High-Temperature Evolution of Point Defect Equilibria in Hydrous Forsterite Synthesized at 1100 °C and up to 4 GPa


Abstract
:1. Introduction
2. Experimental Methods
2.1. Synthesis and Characterization of Synthetic Forsterite
2.2. High-Pressure and High-Temperature Experiments
2.3. Room Temperature FTIR Analysis
3. Results
3.1. Deconvolution of Spectra and Peaks Identification
3.2. Calculation of Total Water Concentration
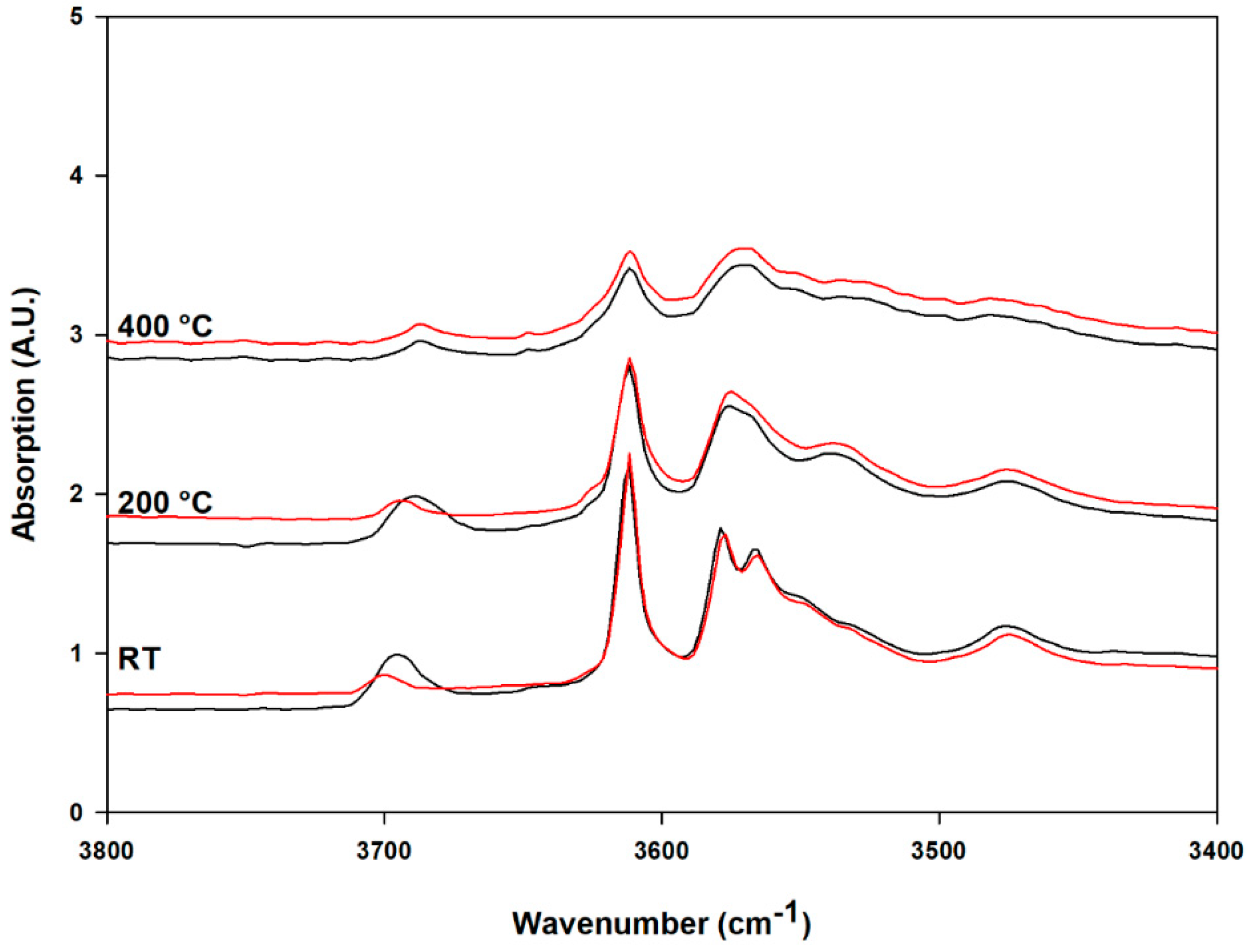
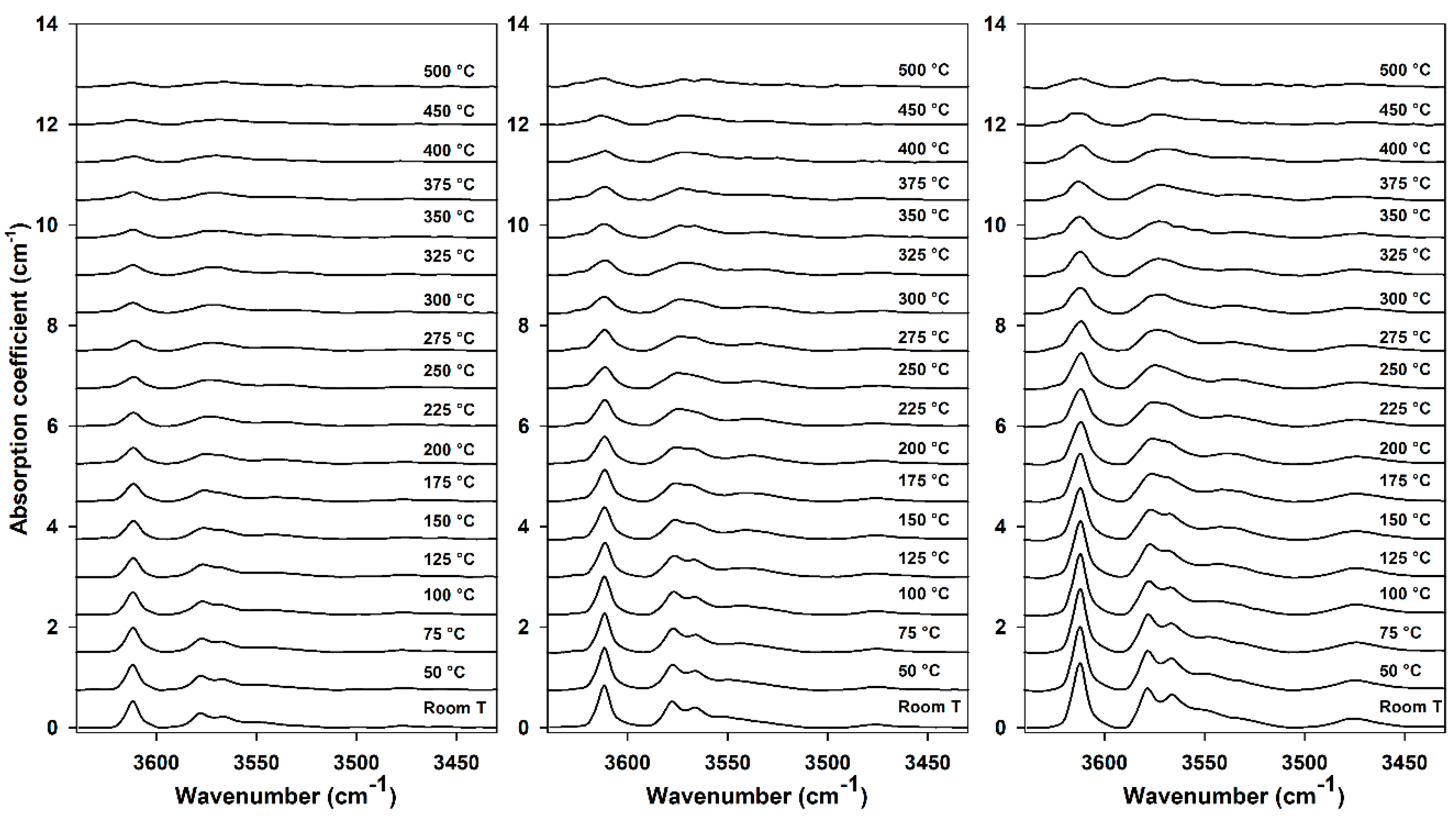
3.3. High-Temperature FTIR Analysis
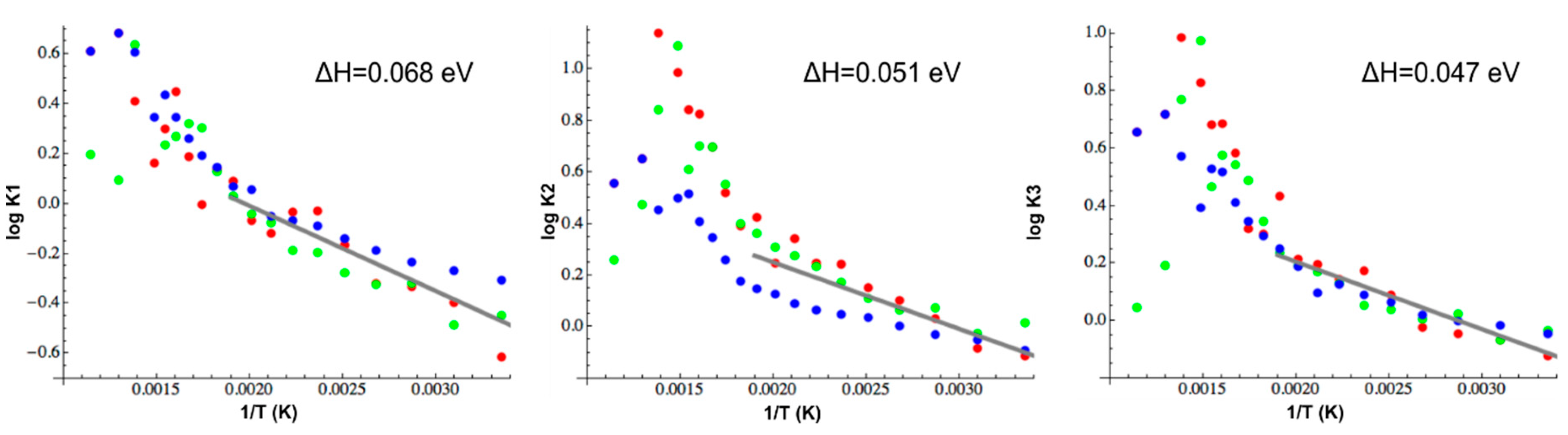
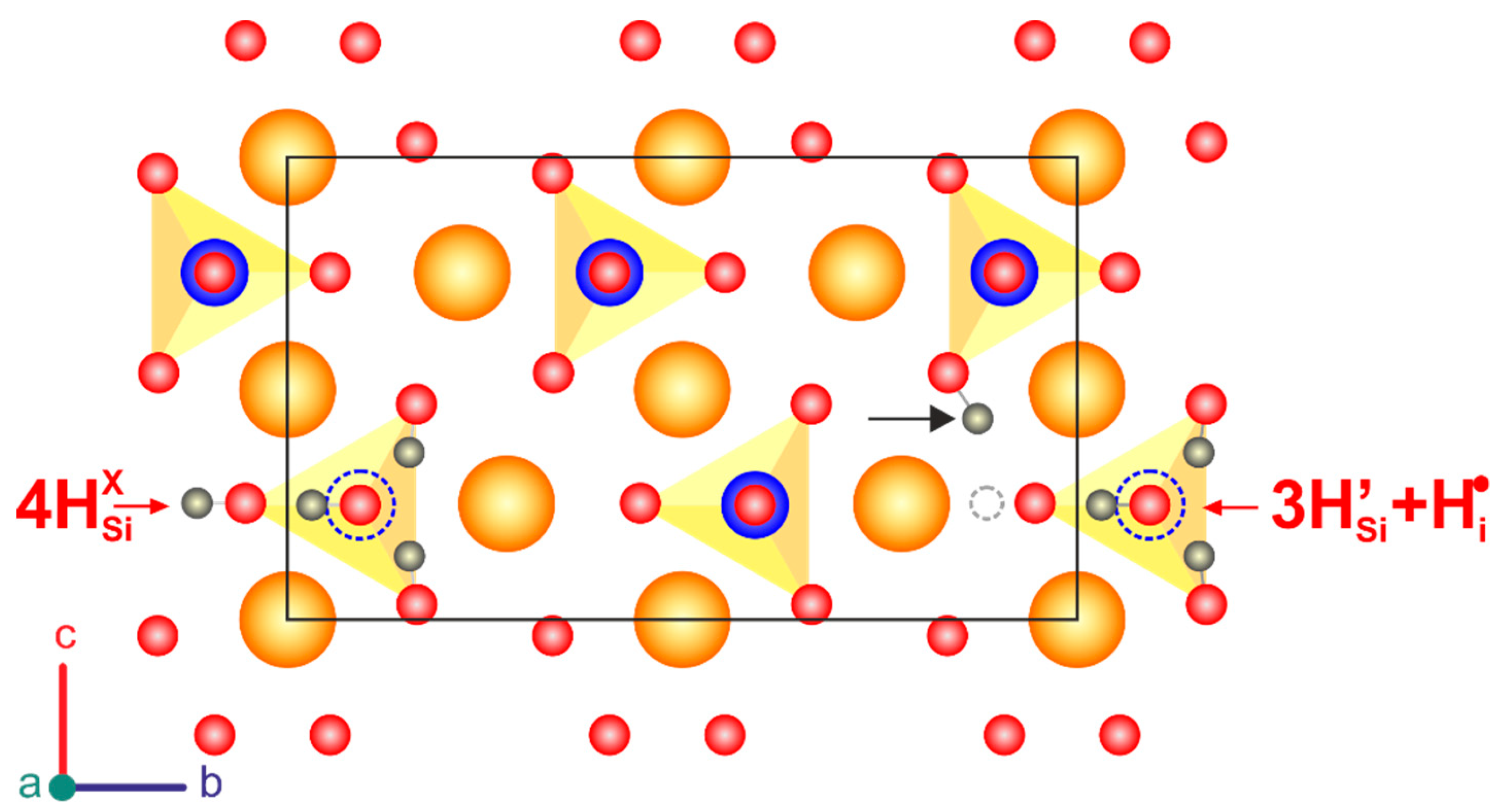
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bai, Q.; Kohlstedt, D. Effects of chemical environment on the solubility and incorporation mechanism for hydrogen in olivine. Phys. Chem. Miner. 1993, 19, 460–471. [Google Scholar] [CrossRef]
- Libowitzky, E.; Beran, A. OH defects in forsterite. Phys. Chem. Miner. 1995, 22, 387–392. [Google Scholar] [CrossRef]
- O’Neill, H.S.C.; Matveev, S.; Ballhaus, C.; Taylor, W.R.; Green, D.H. Effect of Silica Activity on OH- IR Spectra of Olivine: Implications for Low-aSiO2 Mantle Metasomatism. J. Pet. 2001, 42, 721–729. [Google Scholar]
- Matveev, S.; Portnyagin, M.; Ballhaus, C.; Brooker, R.; Geiger, C.A. FTIR Spectrum of Phenocryst Olivine as an Indicator of Silica Saturation in Magmas. J. Petrol. 2005, 46, 603–614. [Google Scholar] [CrossRef]
- Lemaire, C.; Kohn, S.C.; Brooker, R.A. The effect of silica activity on the incorporation mechanisms of water in synthetic forsterite: A polarized infrared spectroscopic study. Contr. Mineral. Petrol. 2004, 147, 48–57. [Google Scholar]
- Berry, A.J.; Hermann, J.; O’Neill, H.S.; Foran, G.J.; Hermann, J. Fingerprinting the water site in mantle olivine. Geology 2005, 33, 869. [Google Scholar] [CrossRef]
- Kudoh, Y.; Kuribayashi, T.; Kagi, H.; Inoue, T. Cation vacancy and possible hydrogen positions in hydrous forsterite, Mg1.985Si0.993H0.06O4, synthesized at 13.5 GPa and 1300 °C. J. Miner. Pet. Sci. 2006, 101, 265–269. [Google Scholar] [CrossRef]
- Deligne, N.I.; Mosenfelder, J.L.; Asimow, P.D.; Rossman, G.R. Hydrogen incorporation in olivine from 2–12 GPa. Am. Miner. 2006, 91, 285–294. [Google Scholar]
- Koch-Müller, M.; Matsyuk, S.S.; Rhede, D.; Wirth, R.; Khisina, N. Hydroxyl in mantle olivine xenocrysts from the Udachnaya kimberlite pipe. Phys. Chem. Miner. 2006, 33, 276–287. [Google Scholar] [CrossRef]
- Pearson, D.G.; Brenker, F.E.; Nestola, F.; McNeill, J.; Nasdala, L.; Hutchison, M.T.; Matveev, S.; Mather, K.; Silversmit, G.; Schmitz, S.; et al. Hydrous mantle transition zone indicated by ringwoodite included within diamond. Nature 2014, 507, 221–224. [Google Scholar] [CrossRef]
- Chopra, P.N.; Paterson, M.S. The role of water in the deformation of dunite. J. Geophys. Res. Space Phys. 1984, 89, 7861–7876. [Google Scholar] [CrossRef]
- Mackwell, S.J.; Kohlstedt, D.L.; Paterson, M.S. The role of water in the deformation of olivine single crystals. J. Geophys. Res. Space Phys. 1985, 90, 11319. [Google Scholar] [CrossRef]
- Mackwell, S.J.; Dimos, D.; Kohlstedt, D.L.; Mackwell, S. Transient creep of olivine: Point-defect relaxation times. Philos. Mag. A 1988, 57, 779–789. [Google Scholar] [CrossRef]
- Mackwell, S.J.; Kohlstedt, D.L. Diffusion of hydrogen in olivine: Implications for water in the mantle. J. Geophys. Res. Space Phys. 1990, 95, 5079. [Google Scholar] [CrossRef]
- Karato, S.-I.; Paterson, M.S.; Fitzgerald, J.D. Rheology of synthetic olivine aggregates: Influence of grain size and water. J. Geophys. Res. Space Phys. 1986, 91, 8151. [Google Scholar] [CrossRef]
- Arndt, N.; Ginibre, C.; Chauvel, C.; Albarède, F.; Cheadle, M.; Herzberg, C.; Jenner, G.; Lahaye, Y. Were komatiites wet? Geology 1998, 26, 739–742. [Google Scholar] [CrossRef]
- Gaetani, G.A.; Grove, T.L. The influence of water on melting of mantle peridotite. Contrib. Miner. Pet. 1998, 131, 323–346. [Google Scholar] [CrossRef]
- Inoue, T. Effect of water on melting phase relations and melt composition in the system Mg2SiO4 MgSiO3 H2O up to 15 GPa. Phys. Earth Planet. Inter. 1994, 85, 237–263. [Google Scholar] [CrossRef]
- Hirth, G.; Kohlstedt, D.L. Water in the oceanic upper mantle: Implications for rheology, melt extraction and the evolution of the lithosphere. Earth Planet. Sci. Lett. 1996, 144, 93–108. [Google Scholar] [CrossRef]
- Karato, S.-I.; Jung, H. Water, partial melting and the origin of the seismic low velocity and high attenuation zone in the upper mantle. Earth Planet. Sci. Lett. 1998, 157, 193–207. [Google Scholar] [CrossRef]
- Karato, S. The role of hydrogen in the electrical conductivity of the upper mantle. Nature 1990, 347, 272–273. [Google Scholar] [CrossRef]
- Hirsch, L.M.; Shankland, T.J.; Duba, A.G. Electrical Conduction and Polaron Mobility In Fe-Bearing Olivine. Geophys. J. Int. 1993, 114, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Poe, B.T.; Shankland, T.J.; Rubie, D.C. Electrical Conductivity of Olivine, Wadsleyite, and Ringwoodite Under Upper-Mantle Conditions. Science 1998, 280, 1415–1418. [Google Scholar] [CrossRef]
- Du Frane, W.L.; Roberts, J.J.; Toffelmier, D.A.; Tyburczy, J.A. Anisotropy of electrical conductivity in dry olivine. Geophys. Res. Lett. 2005, 32, 32. [Google Scholar] [CrossRef]
- Wang, D.; Mookherjee, M.; Xu, Y.; Karato, S.-I. The effect of water on the electrical conductivity of olivine. Nature 2006, 443, 977–980. [Google Scholar] [CrossRef]
- Yoshino, T.; Matsuzaki, T.; Yamashita, S.; Katsura, T. Hydrous olivine unable to account for conductivity anomaly at the top of the astenosphere. Lett. Nat. 2006, 443, 973. [Google Scholar] [CrossRef]
- Poe, B.T.; Romano, C.; Nestola, F.; Smyth, J.R. Electrical conductivity anisotropy of dry and hydrous olivine at 8GPa. Phys. Earth Planet. Inter. 2010, 181, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Du Frane, W.L.; Tyburczy, J.A. Deuterium-hydrogen exchange in olivine: Implications for point defects and electrical conductivity. Geochem. Geophys. Geosystems 2012, 13, 3. [Google Scholar] [CrossRef]
- Novella, D.; Dolejš, D.; Myhill, R.; Pamato, M.G.; Manthilake, G.; Frost, D.J. Melting phase relations in the systems Mg2SiO4–H2O and MgSiO3–H2O and the formation of hydrous melts in the upper mantle. Geochim. et Cosmochim. Acta 2017, 204, 68–82. [Google Scholar] [CrossRef]
- Bell, D.R.; Rossman, G.R. Water in the earth’s mantle: The role of nominally anhydrous minerals. Science 1992, 255, 1391–1397. [Google Scholar] [CrossRef]
- Thompson, A.B. Water in the Earth’s upper mantle. Nature 1992, 358, 295–302. [Google Scholar] [CrossRef]
- Grant, K.; Ingrin, J.; Lorand, J.P.; Dumas, P. Water partitioning between mantle minerals from peridotite xenoliths. Contrib. Miner. Pet. 2007, 154, 15–34. [Google Scholar] [CrossRef]
- Demouchy, S.; Mackwell, S. Mechanisms of hydrogen incorporation and diffusion in iron-bearing olivine. Phys. Chem. Miner. 2006, 33, 347–355. [Google Scholar] [CrossRef]
- Peslier, A.H.; Woodland, A.B.; Wolff, J.A. Fast kimberlite ascent rates estimated from hydrogen diffusion profiles in xenolithic mantle olivines from southern Africa. Geochim. et Cosmochim. Acta 2008, 72, 2711–2722. [Google Scholar] [CrossRef]
- Demouchy, S.; Bolfan-Casanova, N. Distribution and transport of hydrogen in the lithospheric mantle: A review. Lithos 2016, 240–243, 402–425. [Google Scholar] [CrossRef]
- Stalder, R.; Skogby, H. Hydrogen diffusion in natural and synthetic orthopyroxene. Phys. Chem. Miner. 2003, 30, 12–19. [Google Scholar] [CrossRef]
- Huang, X.; Xu, Y.; Karato, S.-I. Water content in the transition zone from electrical conductivity of wadsleyite and ringwoodite. Nature 2005, 434, 746–749. [Google Scholar] [CrossRef]
- Hae, R.; Ohtani, E.; Kubo, T.; Koyama, T.; Utada, H. Hydrogen diffusivity in wadsleyite and water distribution in the mantle transition zone. Earth Planet. Sci. Lett. 2006, 243, 141–148. [Google Scholar] [CrossRef]
- Berry, A.J.; O’Neill, H.S.; Hermann, J.; Scott, D.R.; Hermann, J. The infrared signature of water associated with trivalent cations in olivine. Earth Planet. Sci. Lett. 2007, 261, 134–142. [Google Scholar] [CrossRef]
- Hushur, A.; Manghnani, M.H.; Smyth, J.R.; Nestola, F.; Frost, D.J. Crystal chemistry of hydrous forsterite and its vibrational properties up to 41 GPa. Am. Miner. 2009, 94, 751–760. [Google Scholar] [CrossRef]
- Ringwood, A.E.; Major, A. High-pressure reconnaissance investigations in the system Mg2SiO4–MgO–H2O. Earth Planet. Sci. Lett. 1967, 2, 130–133. [Google Scholar]
- Ohtani, E.; Toma, M.; Litasov, K.; Kubo, T.; Suzuki, A. Stability of dense hydrous magnesium silicate phases and water storage capacity in the transition zone and lower mantle. Phys. Earth Planet. Inter. 2001, 124, 105–117. [Google Scholar] [CrossRef]
- Kohlstedt, D.L.; Keppler, H.; Rubie, D.C. Solubility of water in the α, β and γ phases of (Mg,Fe)2SiO4. Contrib. Miner. Pet. 1996, 123, 345–357. [Google Scholar] [CrossRef]
- Bolfan-Casanova, N. Water in the Earth’s mantle. Mineral. Mag. 2005, 69, 229–257. [Google Scholar] [CrossRef]
- Beran, A.; Libowitzky, E. Water in natural mantle minerals II: Olivine, garnet and accessory minerals. Rev. Mineral. Geochem. 2005, 62, 169–191. [Google Scholar] [CrossRef]
- Tenner, T.J.; Hirschmann, M.M.; Withers, A.C.; Hervig, R.L. Hydrogen partitioning between nominally anhydrous upper mantle minerals and melt between 3 and 5 GPa and applications to hydrous peridotite partial melting. Chem. Geol. 2009, 262, 42–56. [Google Scholar] [CrossRef]
- Ferot, A.; Bolfan-Casanova, N. Water storage capacity in olivine and pyroxene to 14 GPa: Implications for the water content of the Earth’s upper mantle and nature of seismic discontinuities. Earth Planet. Sci. Lett. 2012, 349–350, 218–230. [Google Scholar] [CrossRef]
- Matsyuk, S.S.; Langer, K. Hydroxyl in olivines from mantle xenoliths in kimberlites of the Siberian platform. Contrib. Miner. Pet. 2004, 147, 413–437. [Google Scholar] [CrossRef]
- Walker, A.M.; Hermann, J.; Berry, A.J.; O’Neill, H.S.C.; Walker, A.; Berry, A. Three water sites in upper mantle olivine and the role of titanium in the water weakening mechanism. J. Geophys. Res. Space Phys. 2007, 112, 112. [Google Scholar] [CrossRef]
- O’Neill, H.S.; Hermann, J.; Hauri, E.H.; Kovács, I.; O’Neill, H.S. Site-specific infrared O-H absorption coefficients for water substitution into olivine. Am. Miner. 2010, 95, 292–299. [Google Scholar]
- Brodholt, J.P.; Refson, K. An ab initio study of hydrogen in forsterite and a possible mechanism for hydrolytic weakening. J. Geophys. Res. Space Phys. 2000, 105, 18977–18982. [Google Scholar] [CrossRef]
- Braithwaite, J.S.; Wright, K.; Catlow, C.R.A. A theoretical study of the energetics and IR frequencies of hydroxyl defects in forsterite. J. Geophys. Res. Space Phys. 2003, 108, 2284. [Google Scholar] [CrossRef]
- Verma, A.K.; Karki, B.B. Ab initio investigations of native and protonic point defects in Mg2SiO4 polymorphs under high pressure. Earth Planet. Sci. Lett. 2009, 285, 140–149. [Google Scholar] [CrossRef]
- Balan, E.; Ingrin, J.; Delattre, S.; Kovács, I.; Blanchard, M. Theoretical infrared spectrum of OH-defects in forsterite. Eur. J. Miner. 2011, 23, 285–292. [Google Scholar] [CrossRef]
- Umemoto, K.; Wentzcovitch, R.M.; Hirschmann, M.M.; Kohlstedt, D.L.; Withers, A.C. A first-principles investigation of hydrous defects and IR frequencies in forsterite: The case for Si vacancies. Am. Miner. 2011, 96, 1475–1479. [Google Scholar] [CrossRef]
- Balan, E.; Blanchard, M.; Lazzeri, M.; Ingrin, J. Contribution of interstitial OH groups to the incorporation of water in forsterite. Phys. Chem. Miner. 2014, 41, 105–114. [Google Scholar] [CrossRef]
- Crépisson, C.; Bureau, H.; Blanchard, M.; Ingrin, J.; Balan, E. Theoretical infrared spectrum of partially protonated cationic vacancies in forsterite. Eur. J. Miner. 2014, 26, 203–210. [Google Scholar] [CrossRef]
- Xue, X.; Turner, D.; Loroch, D.; Kanzaki, M. Hydrogen incorporation mechanisms in forsterite: New insights from 1 H and 29 Si NMR spectroscopy and first-principles calculation. Am. Miner. 2017, 102, 519–536. [Google Scholar] [CrossRef]
- Tavangarian, F.; Emadi, R. Synthesis of pure nanocrystalline magnesium silicate powder. Ceramics 2010, 54, 122–127. [Google Scholar]
- Tavangarian, F.; Emadi, R. Mechanochemical synthesis of single phase nanocrystalline forsterite powder. Int. J. Mod. Phys. B 2010, 24, 343–350. [Google Scholar] [CrossRef]
- Chopelas, A. Single crystal Raman spectra of forsterite, fayalite and monticellite. Am. Mineral. 1991, 76, 1101–1109. [Google Scholar]
- Kolesov, B.A.; Geiger, C.A. A Raman spectroscopic study of Fe-Mg olivines. Phys. Chem. Miner. 2004, 31, 142–154. [Google Scholar] [CrossRef]
- McKeown, D.A.; Bell, M.I.; Caracas, R. Theoretical determination of the Raman spectra of single-crystal forsterite (Mg2SiO4). Am. Mineral. 2010, 95, 980–986. [Google Scholar] [CrossRef]
- Bolfan-Casanova, N.; Montagnac, G.; Reynard, B. Measurement of water contents in olivine using Raman spectroscopy. Am. Miner. 2014, 99, 149–156. [Google Scholar] [CrossRef]
- Joachim, B.; Wohlers, A.; Norberg, N.; Gardes, E.; Petrishcheva, E.; Abart, R. Diffusion and solubility of hydrogen and water in periclase. Phys. Chem. Miner. 2013, 40, 19–27. [Google Scholar] [CrossRef]
- Shinoda, K.; Yamakata, M.; Nanba, T.; Kimura, H.; Moriwaki, T.; Kondo, Y.; Kawamoto, T.; Niimi, N.; Miyoshi, N.; Aikawa, N. High-pressure phase transition and behavior of protons in brucite Mg(OH)2: A high-pressure-temperature study using IR synchrotron radiation. Phys. Chem. Miner. 2002, 29, 396–402. [Google Scholar] [CrossRef]
- Frost, R.L.; Kloprogge, J. Infrared emission spectroscopic study of brucite. Spectrochim. Acta Part A: Mol. Biomol. Spectrosc. 1999, 55, 2195–2205. [Google Scholar] [CrossRef]
- Paterson, M.S. The determination of hydroxyl by infrared absorption in quartz, silicate glasses and similar materials. Bull. Minéral 1982, 105, 20–29. [Google Scholar]
- Withers, A.C.; Bureau, H.; Raepsaet, C.; Hirschmann, M.M. Calibration of infrared spectroscopy by elastic recoil detection analysis of H in synthetic olivine. Chem. Geol. 2012, 334, 92–98. [Google Scholar] [CrossRef]
- Bolfan-Casanova, N.; Keppler, H.; Rubie, D.C. Water partitioning between nominally anhydrous minerals in the MgO-SiO2-H2O system up to 24 GPa: Implications for the distribution of water in the Earth’s mantle. Earth Planet. Sci. Lett. 2000, 182, 209–221. [Google Scholar]
- Libowitzky, E.; Rossman, G. Principles of quantitative absorbance measurements in anisotropic crystals. Phys. Chem. Miner. 1996, 23, 319–327. [Google Scholar] [CrossRef]
- Bell, D.R. Hydroxide in olivine: A quantitative determination of the absolute amount and calibration of the IR spectrum. J. Geophys. Res. Space Phys. 2003, 108, 2105–2113. [Google Scholar] [CrossRef]
- Mei, S.; Kohlstedt, D.L. Influence of water on plastic deformation of olivine aggregates: 1. Diffusion creep regime. J. Geophys. Res. Space Phys. 2000, 105, 21457–21469. [Google Scholar] [CrossRef]
- Zhao, Y.-H.; Ginsberg, S.B.; Kohlstedt, D.L. Solubility of hydrogen in olivine: Dependence on temperature and iron content. Contrib. Miner. Pet. 2004, 147, 155–161. [Google Scholar] [CrossRef]
- Bali, E.; Bolfan-Casanova, N.; Koga, K.T. Pressure and temperature dependence of H solubility in forsterite: An implication to water activity in the Earth interior. Earth Planet. Sci. Lett. 2008, 268, 354–363. [Google Scholar] [CrossRef]
- Otsuka, K.; Karato, S.-I. Control of the water fugacity at high pressures and temperatures: Applications to the incorporation mechanisms of water in olivine. Phys. Earth Planet. Inter. 2011, 189, 27–33. [Google Scholar] [CrossRef]
- Withers, A.C.; Blanchard, M.; Ingrin, J.; Balan, E.; Kovács, I. Effect of iron and trivalent cations on OH defects in olivine. Am. Miner. 2017, 102, 302–311. [Google Scholar]
- Walker, A.M.; Demouchy, S.; Wright, K. Computer modelling of the energies and vibrational properties of hydroxyl groups in a and b-Mg2SiO4. Eur. J. Mineral. 2006, 18, 529–543. [Google Scholar] [CrossRef]
- Smyth, J.R.; Frost, D.J.; Nestola, F.; Holl, C.M.; Bromiley, G.; Frost, D. Olivine hydration in the deep upper mantle: Effects of temperature and silica activity. Geophys. Res. Lett. 2006, 33, 33. [Google Scholar] [CrossRef]
- Tollan, P.M.E.; Smith, R.; O’Neill, H.S.; Hermann, J. The responses of the four main substitution mechanisms of H in olivine to H2O activity at 1050 °C and 3 GPa. Prog. Earth Planet. Sci. 2017, 4, 200. [Google Scholar] [CrossRef]
- Ingrin, J.; Liu, J.; Depecker, C.; Kohn, S.C.; Balan, E.; Grant, K.J. Low-temperature evolution of OH bands in synthetic forsterite, implication for the nature of H defects at high pressure. Phys. Chem. Miner. 2013, 40, 499–510. [Google Scholar] [CrossRef]
- Yoshino, T.; Matsuzaki, T.; Shatskiy, A.; Katsura, T. The effect of water on the electrical conductivity of olivine aggregates and its implications for the electrical structure of the upper mantle. Earth Planet. Sci. Lett. 2009, 288, 291–300. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Starting Material | Experimental Conditions 1 | Water Content (ppm wt H2O) 2 |
|---|---|---|---|
| H0203 | forst + ~10 µL H2O | PC/1100/1.5/90 | 131 (5) |
| D1707 | forst + ~10 µL H2O | PC/1100/1.5/120 | 188 (7) |
| D1409 | forst + ~20 µL H2O | PC/1100/1.5/180 | 194 (10) |
| H0303 | forst + ~20 µL H2O | PC/1100/1.5/180 | 239 (6) |
| H1902 | forst + ~10 µL H2O | MA/1100/4.0/300 | 465 (40) |
| H2002 | forst + ~10 µL H2O | MA/1100/4.0/300 | 508 (10) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Del Vecchio, A.; