the iron coatings formed during marcasite and arsenopyrite oxidation at neutral-alkaline conditions. Geol. Acta 2013, 11, 465–481. [Google Scholar]
- Rye, R.O. A review of the stable-isotope geochemistry of sulfate minerals in selected igneous environments and related hydrothermal systems. Chem. Geol. 2005, 215, 5–36. [Google Scholar] [CrossRef]
- Georgieva, S.; Velinova, N. Alunite from the advanced argillic alterations in the Chelopech high-sulphidation epithermal Cu-Au deposit, Bulgaria: Chemistry, morphology and genetic significance. Geochem. Mineral. Petrol. 2012, 49, 17–31. [Google Scholar]
- Kubisz, J. Natural hydronium jarosites. Bull. L’académie Pol. Sci. 1961, 195–200. [Google Scholar]
- Dutrizac, J.E.; Jambor, J.L. Jarosites and their application in hydrometallurgy. In Sulfate Minerals, Crystallography, Geochemistry, and Environmental Significance; Reviews in Mineralogy and Geochemistry; GeoScienceWorld: McLean, VA, USA, 2000; Volume 40, pp. 405–443. [Google Scholar]
- Desborough, G.A.; Smith, K.S.; Lowers, H.A.; Swayze, G.A.; Hammarstrom, J.M.; Diehl, S.F.; Leinz, R.W.; Driscoll, R.L. Mineralogical and chemical characteristics of some natural jarosites. Geochim. Cosmochim. Acta 2010, 74, 1041–1056. [Google Scholar] [CrossRef]
- Rye, R.O.; Bethke, P.M.; Wasserman, M.D. The stable isotope geochemistry of acid sulfate alteration. Econ. Geol. 1992, 87, 225–262. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dulski, M.; Janeczek, J.; Włodyka, R. In-Depth Analysis of Complex Multiphase Oxidative Transformations in Iron Sulfides (Pyrrhotite and Pyrite) Within Migmatitic Gneiss. Minerals 2025, 15, 49. https://doi.org/10.3390/min15010049
Dulski M, Janeczek J, Włodyka R. In-Depth Analysis of Complex Multiphase Oxidative Transformations in Iron Sulfides (Pyrrhotite and Pyrite) Within Migmatitic Gneiss. Minerals. 2025; 15(1):49. https://doi.org/10.3390/min15010049
Chicago/Turabian StyleDulski, Mateusz, Janusz Janeczek, and Roman Włodyka. 2025. "In-Depth Analysis of Complex Multiphase Oxidative Transformations in Iron Sulfides (Pyrrhotite and Pyrite) Within Migmatitic Gneiss" Minerals 15, no. 1: 49. https://doi.org/10.3390/min15010049
APA StyleDulski, M., Janeczek, J., & Włodyka, R. (2025). In-Depth Analysis of Complex Multiphase Oxidative Transformations in Iron Sulfides (Pyrrhotite and Pyrite) Within Migmatitic Gneiss. Minerals, 15(1), 49. https://doi.org/10.3390/min15010049