
Thermodynamic Stability of Clay Minerals in Boreal Forest Soil and Its Relationship to the Properties of Soil Organic Matter


Abstract
1. Introduction
2. Materials and Methods
3. Results
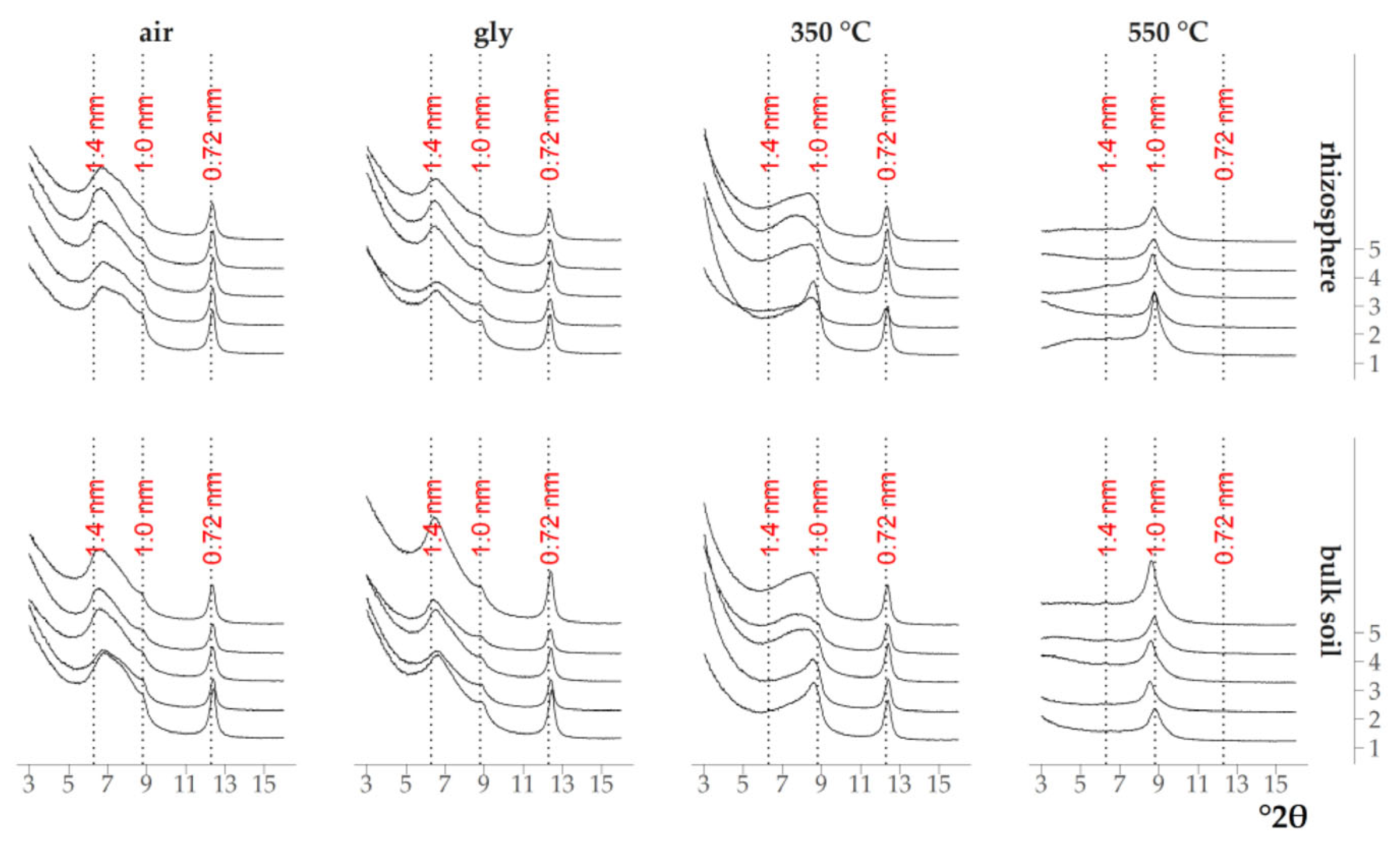
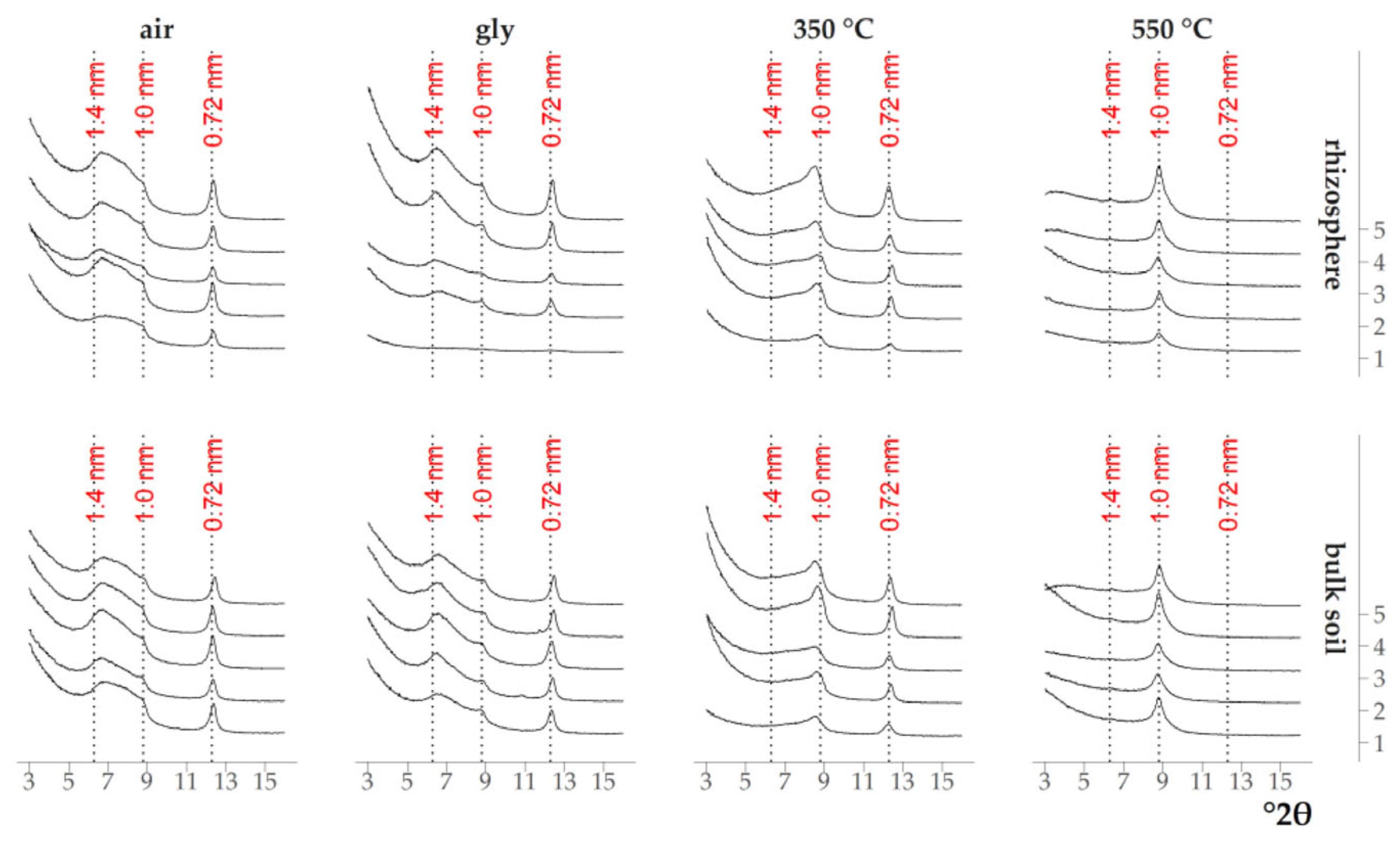
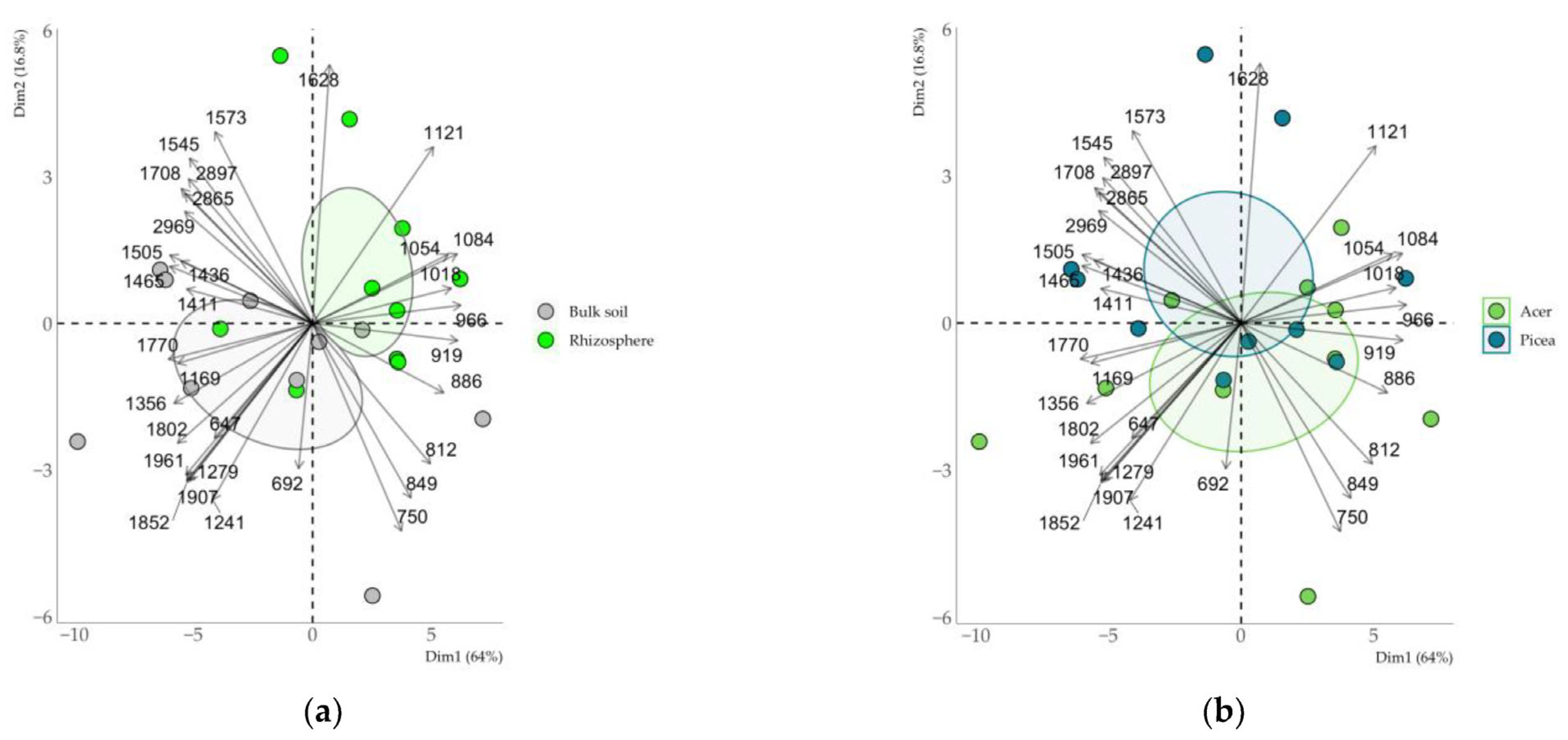
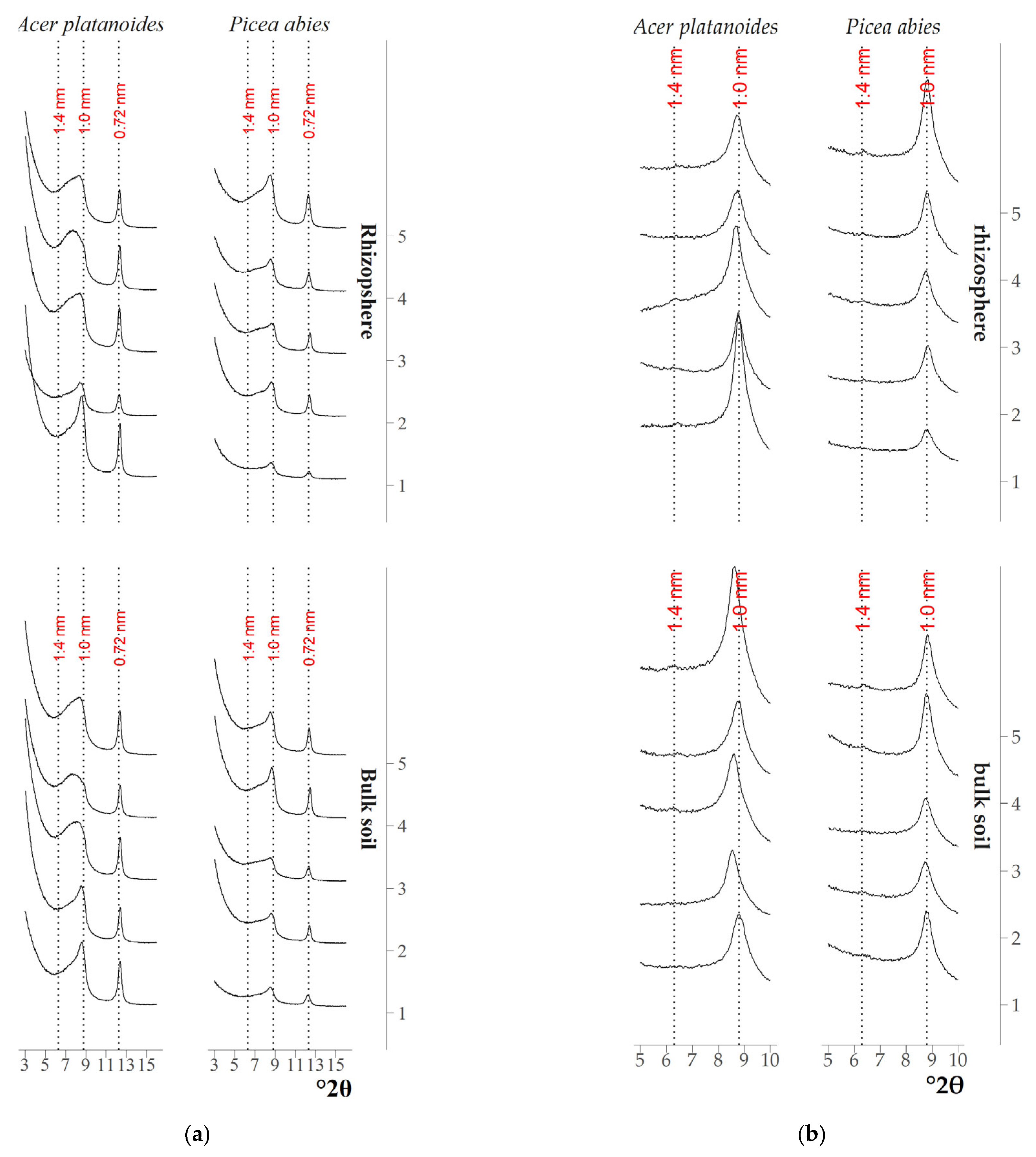
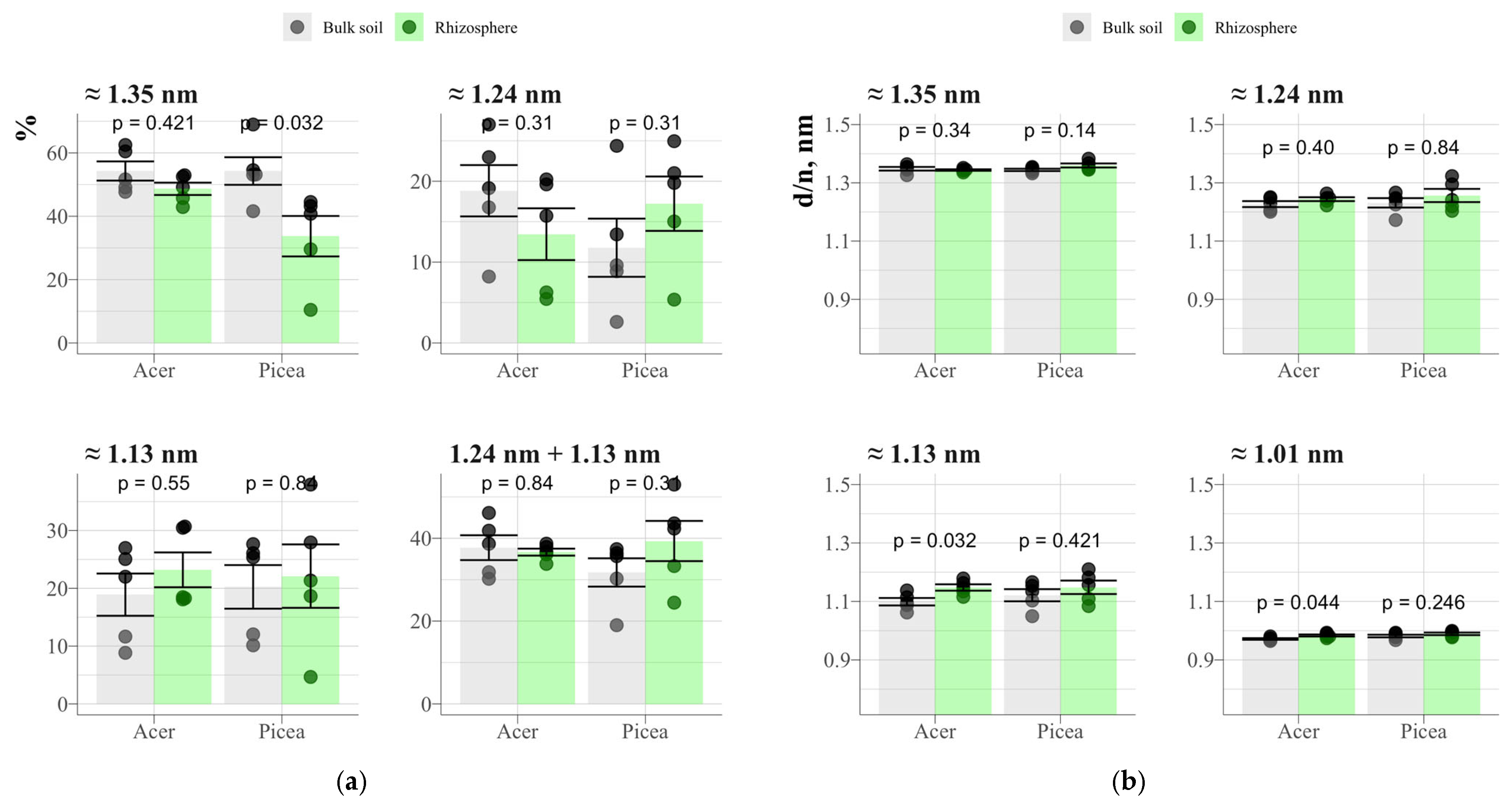
3.1. Qualitative Analysis of the Mineral Composition of Clay Fractions in Soil and Rhizosphere
3.2. Assessment of Chemical Composition of Mineral Phases of Clay Fractions in Soil and Rhizosphere

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mineral | Structural Formula | К | ΔG, kcal/mol | ΔG Theor. Min., kcal/mol | ΔG Theor. Max., kcal/mol | К Theor. Min. | К Theor. Max. |
|---|---|---|---|---|---|---|---|
| Kaolinite | Al2Si2O5(OH)4 | 6.9 | −906.2 | −912.1 | −898.4 | 2.3 | 12.9 |
| Chlorite | (Mg,Fe)5(Al,Si)5O10(OH)8 | 61.3 | −2112.7 | −1974.0 | −1938.2 | 63.1 | 76.8 |
| Illite | (K0.71Ca0.01Na0.01)(Al1.86Mg0.15Fe0.04)(Si3.27Al0.73)O10(OH)2 | 13.3 | −1312.0 | −1313.6 | −1250.5 | 12.9 | 17.1 |
| Vermiculite | (Mg0.38Ca0.03Na0.02)2 (Mg2.46Al0.3Fe0.22Ti0.021)2 ((Si2.83Al1.17)O10)2 OH4 (H2O)3.4 | 33.8 | −1363.2 | −1298.0 | −1364.8 | 20.2 | 45.2 |
| Hydroxy-interlayered vermiculite (HIV) | ND | ND | ND | −1842.7 | −1418.0 | 4.6 | 140.9 |
3.3. Assessment of Mineral Thermodynamic Stability in the Composition of Clay Fractions in Soil and Rhizosphere
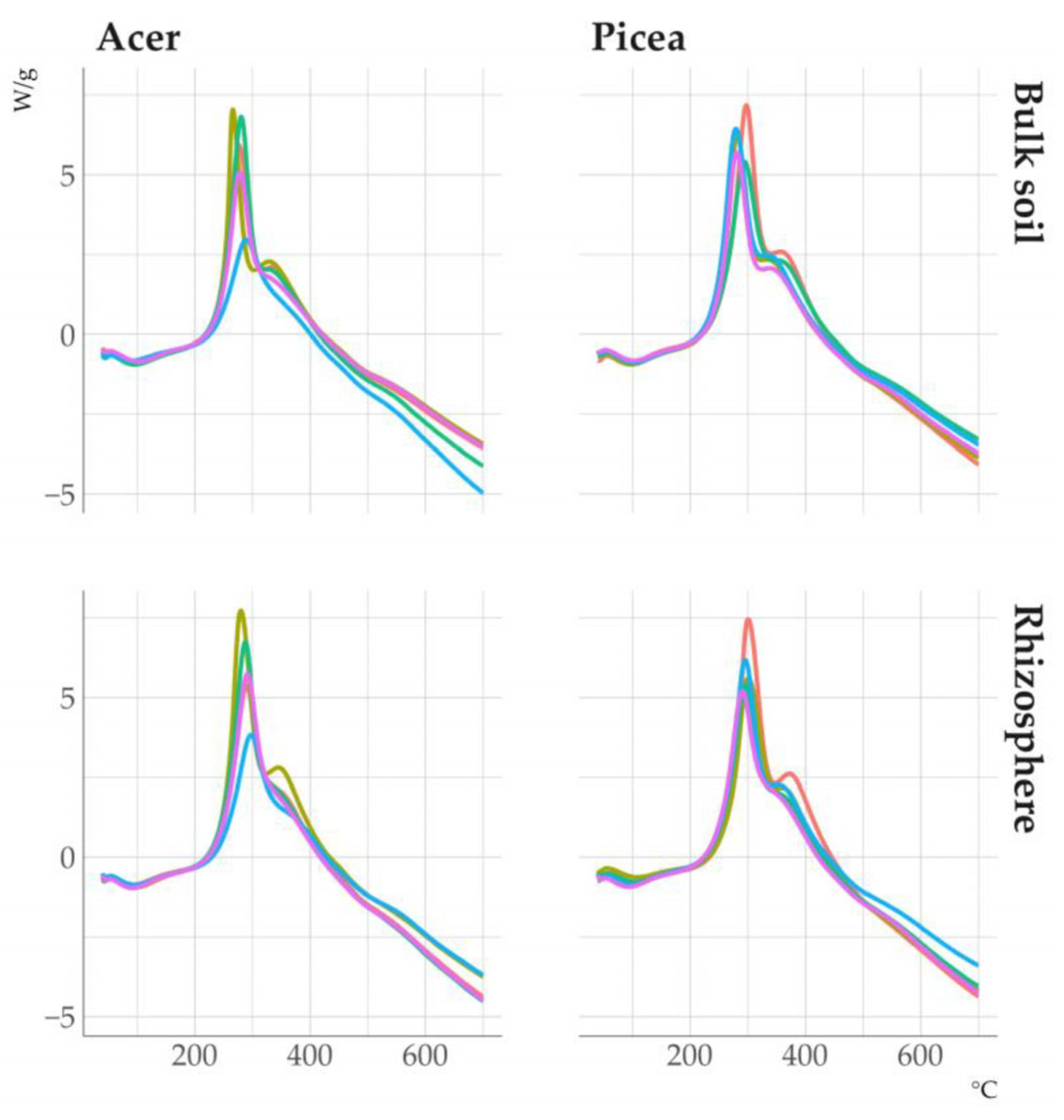
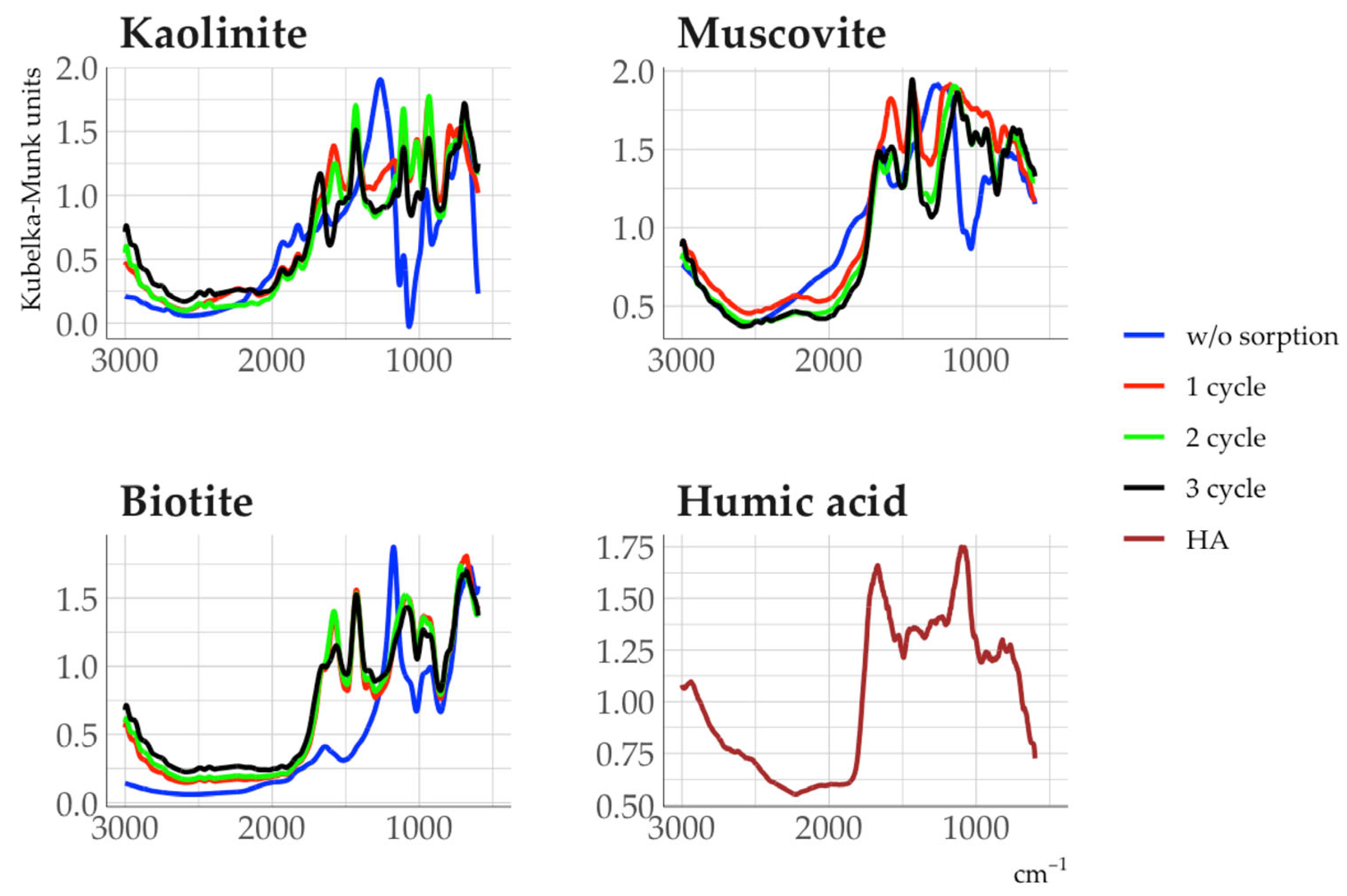
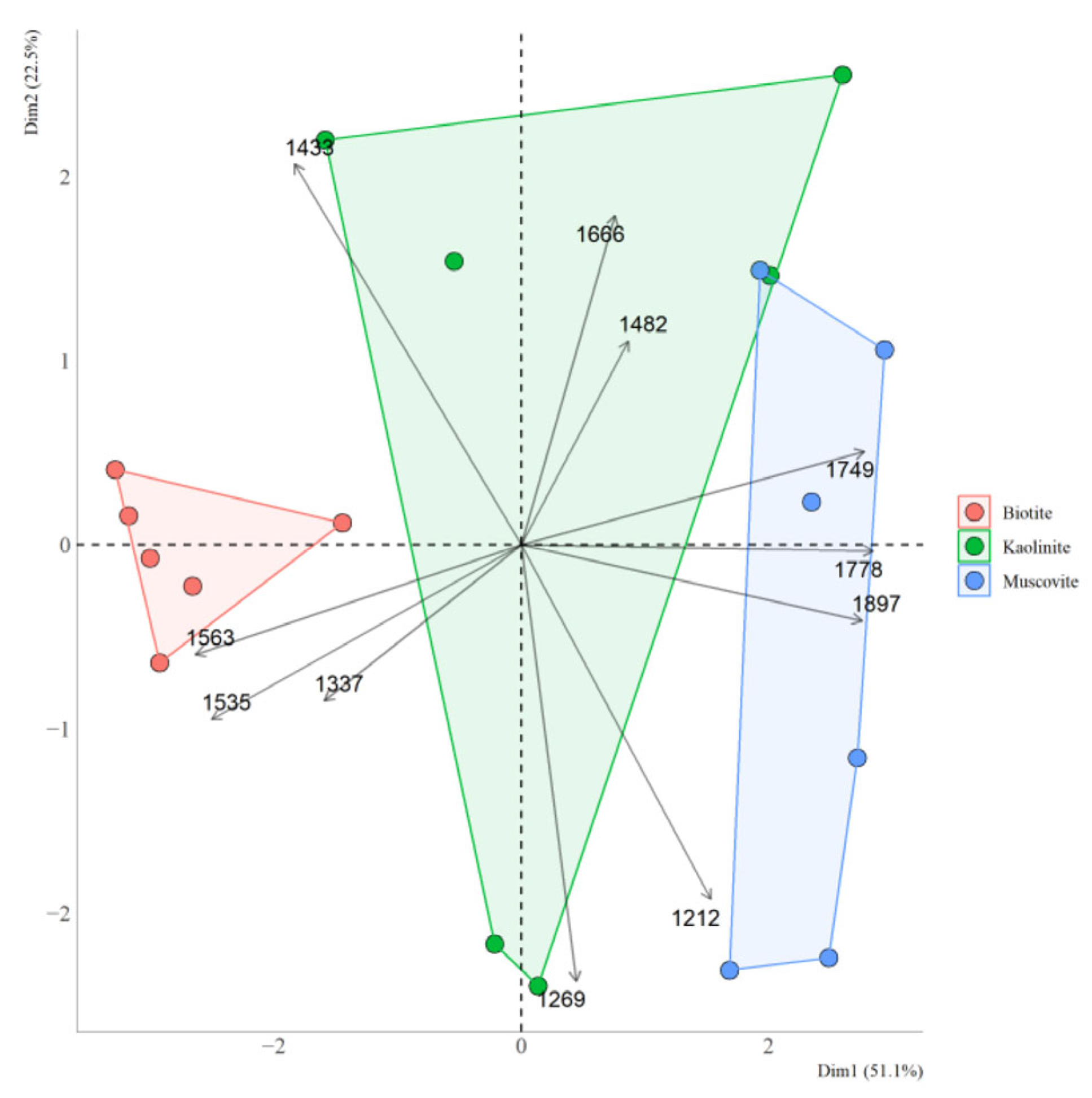
3.4. Organic Matter Properties of Clay Fractions in Soil and Rhizosphere
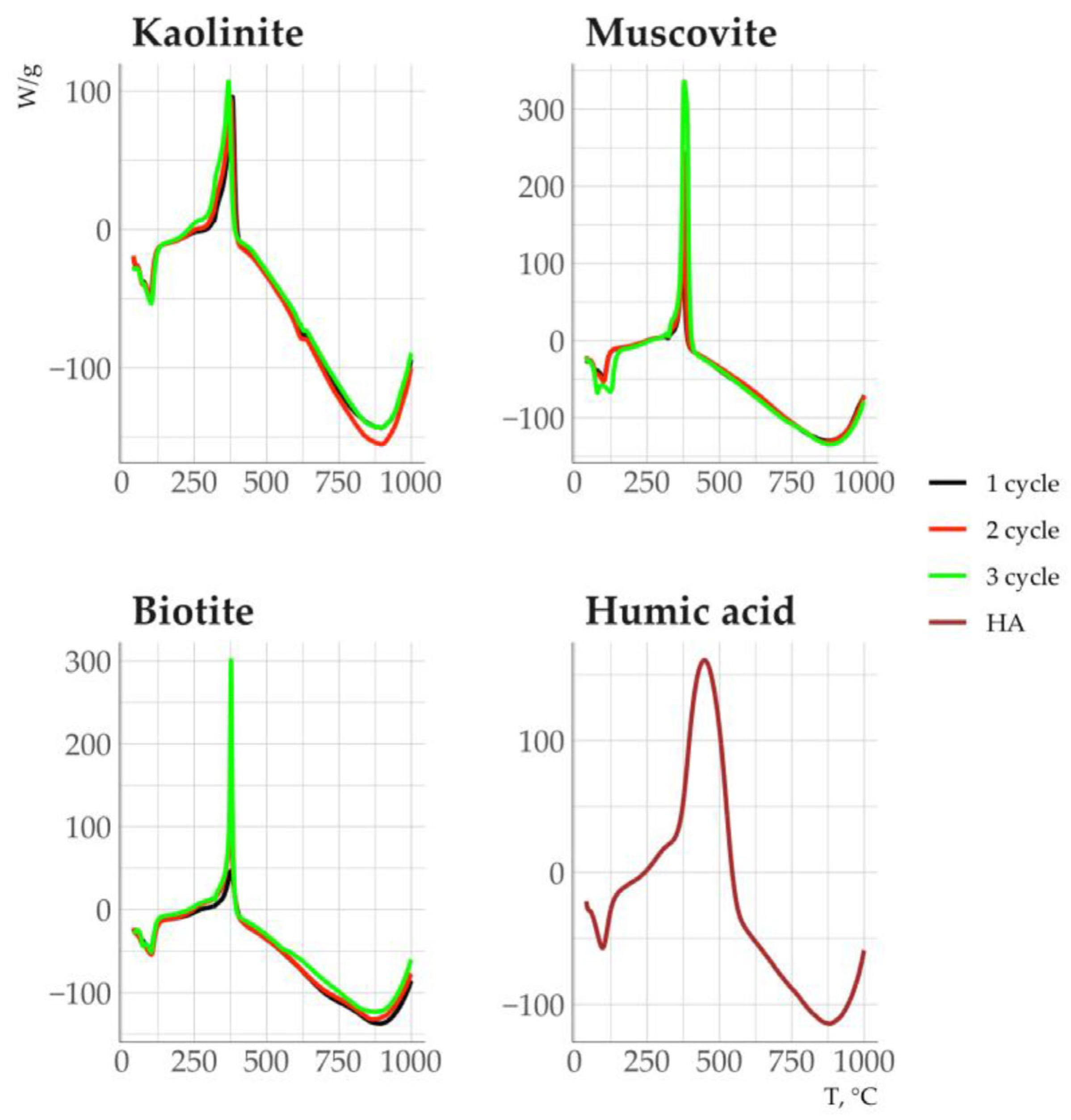
3.5. Properties of Clay Minerals Before and After Humic Acid Treatment
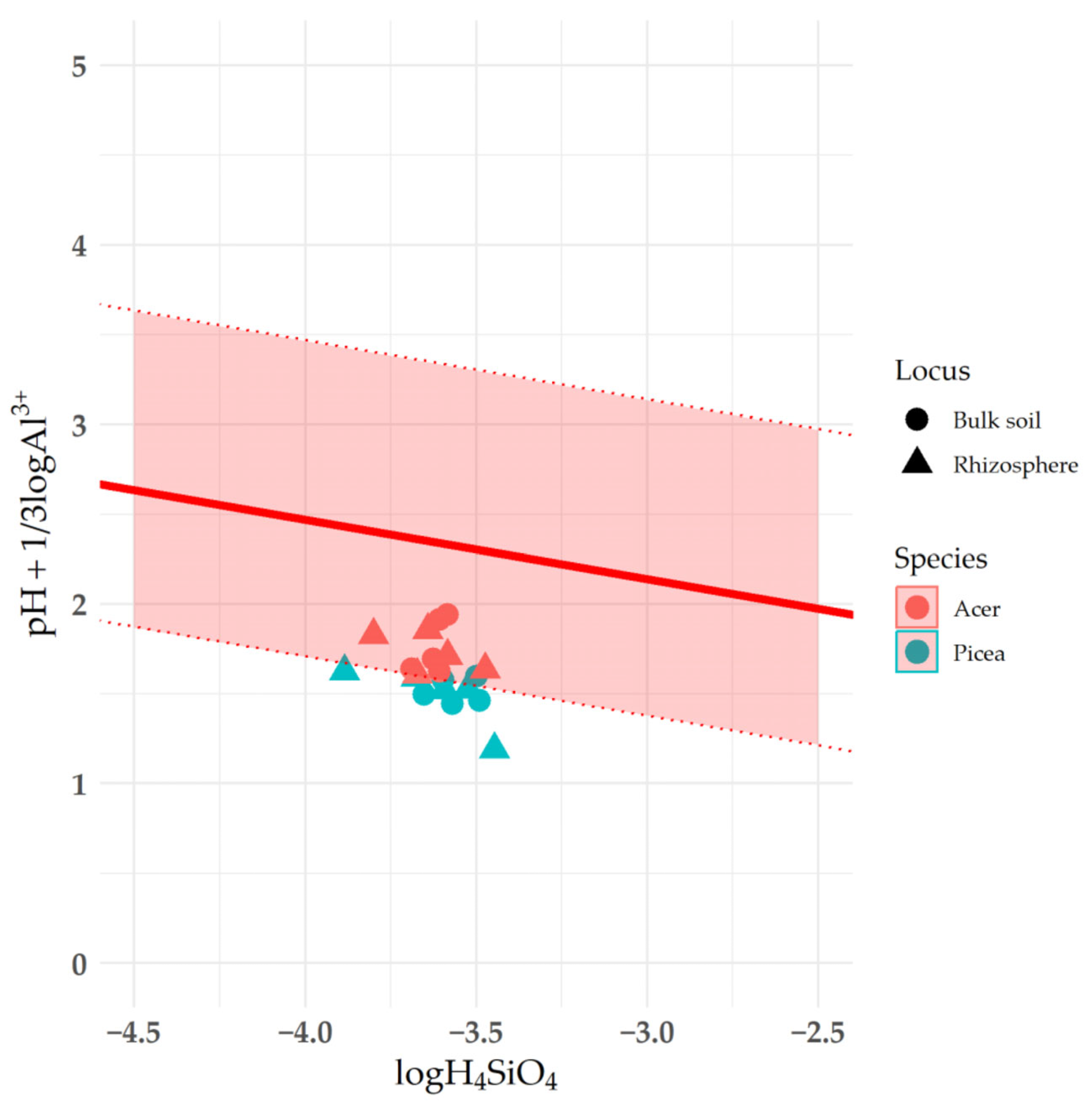
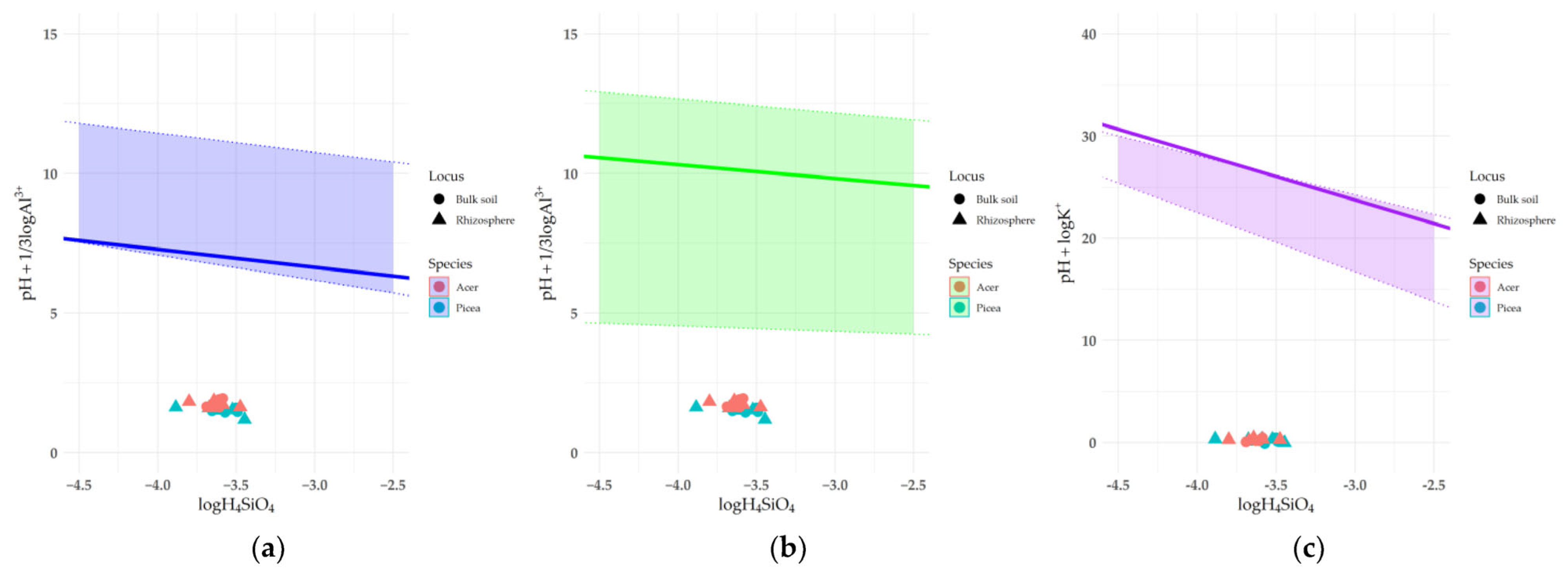
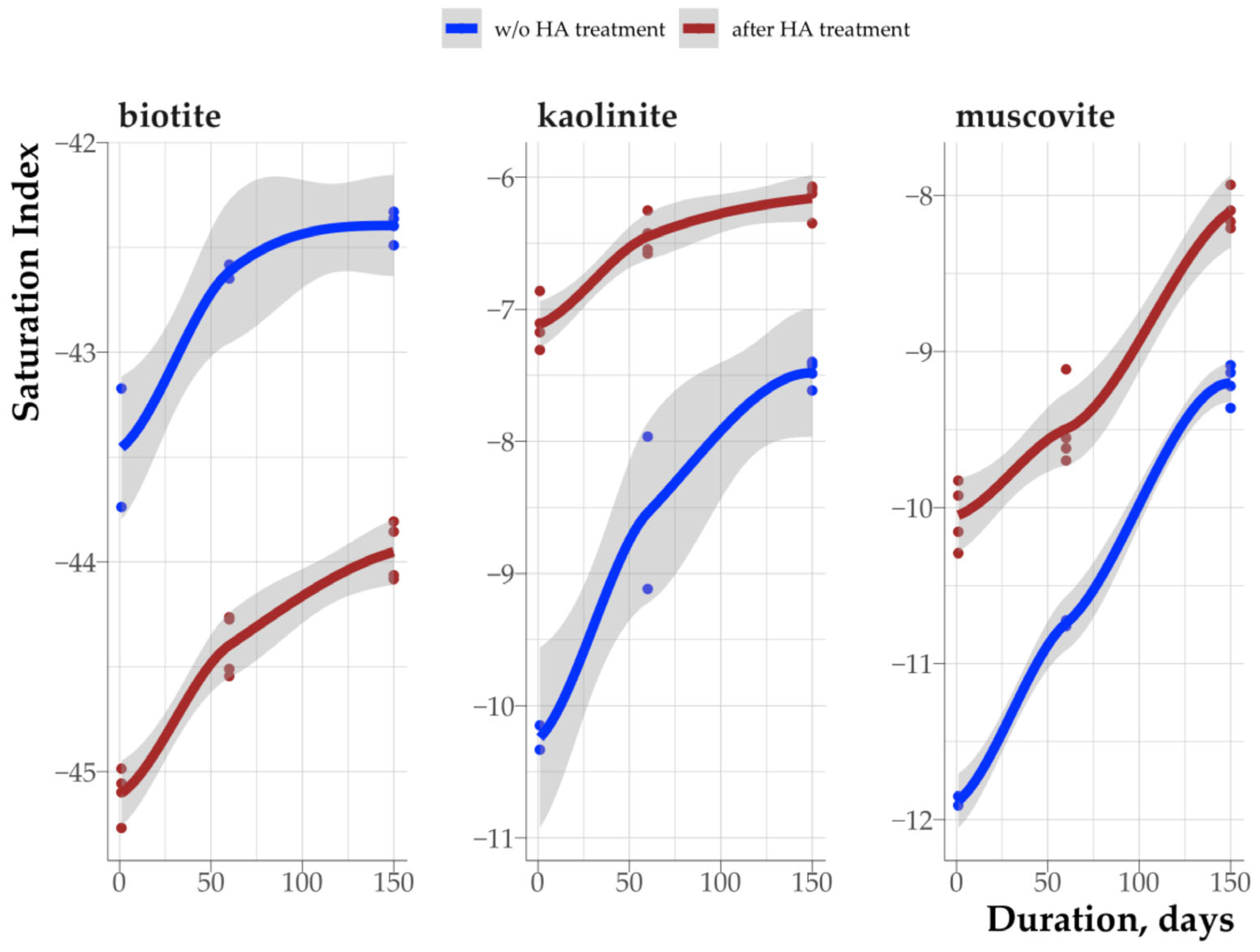
3.6. Effect of Humic Acid on Saturation Indices of Clay Minerals
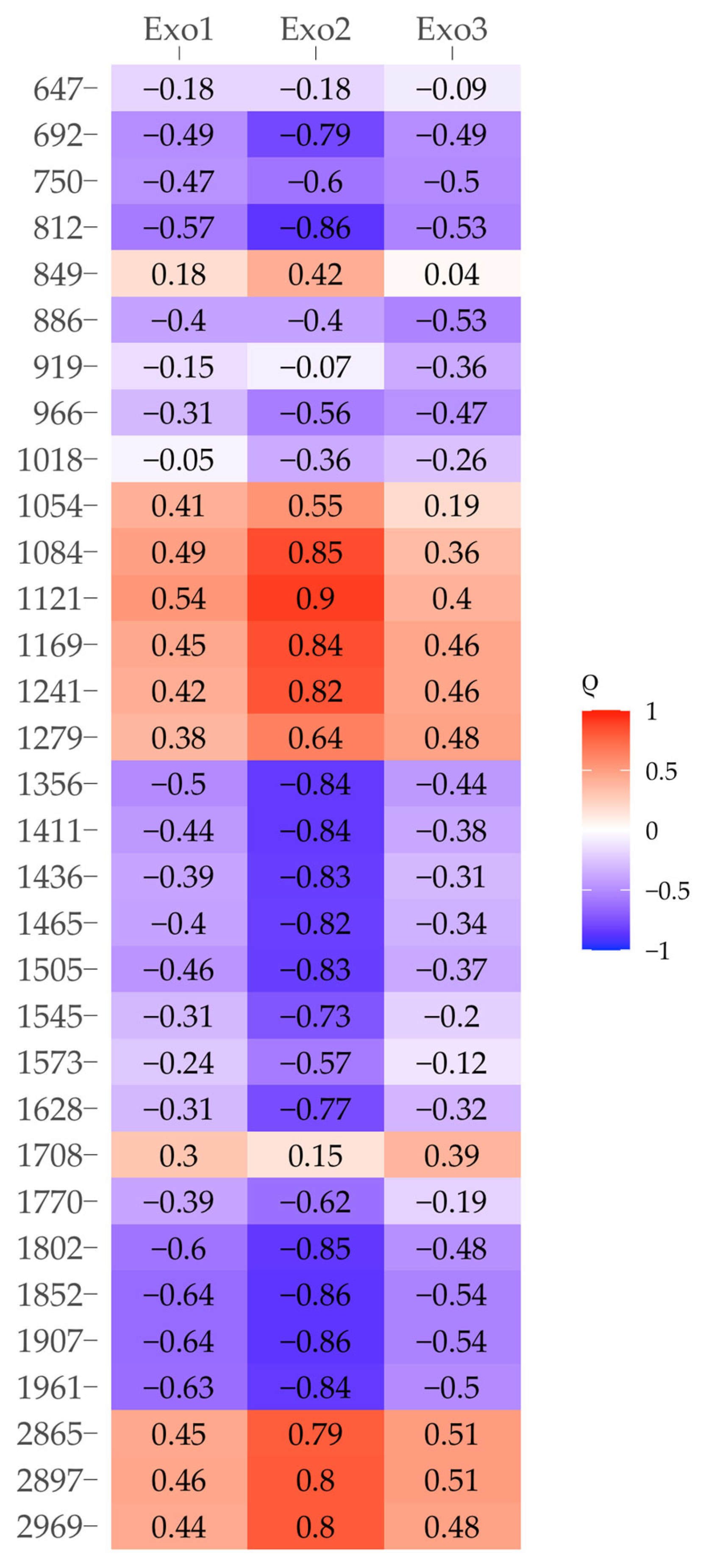
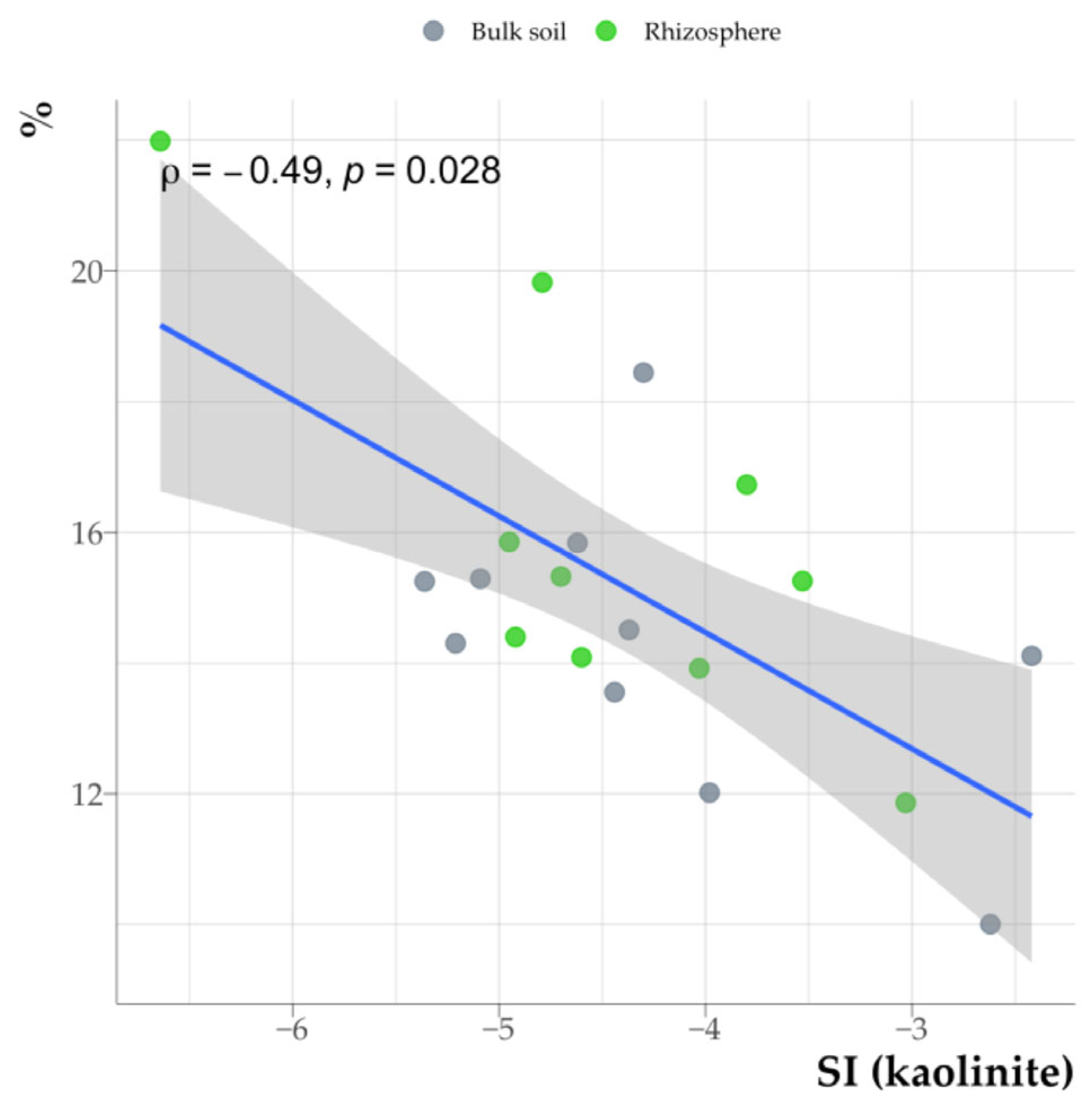
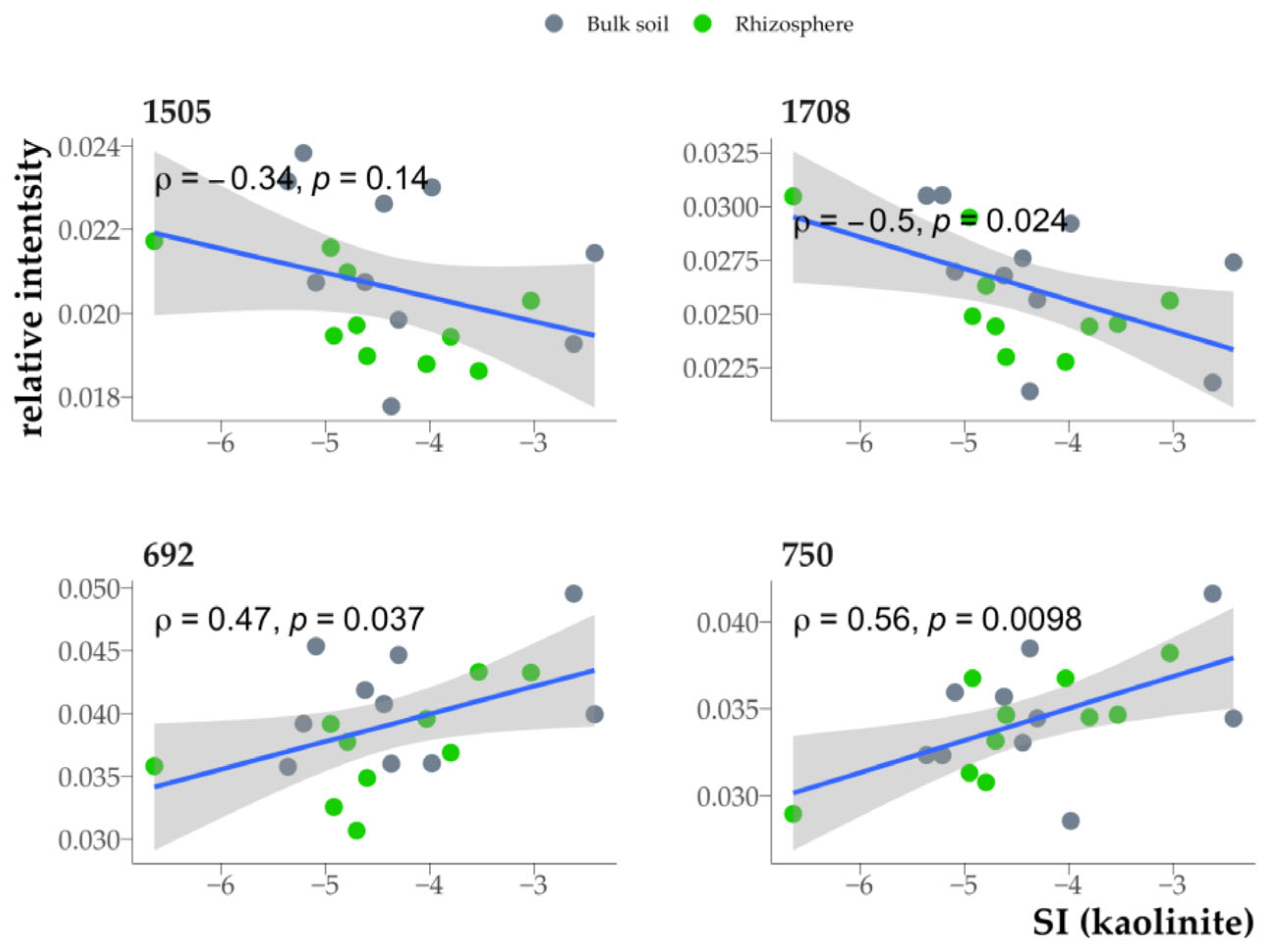
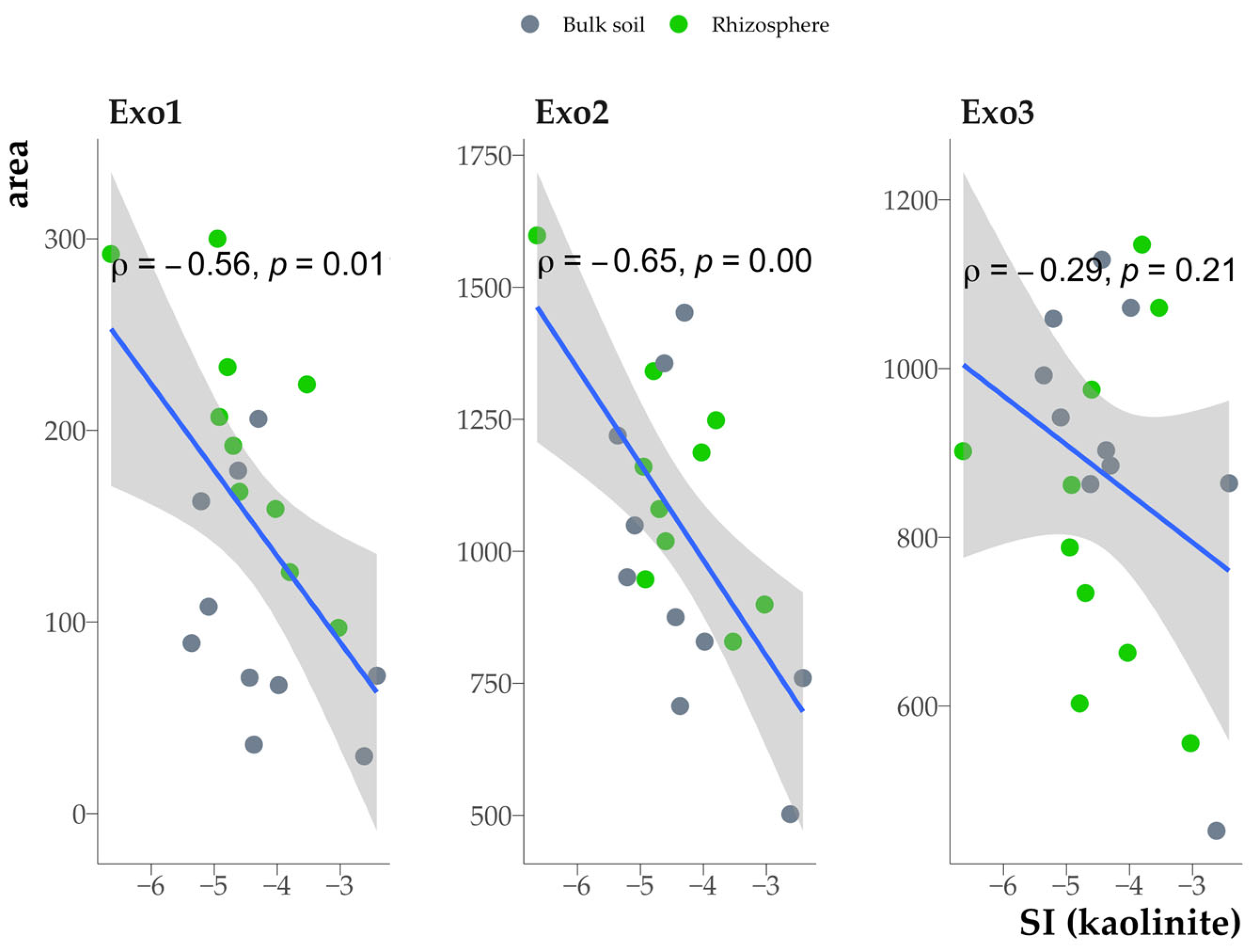
3.7. Relationship Between Organic Matter Properties in the Clay Fraction and Clay Mineral Saturation Indices
4. Discussion
4.1. Transformation of Clay Minerals in the Rhizosphere According to XRD Data
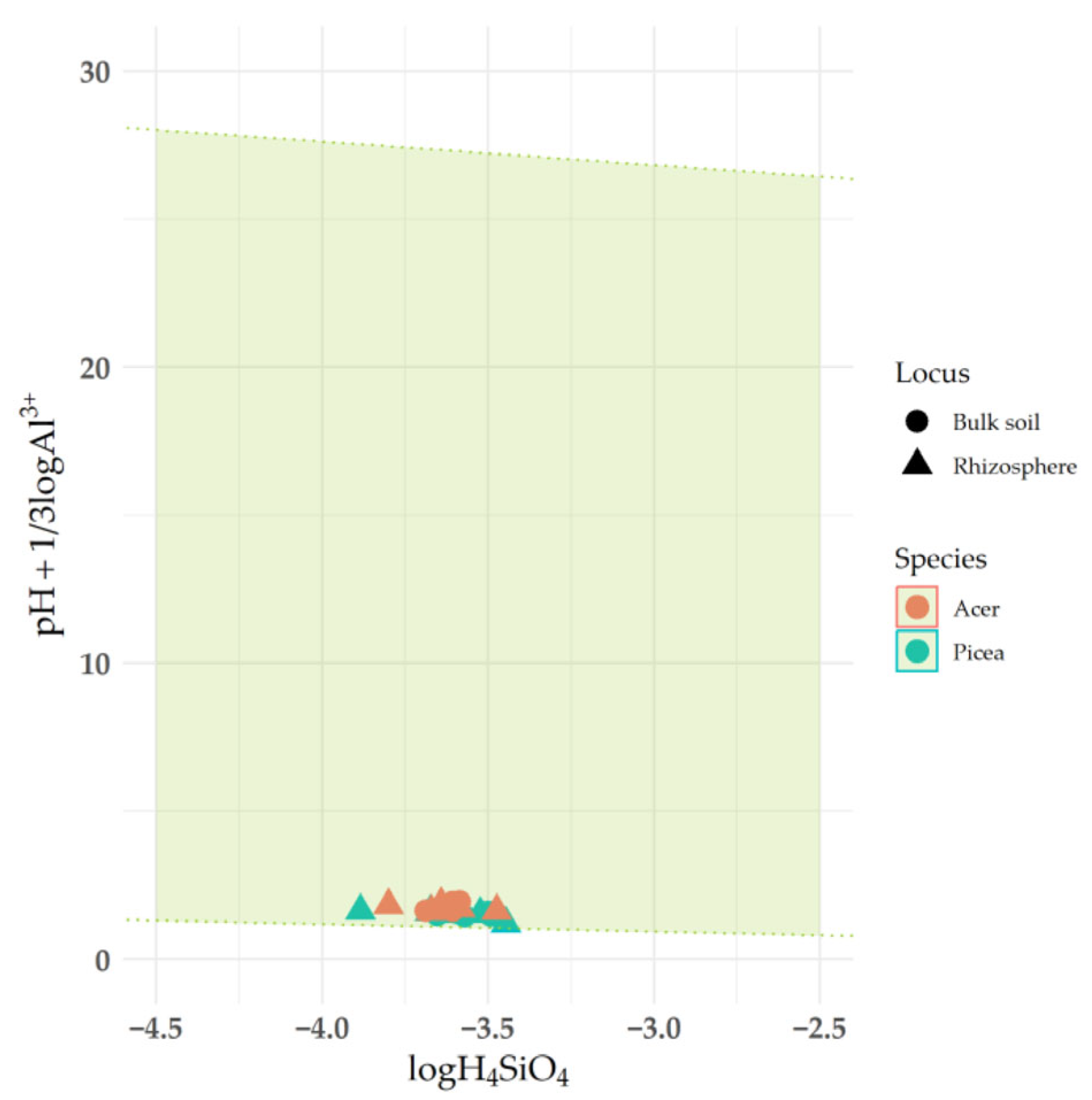
4.2. The Impact of Rhizosphere Processes on the Saturation Index of Clay Minerals
4.3. Dependence of Humic Acid Composition on Sorption in Different Minerals
4.4. Impact of Sorbed Organic Matter on the Thermodynamic Stability of Clay Minerals
4.5. Relationship Between the Organic Matter Properties of Clay Fractions from Soil and Rhizosphere and Saturation Indices of Clay Minerals
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| XRD | X-ray diffraction |
| HIV | Hydroxy-interlayered vermiculite |
| DRIFTS | Diffuse reflectance infrared Fourier-transform spectroscopy |
| DSC | Differential scanning calorimetry |
| TGA | Thermogravimetry |
| HA | Humic acid |
| SOM | Soil organic matter |
| SI | Saturation index |
| LMWOA | Low-molecular weight organic acid |
Appendix A
| Duration, h | pH | Al3+ | Al3+ (pH = 1.0) | Fe2+ | K+ | Mg2+ | Ca2+ | Na+ | TOC |
|---|---|---|---|---|---|---|---|---|---|
| 24 | 3.63 | 6.74 | 7.09 | 0.87 | 8.62 | 3.64 | 12.19 | 2.52 | 117.6 |
| 48 | 3.71 | 7.21 | 8.06 | 0.95 | 9.85 | 4.43 | 16.41 | 2.60 | 102.9 |
| 72 | 3.88 | 7.72 | 8.10 | 1.02 | 10.06 | 4.90 | 18.21 | 3.72 | 107.3 |
| 144 | 4.01 | 7.79 | 8.18 | 0.93 | 10.13 | 5.04 | 18.45 | 3.34 | 264.7 |
| 240 | 4.11 | 7.84 | 8.09 | 1.07 | 9.97 | 4.93 | 18.39 | 3.53 | 558.9 |
| Sample | pH | Al3+ | Ca2+ | Fe2+ | K+ | Mg2+ | Mn2+ | Na+ | H4SiO4 |
|---|---|---|---|---|---|---|---|---|---|
| Rhizosphere (Picea) | 3.71 ± 0.1 | 0.15 ± 0.02 | 10.36 ± 1.51 | 0.47 ± 0.06 | 284.45 ± 23.75 | 7.26 ± 0.76 | 2.3 ± 0.74 | 167.18 ± 31.12 | 250.53 ± 86.44 |
| Bulk soil (Picea) | 3.86 ± 0.1 | 0.25 ± 0.02 | 13.35 ± 2.03 | 0.09 ± 0.01 | 295.52 ± 99.0 | 9.01 ± 1.14 | 7.91 ± 1.16 | 133.53 ± 24.63 | 276.7 ± 42.63 |
| Rhizosphere (Acer) | 3.94 ± 0.1 | 0.24 ± 0.04 | 14.37 ± 2.56 | 0.03 ± 0.01 | 236.09 ± 43.63 | 11.18 ± 1.76 | 3.00 ± 0.29 | 149.06 ± 54.68 | 238.86 ± 65.07 |
| Bulk soil (Acer) | 3.92 ± 0.2 | 0.33 ± 0.05 | 15.3 ± 3.32 | 0.58 ± 0.19 | 193.96 ± 49.69 | 10.87 ± 2.4 | 6.46 ± 0.71 | 103.6 ± 26.26 | 239.01 ± 20.93 |
References
- Kittrick, J.A. Soil Minerals in the Al2O3-SiO2-H2O System and a Theory of Their Formation. Clays Clay Miner. 1969, 17, 157–167. [Google Scholar] [CrossRef]
- Nriagu, J.O. Thermochemical Approximations for Clay Minerals. Am. Mineral. 1975, 60, 834–839. [Google Scholar]
- Karathanasis, A.D. Seasonal Variation in Solution Composition and Mineral Stability of Two Kentucky Alfisols. Soil Sci. Soc. Am. J. 1991, 55, 881–890. [Google Scholar] [CrossRef]
- Blanc, P.; Vieillard, P.; Gailhanou, H.; Gaboreau, S.; Gaucher, É.; Fialips, C.I.; Madé, B.; Giffaut, E. A Generalized Model for Predicting the Thermodynamic Properties of Clay Minerals. Am. J. Sci. 2015, 315, 734–780. [Google Scholar] [CrossRef]
- Gaboreau, S.; Gailhanou, H.; Blanc, P.; Vieillard, P.; Made, B. Clay Mineral Solubility from Aqueous Equilibrium: Assessment of the Measured Thermodynamic Properties. Appl. Geochem. 2020, 113, 104465. [Google Scholar] [CrossRef]
- Gailhanou, H.; Blanc, P.; Rogez, J.; Mikaelian, G.; Kawaji, H.; Olives, J.; Amouric, M.; Denoyel, R.; Bourrelly, S.; Montouillout, V.; et al. Thermodynamic Properties of Illite, Smectite and Beidellite by Calorimetric Methods: Enthalpies of Formation, Heat Capacities, Entropies and Gibbs Free Energies of Formation. Geochim. Cosmochim. Acta 2012, 89, 279–301. [Google Scholar] [CrossRef]
- Arocena, J.M.; Glowa, K.R. Mineral Weathering in Ectomycorrhizosphere of Subalpine Fir (Abies lasiocarpa (Hook.) Nutt.) as Revealed by Soil Solution Composition. For. Ecol. Manag. 2000, 133, 61–70. [Google Scholar] [CrossRef]
- Bakker, M.R.; George, E.; Turpault, M.-P.; Zhang, J.L.; Zeller, B. Impact of Douglas-Fir and Scots Pine Seedlings on Plagioclase Weathering under Acidic Conditions. Plant Soil 2004, 266, 247–259. [Google Scholar] [CrossRef]
- Calvaruso, C.; Mareschal, L.; Turpault, M.-P.; Leclerc, E. Rapid Clay Weathering in the Rhizosphere of Norway Spruce and Oak in an Acid Forest Ecosystem. Soil Sci. Soc. Am. J. 2009, 73, 331–338. [Google Scholar] [CrossRef]
- Gobran, G.R.; Clegg, S.; Courchesne, F. Rhizospheric Processes Influencing the Biogeochemistry of Forest Ecosystems. Biogeochemistry 1998, 42, 107–120. [Google Scholar] [CrossRef]
- Calvaruso, C.; Turpault, M.P.; Leclerc, E.; Ranger, J.; Garbaye, J.; Uroz, S.; Frey-Klett, P. Influence of Forest Trees on the Distribution of Mineral Weathering-Associated Bacterial Communities of the Scleroderma Citrinum Mycorrhizosphere. Appl. Environ. Microbiol. 2010, 76, 4780–4787. [Google Scholar] [CrossRef] [PubMed]
- Mouas Bourbia, S.; Barré, P.; Boudiaf Naït Kaci, M.; Derridj, A.; Velde, B. Potassium Status in Bulk and Rhizospheric Soils of Olive Groves in North Algeria. Geoderma 2013, 197–198, 161–168. [Google Scholar] [CrossRef]
- Uroz, S.; Calvaruso, C.; Turpault, M.P.; Frey-Klett, P. Mineral Weathering by Bacteria: Ecology, Actors and Mechanisms. Trends Microbiol. 2009, 17, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Bormann, B.T.; Wang, D.; Herbert Bormann, F.; Benoit, G.; April, R.; Snyder, M.C. Rapid, Plant-Induced Weathering in an Aggrading Experimental Ecosystem. Biogeochemistry 1998, 43, 129–155. [Google Scholar] [CrossRef]
- Kalinowski, B.E.; Schweda, P. Kinetics of Muscovite, Phlogopite, and Biotite Dissolution and Alteration at pH 1-4, Room Temperature. Geochim. Cosmochim. Acta 1996, 60, 367–385. [Google Scholar] [CrossRef]
- Raulund-Rasmussen, K.; Borggaard, O.K.; Hansen, H.C.B.; Olsson, M. Effect of Natural Organic Soil Solutes on Weathering Rates of Soil Minerals. Eur. J. Soil Sci. 1998, 49, 397–406. [Google Scholar] [CrossRef]
- Bray, A.W.; Oelkers, E.H.; Bonneville, S.; Wolff-Boenisch, D.; Potts, N.J.; Fones, G.; Benning, L.G. The Effect of PH, Grain Size, and Organic Ligands on Biotite Weathering Rates. Geochim. Cosmochim. Acta 2015, 164, 127–145. [Google Scholar] [CrossRef]
- Sokolova, T.A. Low-Molecular-Weight Organic Acids in Soils: Sources, Composition, Concentrations, and Functions: A Review. Eurasian Soil Sci. 2020, 53, 580–594. [Google Scholar] [CrossRef]
- McGahan, D.G.; Southard, R.J.; Zasoski, R.J. Rhizosphere Effects on Soil Solution Composition and Mineral Stability. Geoderma 2014, 226–227, 340–347. [Google Scholar] [CrossRef]
- Fernandez-Marcos, M.L.; Macías, F.; Guitián-Ojea, F. A Contribution to the Study of the Stability of Clay Minerals from the Soil Solution Composition at Different PF Values. Clay Miner 1979, 14, 29–37. [Google Scholar] [CrossRef]
- Pedro, G.; Jamagne, M.; Begon, J.C. Two Routes in Genesis of Strongly Differentiated Acid Soils under Humid, Cool-Temperate Conditions. Geoderma 1978, 20, 173–189. [Google Scholar] [CrossRef]
