Assessment of Bioleaching Microbial Community Structure and Function Based on Next-Generation Sequencing Technologies


Abstract
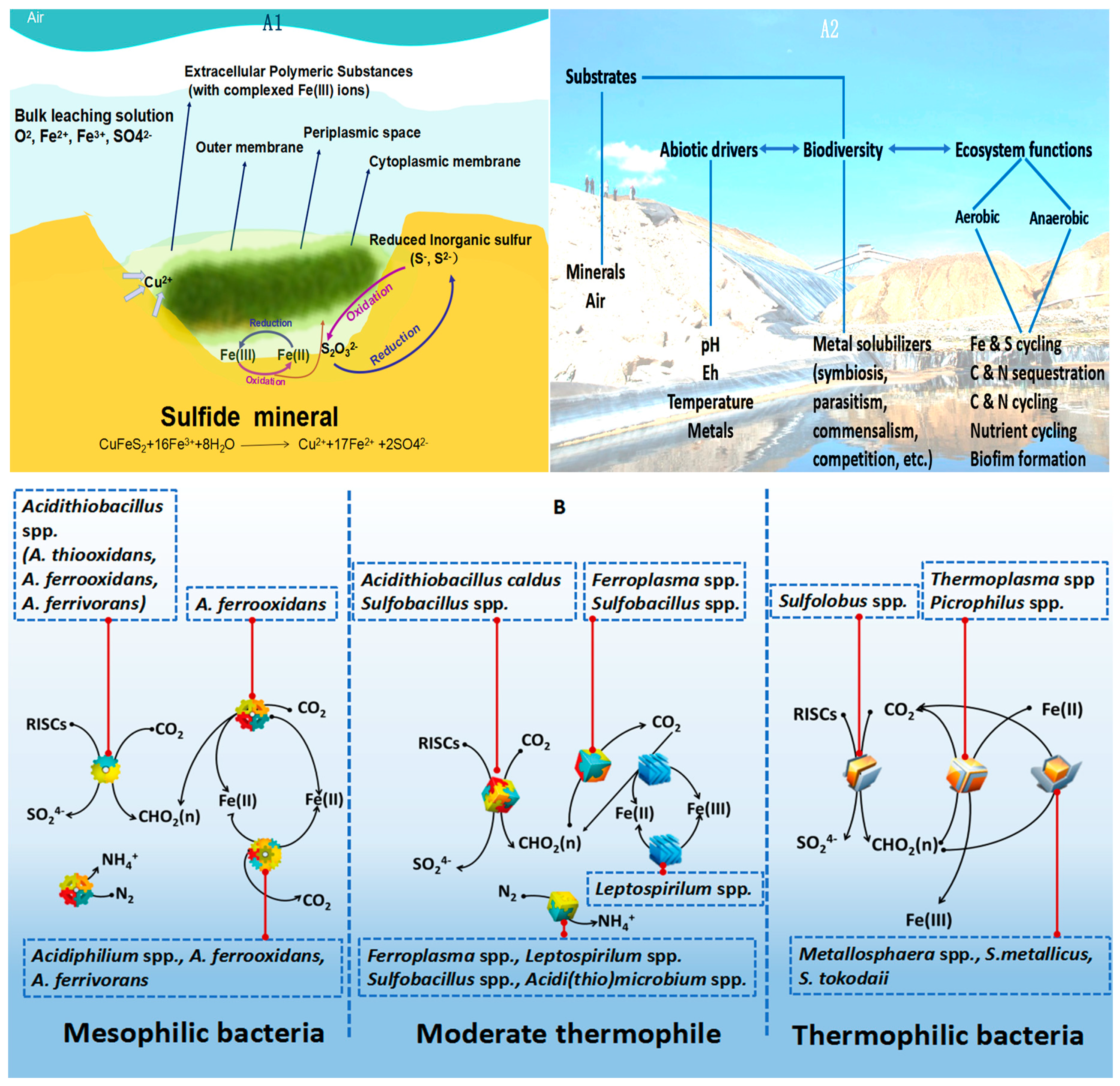
1. Introduction
2. NGS for Addressing the Challenge of Analyzing the Microbial Ecology in Bioleaching Environments
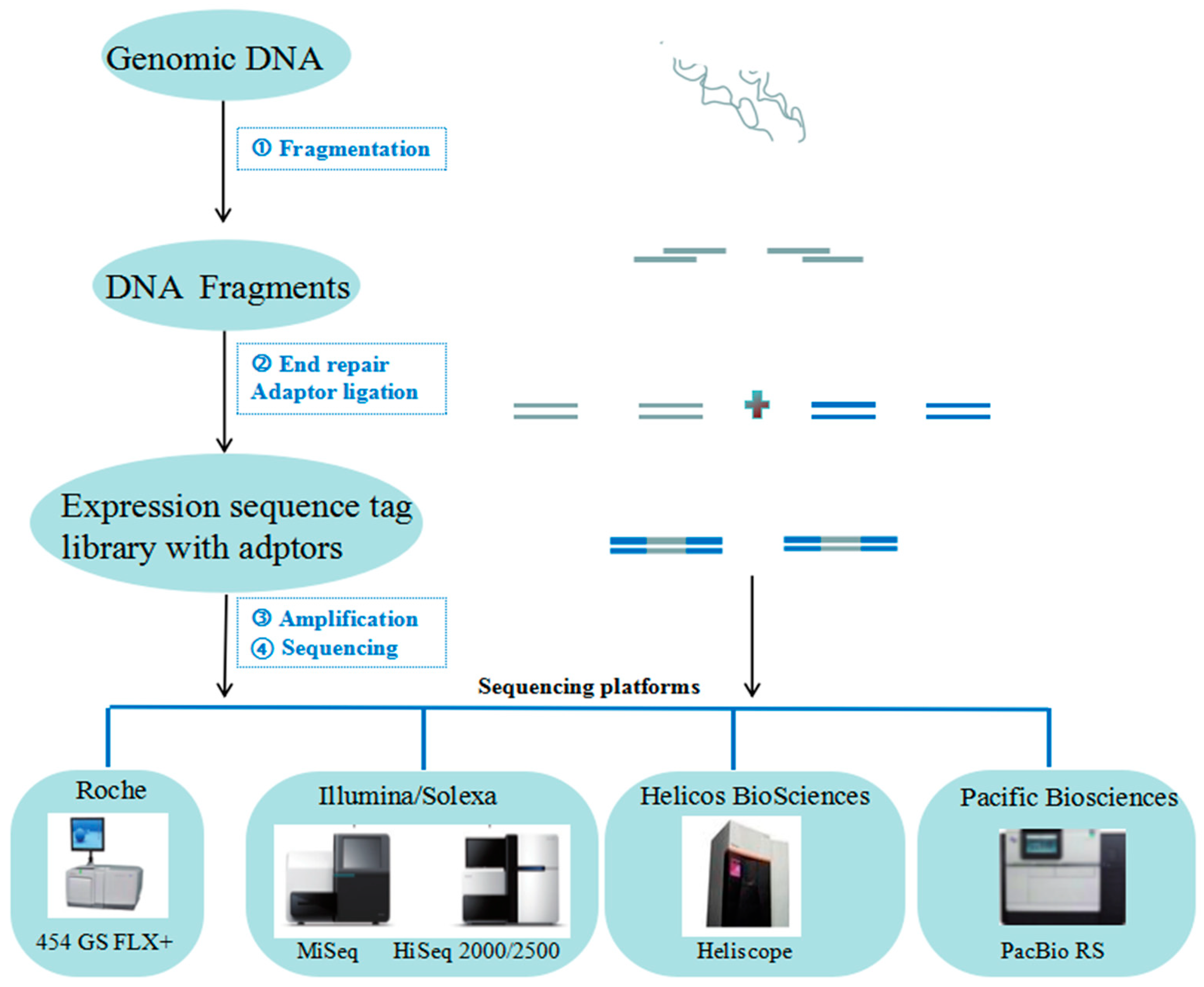
2.1. NGS for Genome Analysis
2.2. NGS for Analysis of Bacterial Diversity Present in the Ore Leaching Environment
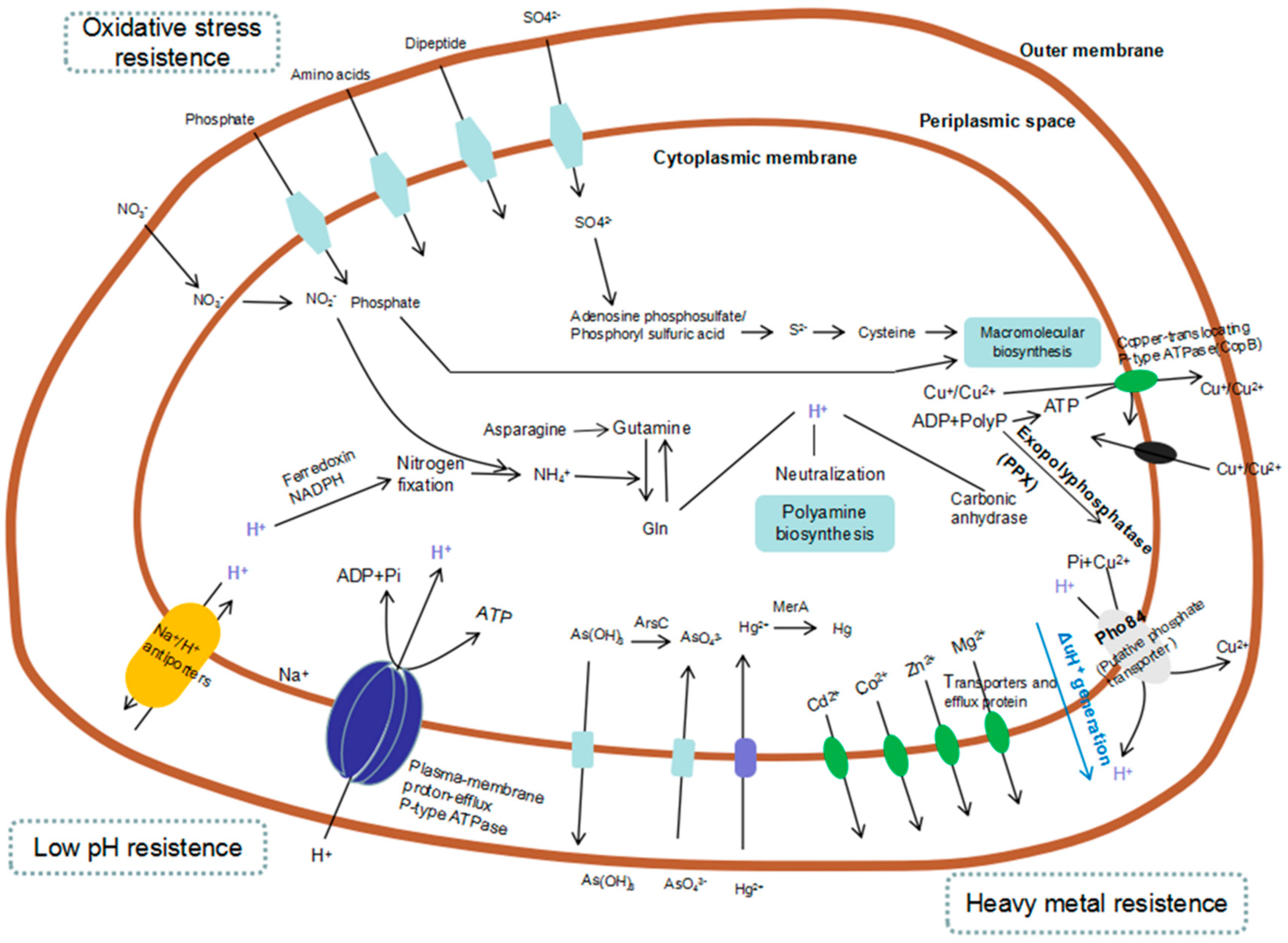
2.3. NGS for Analysis of Gene Expression in Bioleaching Microorganisms
3. Challenges and Prospect
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AMD | Acid Mine Drainage |
| CBB | Calvin–Benson–Bassham cycle |
| DGGE | Denaturing Gradient Gel Electrophoresis |
| FISH | Fluorescence in situ hybridization |
| LH | Leaching Heap |
| LS | Leaching Solution |
| MPS | Massively Parallel Sequencing |
| NanoSIMS | Nano-scale Secondary Ion Mass Spectrometry |
| NGS | Next-generation sequencing |
| PLS | Pregnant Leach Solution |
| qRT-PCR | quantitative Real-Time Polymerase Chain Reaction |
| rTCA | Reductive Citric Acid Cycle |
| RICS | Reduced Inorganic Sulfur Compounds |
| SIP | Stable Isotope Probing |
| SNPs | Single Nucleotide Polymorphisms |
| WGS | Whole Genome Sequencing |
References
- Dopson, M. Physiological Adaptations and Biotechnological Applications of Acidophiles; Horizon Scientific Press: Norwich, UK, 2012; pp. 265–294. [Google Scholar]
- Ai, C.; McCarthy, S.; Liang, Y.; Rudrappa, D.; Qiu, G.; Blum, P. Evolution of copper arsenate resistance for enhanced enargite bioleaching using the extreme thermoacidophile Metallosphaera sedula. J. Ind. Microbiol. Biotechnol. 2017, 44, 1613–1625. [Google Scholar] [CrossRef] [PubMed]
- Pablo Cardenas, J.; Quatrini, R.; Holmes, D.S. Genomic and metagenomic challenges and opportunities for bioleaching: A mini-review. Res. Microbiol. 2016, 167, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Khoshkhoo, M.; Dopson, M.; Engstrom, F.; Sandstrom, A. New insights into the influence of redox potential on chalcopyrite leaching behaviour. Miner. Eng. 2017, 100, 9–16. [Google Scholar] [CrossRef]
- Xiong, X.; Hua, X.; Zheng, Y.; Lu, X.; Li, S.; Cheng, H.; Xu, Q. Oxidation mechanism of chalcopyrite revealed by X-ray photoelectron spectroscopy and first principles studies. Appl. Surf. Sci. 2018, 427, 233–241. [Google Scholar] [CrossRef]
- Wang, J.; Gan, X.; Zhao, H.; Hu, M.; Li, K.; Qin, W.; Qiu, G. Dissolution and passivation mechanisms of chalcopyrite during bioleaching: DFT calculation, XPS and electrochemistry analysis. Miner. Eng. 2016, 98, 264–278. [Google Scholar] [CrossRef]
- Ma, L.; Wang, X.; Feng, X.; Liang, Y.; Xiao, Y.; Hao, X.; Yin, H.; Liu, H.; Liu, X. Co-culture microorganisms with different initial proportions reveal the mechanism of chalcopyrite bioleaching coupling with microbial community succession. Bioresour. Technol. 2017, 223, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Quatrini, R.; Johnson, D.B. Microbiomes in extremely acidic environments: Functionalities and interactions that allow survival and growth of prokaryotes at low pH. Curr. Opin. Microbiol. 2018, 43, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Niu, J.; Liang, Y.; Liu, X.; Yin, H. Metagenome-scale analysis yields insights into the structure and function of microbial communities in a copper bioleaching heap. BMC Genet. 2016, 17, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Liu, X.; Ma, L.; Liang, Y.; Niu, J.; Gu, Y.; Zhang, X.; Hao, X.; Dong, W.; She, S.; et al. Microbial communities from different subsystems in biological heap leaching system play different roles in iron and sulfur metabolisms. Appl. Microbiol. Biotechnol. 2016, 100, 6871–6880. [Google Scholar] [CrossRef]
- Li, Q.; Ding, D.; Sun, J.; Wang, Q.; Hu, E.; Shi, W.; Ma, L.; Guo, X.; Liu, X. Community dynamics and function variation of a defined mixed bioleaching acidophilic bacterial consortium in the presence of fluoride. Ann. Microbiol. 2015, 65, 121–128. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, X.; Liang, Y.; Fan, F.; Zhang, X.; Yin, H. Metabolic diversity and adaptive mechanisms of iron- and/or sulfur-oxidizing autotrophic acidophiles in extremely acidic environments. Environ. Microbiol. Rep. 2016, 8, 738–751. [Google Scholar] [CrossRef] [PubMed]
- Hungate, B.A.; Mau, R.L.; Schwartz, E.; Caporaso, J.G.; Dijkstra, P.; van Gestel, N.; Koch, B.J.; Liu, C.M.; McHugh, T.A.; Marks, J.C.; et al. Quantitative microbial ecology through stable isotope probing. Appl. Environ. Microbiol. 2015, 81, 7570–7581. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Kilburn, M.R.; Decelle, J.; Musat, N. Nanosims chemical imaging combined with correlative microscopy for biological sample analysis. Curr. Opin. Biotechnol. 2016, 41, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Fazzini, R.A.B.; Levican, G.; Parada, P. Acidithiobacillus thiooxidans secretome containing a newly described lipoprotein licanantase enhances chalcopyrite bioleaching rate. Appl. Microbiol. Biotechnol. 2011, 89, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Khaleque, H.N.; Shafique, R.; Kaksonen, A.H.; Boxall, N.J.; Watkin, E.L. Quantitative proteomics using SWATH-MS identifies mechanisms of chloride tolerance in the halophilic acidophile Acidihalobacter prosperus DSM 14174. Res. Microbiol. 2018, 169, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Christel, S.; Herold, M.; Bellenberg, S.; El Hajjami, M.; Buetti-Dinh, A.; Pivkin, I.V.; Sand, W.; Wilmes, P.; Poetsch, A.; Dopson, M. Multi-omics reveals the lifestyle of the acidophilic, mineral-oxidizing model species Leptospirillum ferriphilumT. Appl. Environ. Microbiol. 2018, 84, 1–17. [Google Scholar]
- Mohapatra, B.R.; Gould, W.D.; Dinardo, O.; Koren, D.W. Tracking the prokaryotic diversity in acid mine drainage-contaminated environments: A review of molecular methods. Miner. Eng. 2011, 24, 709–718. [Google Scholar] [CrossRef]
- He, Z.; Deng, Y.; Zhou, J. Development of functional gene microarrays for microbial community analysis. Curr. Opin. Biotechnol. 2012, 23, 49–55. [Google Scholar] [CrossRef]
- Chen, Q.; Yin, H.; Luo, H.; Xie, M.; Qiu, G.; Liu, X. Micro-array based whole-genome hybridization for detection of microorganisms in acid mine drainage and bioleaching systems. Hydrometallurgy 2009, 95, 96–103. [Google Scholar] [CrossRef]
- He, Z. (Ed.) Microarrays: Current Technology, Innovations and Applications; Caister Academic Press: Poole, UK, 2014. [Google Scholar]
- Li, Q.; Li, N.; Liu, X.; Zhou, Z.; Li, Q.; Fang, Y.; Fan, X.; Fu, X.; Liu, Y.; Yin, H. Characterization of the acid stress response of Acidithiobacillus ferrooxidans ATCC 23270 based on the method of microarray. J. Biol. Res. 2012, 17, 3–15. [Google Scholar]
- Vera, M.; Rohwerder, T.; Bellenberg, S.; Sand, W.; Denis, Y.; Bonnefoy, V. Characterization of biofilm formation by the bioleaching acidophilic bacterium Acidithiobacillus ferrooxidans by a microarray transcriptome analysis. Adv. Mater. Res. 2009, 71–73, 175–178. [Google Scholar] [CrossRef]
- Moreno-Paz, M.; Gomez, M.J.; Arcas, A.; Parro, V. Environmental transcriptome analysis reveals physiological differences between biofilm and planktonic modes of life of the iron oxidizing bacteria Leptospirillum spp. In their natural microbial community. BMC Genom. 2010, 11, 404. [Google Scholar] [CrossRef] [PubMed]
- Schena, M.; Shalon, D.; Davis, R.W.; Brown, P.O. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 1995, 270, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Bradford, J.R.; Hey, Y.; Yates, T.; Li, Y.; Pepper, S.D.; Miller, C.J. A comparison of massively parallel nucleotide sequencing with oligonucleotide microarrays for global transcription profiling. BMC Genom. 2010, 11, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Fu, N.; Guo, S.; Yan, Z.; Xu, Y.; Hu, H.; Menzel, C.; Chen, W.; Li, Y.; Zeng, R.; et al. Estimating accuracy of RNA-seq and microarrays with proteomics. BMC Genom. 2009, 10, 161. [Google Scholar] [CrossRef] [PubMed]
- Marioni, J.C.; Mason, C.E.; Mane, S.M.; Stephens, M.; Gilad, Y. Rna-seq: An assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008, 18, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- t’ Hoen, P.A.C.; Ariyurek, Y.; Thygesen, H.H.; Vreugdenhil, E.; Vossen, R.H.A.M.; de Menezes, R.X.; Boer, J.M.; van Ommen, G.-J.B.; den Dunnen, J.T. Deep sequencing-based expression analysis shows major advances in robustness, resolution and inter-lab portability over five microarray platforms. Nucleic Acids Res. 2008, 36, 141. [Google Scholar] [CrossRef] [PubMed]
- Hall, N. Advanced sequencing technologies and their wider impact in microbiology. J. Exp. Biol. 2007, 210, 1518–1525. [Google Scholar] [CrossRef]
- Shendure, J.; Ji, H. Next-generation DNA sequencing. Nat. Biotechnol. 2008, 26, 1135–1145. [Google Scholar] [CrossRef]
- Cardenas, E.; Tiedje, J.M. New tools for discovering and characterizing microbial diversity. Curr. Opin. Biotechnol. 2008, 19, 544–549. [Google Scholar] [CrossRef]
- Beale, D.J.; Crosswell, J.; Karpe, A.V.; Metcalfe, S.S.; Morrison, P.D.; Staley, C.; Ahmed, W.; Sadowsky, M.J.; Palombo, E.A.; Steven, A.D.L. Seasonal metabolic analysis of marine sediments collected from Moreton bay in south east Queensland, Australia, using a multi-omics-based approach. Sci. Total. Environ. 2018, 631–632, 1328–1341. [Google Scholar] [CrossRef] [PubMed]
- Cabral, L.; de Sousa, S.T.P.; Junior, G.V.L.; Hawley, E.; Andreote, F.D.; Hess, M.; de Oliveira, V.M. Microbial functional responses to long-term anthropogenic impact in mangrove soils. Ecotoxicol. Environ. Saf. 2018, 160, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Chen, T.-B.; Zheng, S.-W.; Liu, H.-T.; Zheng, G.-D. Decomposition of lignocellulose and readily degradable carbohydrates during sewage sludge biodrying, insights of the potential role of microorganisms from a metagenomic analysis. Chemosphere 2018, 201, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wu, H.; Dong, S.; Jin, W.; Han, K.; Ren, Y.; Zeng, M. Glycation of fish protein impacts its fermentation metabolites and gut microbiota during in vitro human colonic fermentation. Food Res. Int. 2018, 113, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Poroyko, V.; White, J.R.; Wang, M.; Donovan, S.; Alverdy, J.; Liu, D.C.; Morowitz, M.J. Gut microbial gene expression in mother-fed and formula-fed piglets. PLoS ONE 2010, 5, e12459. [Google Scholar] [CrossRef] [PubMed]
- Lekunberri, I.; Luis Balcazar, J.; Borrego, C.M. Metagenomic exploration reveals a marked change in the river resistome and mobilome after treated wastewater discharges. Environ. Pollut. 2018, 234, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Haas, B.J.; Chin, M.; Nusbaum, C.; Birren, B.W.; Livny, J. How deep is deep enough for RNA-seq profiling of bacterial transcriptomes? BMC Genom. 2012, 13, 734. [Google Scholar] [CrossRef]
- Marguerat, S.; Baehler, J. RNA-seq: From technology to biology. Cell. Mol. Life Sci. 2010, 67, 569–579. [Google Scholar] [CrossRef]
- Zhou, X.; Ren, L.; Meng, Q.; Li, Y.; Yu, Y.; Yu, J. The next-generation sequencing technology and application. Protein Cell 2010, 1, 520–536. [Google Scholar] [CrossRef]
- Metzker, M.L. Applications of next-generation sequencing technologies—The next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef] [PubMed]
- MacLean, D.; Jones, J.D.G.; Studholme, D.J. Application of ‘next-generation’ sequencing technologies to microbial genetics. Nat. Rev. Microbiol. 2009, 7, 287–296. [Google Scholar] [PubMed]
- Wolf, J.B.W. Principles of transcriptome analysis and gene expression quantification: An RNA-seq tutorial. Mol. Ecol. Resour. 2013, 13, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Nagalakshmi, U.; Wang, Z.; Waern, K.; Shou, C.; Raha, D.; Gerstein, M.; Snyder, M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 2008, 320, 1344–1349. [Google Scholar] [CrossRef] [PubMed]
- Lipson, D.; Raz, T.; Kieu, A.; Jones, D.R.; Giladi, E.; Thayer, E.; Thompson, J.F.; Letovsky, S.; Milos, P.; Causey, M. Quantification of the yeast transcriptome by single-molecule sequencing. Nat. Biotechnol. 2009, 27, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Sugarbaker, D.J.; Richards, W.G.; Gordon, G.J.; Dong, L.; De Rienzo, A.; Maulik, G.; Glickman, J.N.; Chirieac, L.R.; Hartman, M.-L.; Taillon, B.E.; et al. Transcriptome sequencing of malignant pleural mesothelioma tumors. Proc. Natl. Acad. Sci. USA 2008, 105, 3521–3526. [Google Scholar] [CrossRef] [PubMed]
- Core, L.J.; Waterfall, J.J.; Lis, J.T. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 2008, 322, 1845–1848. [Google Scholar] [CrossRef] [PubMed]
- Croucher, N.J.; Thomson, N.R. Studying bacterial transcriptomes using RNA-seq. Curr. Opin. Microbiol. 2010, 13, 619–624. [Google Scholar] [CrossRef]
- Denoeud, F.; Aury, J.M.; Da Silva, C.; Noel, B.; Rogier, O.; Delledonne, M.; Morgante, M.; Valle, G.; Wincker, P.; Scarpelli, C.; et al. Annotating genomes with massive-scale RNA sequencing. Genome Biol. 2008, 9, R175. [Google Scholar] [CrossRef]
- Yu, Y.; Fuscoe, J.C.; Zhao, C.; Guo, C.; Jia, M.; Qing, T.; Bannon, D.I.; Lancashire, L.; Bao, W.; Du, T.; et al. A rat RNA-seq transcriptomic bodymap across 11 organs and 4 developmental stages. Nat. Commun. 2014, 5, 3230. [Google Scholar] [CrossRef]
- Yu, Y.; Zhao, C.; Su, Z.; Wang, C.; Fuscoe, J.C.; Tong, W.; Shi, L. Comprehensive RNA-seq transcriptomic profiling across 11 organs, 4 ages, and 2 sexes of fischer 344 rats. Sci. Data 2014, 1, 140013. [Google Scholar] [CrossRef]
- Denef, V.J.; Mueller, R.S.; Banfield, J.F. AMD biofilms: Using model communities to study microbial evolution and ecological complexity in nature. ISME J. 2010, 4, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, D.E.; Johnson, D.B. The microbiology of biomining: Development and optimization of mineral-oxidizing microbial consortia. Microbiology 2007, 153, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Gadd, G.M. Metals, minerals and microbes: Geomicrobiology and bioremediation. Microbiology 2010, 156, 609–643. [Google Scholar] [CrossRef] [PubMed]
- Sand, W.; Gehrke, T.; Jozsa, P.G.; Schippers, A. (Bio)chemistry of bacterial leaching—Direct vs. Indirect bioleaching. Hydrometallurgy 2001, 59, 159–175. [Google Scholar] [CrossRef]
- Bobadilla-Fazzini, R.A.; Pina, P.; Gautier, V.; Brunel, K.; Parada, P. Mesophilic inoculation enhances primary and secondary copper sulfide bioleaching altering the microbial & mineralogical ore dynamics. Hydrometallurgy 2017, 168, 7–12. [Google Scholar]
- Shiers, D.W.; Collinson, D.M.; Watling, H.R. Life in heaps: A review of microbial responses to variable acidity in sulfide mineral bioleaching heaps for metal extraction. Res. Microbiol. 2016, 167, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Boxall, N.J.; Rea, S.M.; Li, J.; Morris, C.; Kaksonen, A.H. Effect of high sulfate concentrations on chalcopyrite bioleaching and molecular characterisation of the bioleaching microbial community. Hydrometallurgy 2017, 168, 32–39. [Google Scholar] [CrossRef]
- Latorre, M.; Paz Cortes, M.; Travisany, D.; Di Genova, A.; Budinich, M.; Reyes-Jara, A.; Hoedar, C.; Gonzalez, M.; Parada, P.; Bobadilla-Fazzini, R.A.; et al. The bioleaching potential of a bacterial consortium. Bioresour. Technol. 2016, 218, 659–666. [Google Scholar] [CrossRef]
- Niu, J.; Deng, J.; Xiao, Y.; He, Z.; Zhang, X.; Van Nostrand, J.D.; Liang, Y.; Deng, Y.; Liu, X.; Yin, H. The shift of microbial communities and their roles in sulfur and iron cycling in a copper ore bioleaching system. Sci. Rep. 2016, 6, 34744. [Google Scholar] [CrossRef]
- Pablo Cardenas, J.; Valdes, J.; Quatrini, R.; Duarte, F.; Holmes, D.S. Lessons from the genomes of extremely acidophilic bacteria and archaea with special emphasis on bioleaching microorganisms. Appl. Microbiol. Biotechnol. 2010, 88, 605–620. [Google Scholar] [CrossRef] [PubMed]
- Mao, D.; Grogan, D. Genomic evidence of rapid, global-scale gene flow in a Sulfolobus species. ISME J. 2012, 6, 1613–1616. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, S.R.; Poehlein, A.; Voget, S.; Hoppert, M.; Daniel, R.; Leimbach, A.; Tischler, J.S.; Schloemann, M.; Muehling, M. Permanent draft genome sequence of Acidiphilium sp. JA12-A1. Stand. Genom. Sci. 2015, 10, 56. [Google Scholar] [CrossRef] [PubMed]
- Valdes, J.; Quatrini, R.; Hallberg, K.; Dopson, M.; Valenzuela, P.D.T.; Holmes, D.S. Draft genome sequence of the extremely acidophilic bacterium Acidithiobacillus caldus ATCC 51756 reveals metabolic versatility in the genus Acidithiobacillus. J. Bacteriol. 2009, 191, 5877–5878. [Google Scholar] [CrossRef] [PubMed]
- You, X.-Y.; Guo, X.; Zheng, H.-J.; Zhang, M.-J.; Liu, L.-J.; Zhu, Y.-Q.; Zhu, B.; Wang, S.-Y.; Zhao, G.-P.; Poetsch, A.; et al. Unraveling the Acidithiobacillus caldus complete genome and its central metabolisms for carbon assimilation. J. Genet. Genom. 2011, 38, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Talla, E.; Hedrich, S.; Mangenot, S.; Ji, B.; Johnson, D.B.; Barbe, V.; Bonnefoy, V. Insights into the pathways of iron- and sulfur-oxidation, and biofilm formation from the chemolithotrophic acidophile Acidithiobacillus ferrivorans CF27. Res. Microbiol. 2014, 165, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Liljeqvist, M.; Valdes, J.; Holmes, D.S.; Dopson, M. Draft genome of the psychrotolerant acidophile Acidithiobacillus ferrivorans SS3. J. Bacteriol. 2011, 193, 4304–4305. [Google Scholar] [CrossRef]
- Valdes, J.; Pedroso, I.; Quatrini, R.; Dodson, R.J.; Tettelin, H.; Blake, R., II; Eisen, J.A.; Holmes, D.S. Acidithiobacillus ferrooxidans metabolism: From genome sequence to industrial applications. BMC Genom. 2008, 9, 597. [Google Scholar] [CrossRef]
- Yin, H.; Zhang, X.; Liang, Y.; Xiao, Y.; Niu, J.; Liu, X. Draft genome sequence of the extremophile Acidithiobacillus thiooxidans A01, isolated from the wastewater of a coal dump. Genome Announc. 2014, 2, e00222-14. [Google Scholar] [CrossRef]
- Valdes, J.; Ossandon, F.; Quatrini, R.; Dopson, M.; Holmes, D.S. Draft genome sequence of the extremely acidophilic biomining bacterium Acidithiobacillus thiooxidans ATCC 19377 provides insights into the evolution of the Acidithiobacillus genus. J. Bacteriol. 2011, 193, 7003–7004. [Google Scholar] [CrossRef]
- Travisany, D.; Paz Cortes, M.; Latorre, M.; Di Genova, A.; Budinich, M.; Bobadilla-Fazzini, R.A.; Parada, P.; Gonzalez, M.; Maass, A. A new genome of Acidithiobacillus thiooxidans provides insights into adaptation to a bioleaching environment. Res. Microbiol. 2014, 165, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Eisen, S.; Poehlein, A.; Johnson, D.B.; Daniel, R.; Schlomann, M.; Muhling, M. Genome sequence of the Acidophilic ferrous iron-oxidizing isolate Acidithrix ferrooxidans Strain Py-F3, the proposed type strain of the novel Actinobacterial genus Acidithrix. Genome Announc. 2015, 3, e00382-15. [Google Scholar] [CrossRef] [PubMed]
- Eisen, S.; Poehlein, A.; Johnson, D.B.; Daniel, R.; Schlomann, M.; Muhling, M. Genome sequence of the acidophilic iron oxidizer Ferrimicrobium acidiphilum strain T23T. Genome Announc. 2015, 3, e00383-15. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, J.P.; Lazcano, M.; Ossandon, F.J.; Corbett, M.; Holmes, D.S.; Watkin, E. Draft genome sequence of the iron-oxidizing acidophile Leptospirillum ferriphilum type strain DSM 14647. Genome Announc. 2014, 2, e01153-14. [Google Scholar] [CrossRef] [PubMed]
- Issotta, F.; Galleguillos, P.A.; Moya-Beltran, A.; Davis-Belmar, C.S.; Rautenbach, G.; Covarrubias, P.C.; Acosta, M.; Ossandon, F.J.; Contador, Y.; Holmes, D.S.; et al. Draft genome sequence of chloride-tolerant Leptospirillum ferriphilum Sp-Cl from industrial bioleaching operations in northern Chile. Stand. Genom. Sci. 2016, 11, 19. [Google Scholar] [CrossRef] [PubMed]
- Moya-Beltran, A.; Cardenas, J.P.; Covarrubias, P.C.; Issotta, F.; Ossandon, F.J.; Grail, B.M.; Holmes, D.S.; Quatrini, R.; Johnson, D.B. Draft genome sequence of the nominated type strain of “Ferrovum myxofaciens,” an acidophilic, iron-oxidizing betaproteobacterium. Genome Announc. 2014, 2, e00834-14. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, S.R.; Poehlein, A.; Tischler, J.S.; Gonzalez, C.; Ossandon, F.J.; Daniel, R.; Holmes, D.S.; Schloemann, M.; Muehling, M. Genome analysis of the biotechnologically relevant acidophilic iron oxidising strain JA12 indicates phylogenetic and metabolic diversity within the novel genus “Ferrovum”. PLoS ONE 2016, 11, e0146832. [Google Scholar] [CrossRef]
- Dall’Agnol, H.; Nancucheo, I.; Johnson, D.B.; Oliveira, R.; Leite, L.; Pylro, V.S.; Holanda, R.; Grail, B.; Carvalho, N.; Nunes, G.L.; et al. Draft genome sequence of “Acidibacillus ferrooxidans” ITV01, a novel acidophilic firmicute isolated from a chalcopyrite mine drainage site in Brazil. Genome Announc. 2016, 4, e01748-15. [Google Scholar]
- Anderson, I.; Chertkov, O.; Chen, A.; Saunders, E.; Lapidus, A.; Nolan, M.; Lucas, S.; Hammon, N.; Deshpande, S.; Cheng, J.-F.; et al. Complete genome sequence of the moderately thermophilic mineral-sulfide-oxidizing firmicute Sulfobacillus acidophilus type strain (NALT). Stand. Genom. Sci. 2012, 6, 293–303. [Google Scholar] [CrossRef]
- Travisany, D.; Di Genova, A.; Sepulveda, A.; Bobadilla-Fazzini, R.A.; Parada, P.; Maass, A. Draft genome sequence of the Sulfobacillus thermosulfidooxidans cutipay strain, an indigenous bacterium isolated from a naturally extreme mining environment in northern Chile. J. Bacteriol. 2012, 194, 6327–6328. [Google Scholar] [CrossRef]
- Chen, L.-X.; Huang, L.-N.; Mendez-Garcia, C.; Kuang, J.-L.; Hua, Z.-S.; Liu, J.; Shu, W.-S. Microbial communities, processes and functions in acid mine drainage ecosystems. Curr. Opin. Biotechnol. 2016, 38, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Quatrini, R.; Escudero, L.V.; Moya-Beltran, A.; Galleguillos, P.A.; Issotta, F.; Acosta, M.; Pablo Cardenas, J.; Nunez, H.; Salinas, K.; Holmes, D.S.; et al. Draft genome sequence of Acidithiobacillus thiooxidans CLST isolated from the acidic hypersaline Gorbea salt flat in northern Chile. Stand. Genom. Sci. 2017, 12, 84. [Google Scholar] [CrossRef] [PubMed]
- Selkov, E.; Overbeek, R.; Kogan, Y.; Chu, L.; Vonstein, V.; Holmes, D.; Silver, S.; Haselkorn, R.; Fonstein, M. Functional analysis of gapped microbial genomes: Amino acid metabolism of Thiobacillus ferrooxidans. Proc. Natl. Acad. Sci. USA 2000, 97, 3509–3514. [Google Scholar] [CrossRef] [PubMed]
- Valdes, J.; Veloso, F.; Jedlicki, E.; Holmes, D. Metabolic reconstruction of sulfur assimilation in the extremophile Acidithiobacillus ferrooxidans based on genome analysis. BMC Genom. 2003, 4, 51. [Google Scholar] [CrossRef] [PubMed]
- Osorio, H.; Martinez, V.; Nieto, P.A.; Holmes, D.S.; Quatrini, R. Microbial iron management mechanisms in extremely acidic environments: Comparative genomics evidence for diversity and versatility. BMC Microbiol. 2008, 8, 203. [Google Scholar] [CrossRef] [PubMed]
- Quatrini, R.; Lefimil, C.; Veloso, F.A.; Pedroso, I.; Holmes, D.S.; Jedlicki, E. Bioinformatic prediction and experimental verification of fur-regulated genes in the extreme acidophile Acidithiobacillus ferrooxidans. Nucleic Acids Res. 2007, 35, 2153–2166. [Google Scholar] [CrossRef] [PubMed]
- Rivas, M.; Seeger, M.; Jedlicki, E.; Holmes, D.S. Second acyl homoserine lactone production system in the extreme acidophile Acidithiobacillus ferrooxidans. Appl. Environ. Microbiol. 2007, 73, 3225–3231. [Google Scholar] [CrossRef]
- Rivas, M.; Seeger, M.; Holmes, D.S.; Jedlicki, E. A lux-like quorum sensing system in the extreme acidophile Acidithiobacillus ferrooxidans. Biol. Res. 2005, 38, 283–297. [Google Scholar] [CrossRef]
- Farah, C.; Vera, M.; Morin, D.; Haras, D.; Jerez, C.A.; Guiliani, N. Evidence for a functional quorum-sensing type AI-1 system in the extremophilic bacterium Acidithiobacillus ferrooxidans. Appl. Environ. Microbiol. 2005, 71, 7033–7040. [Google Scholar] [CrossRef]
- Barreto, M.; Jedlicki, E.; Holmes, D.S. Identification of a gene cluster for the formation of extracellular polysaccharide precursors in the chemolithoautotroph Acidithiobacillus ferrooxidans. Appl. Environ. Microbiol. 2005, 71, 2902–2909. [Google Scholar] [CrossRef]
- Appia-Ayme, C.; Quatrini, R.; Denis, Y.; Denizot, F.; Silver, S.; Roberto, F.; Veloso, F.; Valdes, J.; Cardenas, J.P.; Esparza, M.; et al. Microarray and bioinformatic analyses suggest models for carbon metabolism in the autotroph Acidithiobacillus ferrooxidans. Hydrometallurgy 2006, 83, 273–280. [Google Scholar] [CrossRef]
- Quatrini, R.; Appia-Ayme, C.; Denis, Y.; Ratouchniak, J.; Veloso, F.; Valdes, J.; Lefimil, C.; Silver, S.; Roberto, F.; Orellana, O.; et al. Insights into the iron and sulfur energetic metabolism of Acidithiobacillus ferrooxidans by microarray transcriptome profiling. Hydrometallurgy 2006, 83, 263–272. [Google Scholar] [CrossRef]
- Quatrini, R.; Appia-Ayme, C.; Denis, Y.; Jedlicki, E.; Holmes, D.S.; Bonnefoy, V. Extending the models for iron and sulfur oxidation in the extreme acidophile Acidithiobacillus ferrooxidans. BMC Genom. 2009, 10, 394. [Google Scholar] [CrossRef] [PubMed]
- Esparza, M.; Pablo Cardenas, J.; Bowien, B.; Jedlicki, E.; Holmes, D.S. Genes and pathways for CO2 fixation in the obligate, chemolithoautotrophic acidophile, Acidithiobacillus ferrooxidans, carbon fixation in A. ferrooxidans. BMC Microbiol. 2010, 10, 229. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-X.; Li, J.-T.; Chen, Y.-T.; Huang, L.-N.; Hua, Z.-S.; Hu, M.; Shu, W.-S. Shifts in microbial community composition and function in the acidification of a lead/zinc mine tailings. Environ. Microbiol. 2013, 15, 2431–2444. [Google Scholar] [CrossRef] [PubMed]
- Woese, C.R.; Kandler, O.; Wheelis, M.L. Towards a natural system of organisms: Proposal for the domains archaea, bacteria, and eucarya. Proc. Natl. Acad. Sci. USA 1990, 87, 4576–4579. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the illumina hiseq and miseq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Sun, H.; Chen, D.; Gao, H.; Ruan, R. Characterization of microbial community in industrial bioleaching heap of copper sulfide ore at monywa mine, myanmar. Hydrometallurgy 2016, 164, 355–361. [Google Scholar] [CrossRef]
- Acosta, M.; Galleguillos, P.A.; Guajardo, M.; Demergasso, C. Microbial survey on industrial bioleaching heap by high-throughput 16S sequencing and metagenomics analysis. Solid State Phenom. 2017, 262, 219–223. [Google Scholar] [CrossRef]
- Baker, B.J.; Banfield, J.F. Metagenomics of acid mine drainage at iron mountain California: Expanding our view from individual genes and cultures to entire communities. In Acidophiles. Life in Extremely Acidic Environments; Caister Academic Press: Poole, UK, 2016; pp. 221–232. [Google Scholar]
- Hu, Q.; Guo, X.; Liang, Y.; Hao, X.; Ma, L.; Yin, H.; Liu, X. Comparative metagenomics reveals microbial community differentiation in a biological heap leaching system. Res. Microbiol. 2015, 166, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Javier Jimenez, D.; Andreote, F.D.; Chaves, D.; Salvador Montana, J.; Osorio-Forero, C.; Junca, H.; Mercedes Zambrano, M.; Baena, S. Structural and functional insights from the metagenome of an acidic hot spring microbial planktonic community in the Colombian Andes. PLoS ONE 2012, 7, e52069. [Google Scholar]
- Nunez, H.; Covarrubias, P.C.; Moya-Beltran, A.; Issotta, F.; Atavales, J.; Acuna, L.G.; Johnson, D.B.; Quatrini, R. Detection, identification and typing of Acidithiobacillus species and strains: A review. Res. Microbiol. 2016, 167, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, C.; Yanquepe, M.; Cardenas, J.P.; Valdes, J.; Quatrini, R.; Holmes, D.S.; Dopson, M. Genetic variability of psychrotolerant Acidithiobacillus ferrivorans revealed by (meta)genomic analysis. Res. Microbiol. 2014, 165, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Schippers, A.; Breuker, A.; Blazejak, A.; Bosecker, K.; Kock, D.; Wright, T.L. The biogeochemistry and microbiology of sulfidic mine waste and bioleaching dumps and heaps, and novel Fe(II)-oxidizing bacteria. Hydrometallurgy 2010, 104, 342–350. [Google Scholar] [CrossRef]
- Peng, T.; Ma, L.; Feng, X.; Tao, J.; Nan, M.; Liu, Y.; Li, J.; Shen, L.; Wu, X.; Yu, R.; et al. Genomic and transcriptomic analyses reveal adaptation mechanisms of an Acidithiobacillus ferrivorans strain YL15 to alpine acid mine drainage. PLoS ONE 2017, 12, e0178008. [Google Scholar] [CrossRef] [PubMed]
- Galleguillos, P.; Remonsellez, F.; Galleguillos, F.; Guiliani, N.; Castillo, D.; Demergasso, C. Identification of differentially expressed genes in an industrial bioleaching heap processing low-grade copper sulphide ore elucidated by RNA arbitrarily primed polymerase chain reaction. Hydrometallurgy 2008, 94, 148–154. [Google Scholar] [CrossRef]
- Remonsellez, F.; Galleguillos, F.; Moreno-Paz, M.; Parro, V.; Acosta, M.; Demergasso, C. Dynamic of active microorganisms inhabiting a bioleaching industrial heap of low-grade copper sulfide ore monitored by real-time PCR and oligonucleotide prokaryotic acidophile microarray. Microb. Biotechnol. 2009, 2, 613–624. [Google Scholar] [CrossRef]
- Marin, S.; Acosta, M.; Galleguillos, P.; Chibwana, C.; Strauss, H.; Demergasso, C. Is the growth of microorganisms limited by carbon availability during chalcopyrite bioleaching? Hydrometallurgy 2017, 168, 13–20. [Google Scholar] [CrossRef]
- Hansen, K.D.; Brenner, S.E.; Dudoit, S. Biases in illumina transcriptome sequencing caused by random hexamer priming. Nucleic Acids Res. 2010, 38, e131. [Google Scholar] [CrossRef] [PubMed]
- Ozsolak, F.; Milos, P.M. Rna sequencing: Advances, challenges and opportunities. Nat. Rev. Genet. 2011, 12, 87–98. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organism | NCBI Accession | Source | Reference |
|---|---|---|---|
| Acidiplasma cupricumulans BH2 | LKBH00000000 | Mineral sulfide ore, Myanmar | not available |
| Acidiplasma cupricumulans JCM 13668 | BBDK00000000 | Industrial-scale chalcocite bioleach heap, Myanmar | not available |
| Acidiplasma sp. MBA-1 | JYHS00000000 | Bioleaching bioreactor pulp, Russia | not available |
| Sulfolobus acidocaldarius Ron12/I | NC_020247 | Uranium mine heaps, Germany | [64] |
| Acidiphilium angustum ATCC 35903T | JNJH00000000 | Waste coal mine waters, USA | not available |
| Acidiphilium cryptum JF-5 | NC_009484 | Acidic coal mine lake sediment, Germany | not available |
| Acidiphilium sp. JA12-A1 | JFHO00000000 | Pilot treatment plant water, Germany | [65] |
| Acidithiobacillus caldus ATCC 51756T | CP005986 | Coal spoil enrichment culture, UK | [66] |
| Acidithiobacillus caldus SM-1 | NC_015850 | Pilot bioleaching reactor, China | [67] |
| Acidithiobacillus ferrivorans CF27 | CCCS000000000 | Abandoned copper/cobalt mine drainage, USA | [68] |
| Acidithiobacillus ferrivorans SS3 | NC_015942 | Enrichment culture from mine-impacted soil samples, Russia | [69] |
| Acidithiobacillus ferrooxidans ATCC 23270T | NC_011761 | Acid, bituminous coal mine effluent, USA | [70] |
| Acidithiobacillus ferrooxidans ATCC 53993 | NC_011206 | Copper deposits, Armenia | not available |
| Acidithiobacillus sp. GGI-221 | AEFB00000000 | Mine water, India | not available |
| Acidithiobacillus thiooxidans A01 | AZMO00000000 | Wastewater of coal dump, China | [71] |
| Acidithiobacillus thiooxidans ATCC 19377T | AFOH00000000 | Kimmeridge clay, UK | [72] |
| Acidithiobacillus thiooxidans Licanantay | JMEB00000000 | Copper mine, Chile | [73] |
| Acidithrix ferrooxidans DSM 28176T | JXYS00000000 | acidic stream draining in abandoned copper mine, UK | [74] |
| Ferrimicrobium acidiphilum DSM 19497T | JQKF00000000 | Mine water, UK | [75] |
| Leptospirillum ferriphilum DSM 14647T | JPGK00000000 | Enrichment culture, Peru | [76] |
| Leptospirillum sp. Sp-Cl | LGSH00000000 | Industrial bioleaching solution, Chile | [77] |
| “Ferrovum myxofaciens” P3GT | JPOQ00000000 | Stream draining an abandoned copper mine, UK | [78] |
| Ferrovum sp. JA12 | LJWX00000000 | Pilot treatment plant water, Germany | [79] |
| Ferrovum sp. Z-31 | LRRD00000000 | Acid mine drainage water, Germany | not available |
| Ferrovum sp. PN-J185 | LQZA00000000 | Acid mine drainage water, Germany | not available |
| “Acidibacillus ferrooxidans” DSM 5130T | LPVJ00000000 | Neutral drainage from copper mine, Brazil | [80] |
| Sulfobacillus acidophilus DSM 10332T | NC_016884 | Coal spoil heap, UK | [81] |
| Sulfobacillus thermosulfidooxidans CBAR13 | LGRO00000000 | Percolate solution of a bioleaching heap in copper mine, Chile | not available |
| Sulfobacillus thermosulfidooxidans Cutipay | ALWJ00000000 | Naturally mining environment, Chile | [82] |
| Sulfobacillus thermosulfidooxidans DSM 9293T | (2506210005) * | Spontaneously heated ore deposit, Kazakhstan | not available |
| Bioleaching heap surface Metagenome | (4664533.3) # | Dexing Copper Mine, China | [9] |
| Bioleaching heap PLS sample Metagenome | (4554868.3) # | Dexing Copper Mine, China | [83] |
| Bioleaching heap sample Metagenome | (4554867.3) # | Dexing Copper Mine, China | [83] |
| Acidithiobacillus thiooxidans CLST | NZ_LGYM01000020.1 | Gorbea salt flat, northern Chile. | [84] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, S.; Gan, M.; Zhu, J.; Liu, X.; Qiu, G. Assessment of Bioleaching Microbial Community Structure and Function Based on Next-Generation Sequencing Technologies. Minerals 2018, 8, 596. https://doi.org/10.3390/min8120596
Zhou S, Gan M, Zhu J, Liu X, Qiu G. Assessment of Bioleaching Microbial Community Structure and Function Based on Next-Generation Sequencing Technologies. Minerals. 2018; 8(12):596. https://doi.org/10.3390/min8120596
Chicago/Turabian StyleZhou, Shuang, Min Gan, Jianyu Zhu, Xinxing Liu, and Guanzhou Qiu. 2018. "Assessment of Bioleaching Microbial Community Structure and Function Based on Next-Generation Sequencing Technologies" Minerals 8, no. 12: 596. https://doi.org/10.3390/min8120596
APA StyleZhou, S., Gan, M., Zhu, J., Liu, X., & Qiu, G. (2018). Assessment of Bioleaching Microbial Community Structure and Function Based on Next-Generation Sequencing Technologies. Minerals, 8(12), 596. https://doi.org/10.3390/min8120596