- IUSS Working Group WRB World Reference Base for Soil Resources 2014, Update 2015: International Soil Classification System for Naming Soils and Creating Legends for Soil Maps; FAO: Rome, Italy, 2015.
- Tolpeshta, I.I.; Leman, M. Spatial Variation and Evaluation of Additivity of Parameters of the Acid-Base State of Pale-Podzolic Soils of the Central Forest Nature Reserve. Vestn. Mosk. Univ. Ser. 17 Pochvoved. 2000, 3, 12–19. [Google Scholar]
- Tolpeshta, I.I.; Sokolova, T.A. Extractable Aluminum Compounds in Soils of the Southern Taiga (Soils of the Central Forest Reserve as an Example). Eurasian Soil Sci. 2010, 43, 893–904. [Google Scholar] [CrossRef]
- Alekseeva, S.A.; Dronova, T.Y.; Sokolova, T.A. Chemical and Mineralogical Characteristics of Podzolic and Bog-Podzolic Soils Developed from Twolayered Deposits in the Central Forest State Biospheric Reserve. Moscow Univ. Soil Sci. Bull. 2007, 62, 140–148. [Google Scholar] [CrossRef]
- Tolpeshta, I.I.; Sokolova, T.A. Aluminum Compounds in Soil Solutions and Their Migration in Podzolic Soils on Two-Layered Deposits. Eurasian Soil Sci. 2009, 42, 24–34. [Google Scholar] [CrossRef]
- Bonifacio, E.; Falsone, G.; Simonov, G.; Sokolova, T.; Tolpeshta, I. Pedogenic Processes and Clay Transformations in Bisequal Soils of the Southern Taiga Zone. Geoderma 2003, 149, 66–75. [Google Scholar] [CrossRef]
- Menzies, N.; Bell, L. Evaluation of the Influence of Sample Preparation and Extraction Technique on Soil Solution Composition. Soil Res. 1988, 26, 451–464. [Google Scholar] [CrossRef]
- Álvarez, E.; Fernández-Sanjurjo, M.J.; Núñez, A.; Seco, N.; Corti, G. Aluminium Fractionation and Speciation in Bulk and Rhizosphere of a Grass Soil Amended with Mussel Shells or Lime. Geoderma 2012, 173–174, 322–329. [Google Scholar] [CrossRef]
- Barreto, M.S.C.; Ramlogan, M.; Oliveira, D.M.S.; Verburg, E.E.J.; Elzinga, E.J.; Rouff, A.A.; Jemo, M.; Alleoni, L.R.F. Thermal Stability of Soil Organic Carbon after Long-Term Manure Application across Land Uses and Tillage Systems in an Oxisol. Catena 2021, 200, 105164. [Google Scholar] [CrossRef]
- Moore, D.M.; Reynolds, R.C. X-Ray Diffraction and Identification and Analysis of Clay Minerals, 2nd ed.; Oxford University Press: Oxford, UK, 1997. [Google Scholar]
- Vieillard, P.; Gailhanou, H.; Lassin, A.; Blanc, P.; Bloch, E.; Gaboreau, S.; Fialips, C.I.; Made, B. A Predictive Model of Thermodynamic Entities of Hydration for Smectites: Application to the Formation Properties of Smectites. Appl. Geochem. 2019, 110, 104423. [Google Scholar] [CrossRef]
- Karathanasis, A.D. Compositional and Solubility Relationships Between Aluminum-Hydroxyinterlayered Soil-Smectites and Vermiculites. Soil Sci. Am. J. 1988, 52, 1500–1508. [Google Scholar] [CrossRef]
- Kittrick, J.A. Solubility Measurments of Phases in Three Illites. Clays Clay Miner. 1984, 32, 115–124. [Google Scholar] [CrossRef]
- Misra, U.K.; Upchurcht, W. Free Energy of Formation of Beidellite from Apparent Solubility Measurments. Clays Alld Clay Miller. 1976, 24, 327–360. [Google Scholar] [CrossRef]
- Varadachari, C. Clay Mineral Equilibria: Fuzzy Phase Diagrams. Clay Res. 2017, 36, 1–16. [Google Scholar]
- Varadachari, C.; Kudrat, M.; Ghosh, K. Evaluation of Standard Free Energies of Formation of Clay Minerals by an Improved Regression Method. Clays Clay Miner. 1994, 42, 298–307. [Google Scholar] [CrossRef]
- Vieillard, P. A New Method for the Prediction of Gibbs Free Energies of Formation of Phyllosilcates (10 A and 14 A) Based on the Electronegativity Scale. Clays Clay Miner. 2002, 50, 352–363. [Google Scholar] [CrossRef]
- Volkov, D.S.; Rogova, O.B.; Proskurnin, M.A.; Farkhodov, Y.R.; Markeeva, L.B. Thermal Stability of Organic Matter of Typical Chernozems under Different Land Uses. Soil Tillage Res. 2020, 197, 104500. [Google Scholar] [CrossRef]
- Margenot, A.J.; Parikh, S.J.; Calderón, F.J. Fourier-Transform Infrared Spectroscopy for Soil Organic Matter Analysis. Soil Sci. Soc. Am. J. 2023, 87, 1503–1528. [Google Scholar] [CrossRef]
- Danilin, I.; Tolpeshta, I.; Izosimova, Y.; Pozdnyakov, L.; Stepanov, A.; Salimgareeva, O. Thermal Stability and Resistance to Biodegradation of Humic Acid Adsorbed on Clay Minerals. Minerals 2023, 13, 101310. [Google Scholar] [CrossRef]
- Tolpeshta, I.I.; Sokolova, T.A.; Bonifacio, E.; Falcone, G. Pedogenic Chlorites in Podzolic Soils with Different Intensities of Hydromorphism: Origin, Properties, and Conditions of Their Formation. Eurasian Soil Sci. 2010, 43, 777–787. [Google Scholar] [CrossRef]
- Bain, D.C. Chemistry, Mineralogy and Morphology of Spodosols at Two Swedish Sites Used to Assess Methods of Counteracting Acidification. Water Air Soil Pollut. Focus 2003, 3, 29–47. [Google Scholar] [CrossRef]
- Goh, T.B.; Huang, P.M. Formation of Hydroxy-Al-Montmorillonite Complexes Influenced by Citric Acid. Can. J. Soil Sci. 1984, 411–421. [Google Scholar] [CrossRef]
- Huang, L.; Tan, W.; Liu, F.; Hu, H.; Huang, Q. Composition and Transformation of 1.4 Nm Minerals in Cutan and Matrix of Alfisols in Central China. J. Soils Sediments 2007, 7, 240–246. [Google Scholar] [CrossRef]
- Sartori, F.; Riffaldi, R.; Levi-Minzi, R. Occurrence of Chloritic Intergrades in the Recent Sediments of the Arno River (Italy). Clay Miner. 1979, 14, 47–65. [Google Scholar] [CrossRef]
- Dijkstra, F.A.; Fitzhugh, R.D. Aluminum Solubility and Mobility in Relation to Organic Carbon in Surface Soils Affected by Six Tree Species of the Northeastern United States. Geoderma 2003, 114, 33–47. [Google Scholar] [CrossRef]
- Kluber, L.A.; Tinnesand, K.M.; Caldwell, B.A.; Dunham, S.M.; Yarwood, R.R.; Bottomley, P.J.; Myrold, D.D. Ectomycorrhizal Mats Alter Forest Soil Biogeochemistry. Soil Biol. Biochem. 2010, 42, 1607–1613. [Google Scholar] [CrossRef]
- Augusto, L.; Ranger, J.; Turpault, M.-P.; Bonnaud, P. Experimental in Situ Transformation of Vermiculites to Study the Weathering Impact of Tree Species on the Soil. Eur. J. Soil Sci. 2001, 81–92. [Google Scholar] [CrossRef]
- Hagen-Thorn, A.; Callesen, I.; Armolaitis, K.; Nihlgård, B. The Impact of Six European Tree Species on the Chemistry of Mineral Topsoil in Forest Plantations on Former Agricultural Land. For. Ecol. Manag. 2004, 195, 373–384. [Google Scholar] [CrossRef]
- Ndayiragije, S.; Delvaux, B. Coexistence of Allophane, Gibbsite, Kaolinite and Hydroxy-Al-Interlayered 2:1 Clay Minerals in a Perudic Andosol. Geoderma 2003, 117, 203–214. [Google Scholar] [CrossRef]
- Vicente, M.A.; Razzaghe, M.; Robert, M. Formation of Aluminium Hydroxy Vermiculite (Intergrade) and Smectite from Mica under Acidic Conditions. Clay Miner. 1977, 12, 101–112. [Google Scholar] [CrossRef]
- Sawhney, B.L. Aluminum Interlayers in Layer Silicates Effect of OH/Al Ratio of Al Solution, Time of Reaction, and Type of Structure. Clays Clay Miner. 1968, 16, 157–163. [Google Scholar] [CrossRef]
- Glowa, K.R.; Arocena, J.M.; Massicotte, H.B. Properties of Soils Influenced by Ectomycorrhizal Fungi in Hybrid Spruce [Picea glauca × Engelmannii (Moench.) Voss]. Can. J. Soil Sci. 2004, 84, 91–102. [Google Scholar] [CrossRef]
- Leyval, C.; Berthelin, J. Weathering of a Mica by Roots and Rhizospheric Microorganisms of Pine. Soil Sci. Soc. Am. J. 1991, 55, 1009–1016. [Google Scholar] [CrossRef]
- Sokolova, T.A. Decomposition of Clay Minerals in Model Experiments and in Soils: Possible Mechanisms, Rates, and Diagnostics (Analysis of Literature). Eurasian Soil Sci. 2013, 46, 182–197. [Google Scholar] [CrossRef]
- Wilson, M.J. The Origin and Formation of Clay Minerals in Soils: Past, Present and Future Perspectives. Clay Miner. 1999, 34, 7–25. [Google Scholar] [CrossRef]
- Gagarina, E.I.; Sokolova, T.A.; Sukhacheva, E.Y. Clay Minerals in Weakly Podzolised Soils of Abraded Moraine Plains in Northwestern Russia. Mosc. Univ. Soil Sci. Bull. 2014, 69, 147–155. [Google Scholar] [CrossRef]
- Wilson, M.J. Weathering of the Primary Rock-Forming Minerals: Processes, Products and Rates. Clay Miner. 2004, 39, 233–266. [Google Scholar] [CrossRef]
- Allen, B.L.; Hajek, B.F. Mineral Occurrence in Soil Environments. In Minerals in Soil Environments; Dixon, B., Weed, S.B., Eds.; Soil Science Society of America: Madison, Wisconsin, 1989; Volume 1, pp. 199–278. [Google Scholar]
- Danilin, I.V.; Tolpeshta, I.I.; Izosimova, Y.G. The Mineral Composition of Clay Subfractions in the Rhizosphere of Norway Maple (Acer Platanoids L.) and in the Enclosing Soil. Mosc. Univ. Soil Sci. Bull. 2025, 80, 1–7. [Google Scholar] [CrossRef]
- Tolpeshta, I.I.; Sokolova, T.A.; Vorob’eva, A.A.; Izosimova, Y.G. Transformation of Trioctahedral Mica in the Upper Mineral Horizon of Podzolic Soil during the Two-Year-Long Field Experiment. Eurasian Soil Sci. 2018, 2018, 843–856. [Google Scholar] [CrossRef]
- Drever, J.I.; Vance, F.V. Role of Soil Organic Acids in Mineral Weathering Processes. In Organic Acids in Geological Processes; Pittman, E.D., Lewan, M.D., Eds.; Springer: Berlin/Heidelberg, Germany, 1994; pp. 138–161. [Google Scholar]
- Sokolova, T.A. Specificity of Soil Properties in the Rhizosphere: Analysis of Literature Data. Eurasian Soil Sci. 2015, 48, 968–980. [Google Scholar] [CrossRef]
- Karathanasis, A.D. Mineral Equilibria in Environmental Soil Systems. In Soil Mineralogy with Environmental Applications; Dixon, J.B., Schulze, D.G., Eds.; Soil Science Society of America Inc.: Madison, WI, USA, 2002; pp. 109–151. [Google Scholar]
- Canarini, A.; Kaiser, C.; Merchant, A.; Richter, A.; Wanek, W. Root Exudation of Primary Metabolites: Mechanisms and Their Roles in Plant Responses to Environmental Stimuli. Front. Plant Sci. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, K.; Guggenberger, G. Mineral Surfaces and Soil Organic Matter. Eur. J. Soil Sci. 2003, 54, 219–236. [Google Scholar] [CrossRef]
- Lützow, M.V.; Kögel-Knabner, I.; Ekschmitt, K.; Matzner, E.; Guggenberger, G.; Marschner, B.; Flessa, H. Stabilization of Organic Matter in Temperate Soils: Mechanisms and Their Relevance under Different Soil Conditions—A Review. Eur. J. Soil Sci. 2006, 57, 426–445. [Google Scholar] [CrossRef]
- Kleber, M.; Sollins, P.; Sutton, R. A Conceptual Model of Organo-Mineral Interactions in Soils: Self-Assembly of Organic Molecular Fragments into Zonal Structures on Mineral Surfaces. Biogeochemistry 2007, 85, 9–24. [Google Scholar] [CrossRef]
- Zhu, X.; He, J.; Su, S.; Zhang, X.; Wang, F. Concept Model of the Formation Process of Humic Acid-Kaolin Complexes Deduced by Trichloroethylene Sorption Experiments and Various Characterizations. Chemosphere 2016, 151, 116–123. [Google Scholar] [CrossRef]
- Tan, K.H. The Catalytic Decomposition of Clay Mineral by Complex Reaction with Humic and Fulvic Acid. Soil Sci. 1975, 120, 188–194. [Google Scholar] [CrossRef]
- Ochs, M. Influence of Humified and Non-Humified Natural Ogranic Compounds on Mineral Dissolution. Chem. Geol. 1996, 132, 119–124. [Google Scholar] [CrossRef]
- Kodama, H.; Schnitzer, M.; Jaakkimainen, M. Chlorite and Biotite Weathering by Fulvic Acid Solutions in Clodes and Open Systems. Can. J. Soil Sci. 1983, 63, 619–629. [Google Scholar] [CrossRef]
- Das, S.; Ghosh, K.; Varadachari, C. Weathering of Silicate Minerals by Humic Acids: II. Nature of Cation Solubilisation from Hornblende and Biotite and Characteristics of the Residual Products. Clay Res. 2014, 33, 46–63. [Google Scholar]
- Zhang, L.; Luo, L.; Zhang, S. Integrated Investigations on the Adsorption Mechanisms of Fulvic and Humic Acids on Three Clay Minerals. Colloids Surf. A Physicochem. Eng. Asp. 2012, 406, 84–90. [Google Scholar] [CrossRef]
- Gouré-Doubi, H.; Martias, C.; Smith, A.; Villandier, N.; Sol, V.; Gloaguen, V.; Feuillade, G. Adsorption of Fulvic and Humic like Acids on Surfaces of Clays: Relation with SUVA Index and Acidity. Appl. Clay Sci. 2018, 154, 83–90. [Google Scholar] [CrossRef]
- Ehrlich, H.L. How Microbes Influence Mineral Growth and Dissolution. Chem. Geol. 1996, 132, 5–9. [Google Scholar] [CrossRef]
- Banfield, J.F.; Barker, W.W.; Welch, S.A.; Taunton, A. Biological Impact on Mineral Dissolution: Application of the Lichen Model to Understanding Mineral Weathering in the Rhizosphere. Proc. Natl. Acad. Sci. USA 1999, 96, 3404–3411. [Google Scholar] [CrossRef] [PubMed]
- Bonneville, S.; Morgan, D.J.; Schmalenberger, A.; Bray, A.; Brown, A.; Banwart, S.A.; Benning, L.G. Tree-Mycorrhiza Symbiosis Accelerate Mineral Weathering: Evidences from Nanometer-Scale Elemental Fluxes at the Hypha-Mineral Interface. Geochim. Cosmochim. Acta 2011, 75, 6988–7005. [Google Scholar] [CrossRef]
- Finlay, R.D.; Mahmood, S.; Rosenstock, N.; Bolou-Bi, E.B.; Köhler, S.J.; Fahad, Z.; Rosling, A.; Wallander, H.; Belyazid, S.; Bishop, K.; et al. Reviews and Syntheses: Biological Weathering and Its Consequences at Different Spatial Levels—From Nanoscale to Global Scale. Biogeosciences 2020, 17, 1507–1533. [Google Scholar] [CrossRef]
- Saidy, A.R.; Smernik, R.J.; Baldock, J.A.; Kaiser, K.; Sanderman, J. Microbial Degradation of Organic Carbon Sorbed to Phyllosilicate Clays with and without Hydrous Iron Oxide Coating. Eur. J. Soil Sci. 2015, 66, 83–94. [Google Scholar] [CrossRef]
- Schmidt, M.W.I.; Kögel-Knaber, I. Organic Matter in Particle-Size Fractions from A and B Horizons of a Haplic Alisol. Eur. J. Soil Sci. 2002, 53, 383391. [Google Scholar] [CrossRef]

| Kaolinite | Illite | Vermiculite | Chlorite | |
|---|---|---|---|---|
| Rhizosphere (Picea) | −5.20 ±1.1 | −12.56 ± 1.6 | −25.28 ± 4.4 | −45.09 ± 8.1 |
| Bulk soil (Picea) | −4.92 ± 1.3 | −12.19 ± 1.8 | −25.07 ± 2.3 | −44.51 ± 3.9 |
| Rhizosphere (Acer) | −3.80 ± 0.9 | −10.59 ± 1.3 | −22.52 ± 1.7 | −40.52 ± 3.1 |
| Bulk soil (Acer) | −3.57 ±1.6 | −10.35 ± 2.2 | −22.4 ±2.5 | −40.03 ± 5.0 |
| Sample | С, % | N, % | C/N | TG50 | DSC50 |
|---|---|---|---|---|---|
| Kaolinite | 0.12 | 0.09 | - | - | - |
| Muscovite | 0.18 | 0.05 | - | - | - |
| Biotite | 0.12 | 0.06 | - | - | - |
| Kaolinite + HA | 5.46 | 0.28 | 19.7 | 378 | 365 |
| Muscovite + HA | 4.56 | 0.32 | 14.3 | 402 | 410 |
| Biotite + HA | 4.39 | 0.38 | 11.6 | 397 | 395 |
| HA | 27.63 | 2.30 | 12.0 | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Danilin, I.V.; Izosimova, Y.G.; Aimaletdinov, R.A.; Tolpeshta, I.I. Thermodynamic Stability of Clay Minerals in Boreal Forest Soil and Its Relationship to the Properties of Soil Organic Matter. Minerals 2025, 15, 430. https://doi.org/10.3390/min15040430
Danilin IV, Izosimova YG, Aimaletdinov RA, Tolpeshta II. Thermodynamic Stability of Clay Minerals in Boreal Forest Soil and Its Relationship to the Properties of Soil Organic Matter. Minerals. 2025; 15(4):430. https://doi.org/10.3390/min15040430
Chicago/Turabian StyleDanilin, Igor V., Yulia G. Izosimova, Ruslan A. Aimaletdinov, and Inna I. Tolpeshta. 2025. "Thermodynamic Stability of Clay Minerals in Boreal Forest Soil and Its Relationship to the Properties of Soil Organic Matter" Minerals 15, no. 4: 430. https://doi.org/10.3390/min15040430
APA StyleDanilin, I. V., Izosimova, Y. G., Aimaletdinov, R. A., & Tolpeshta, I. I. (2025). Thermodynamic Stability of Clay Minerals in Boreal Forest Soil and Its Relationship to the Properties of Soil Organic Matter. Minerals, 15(4), 430. https://doi.org/10.3390/min15040430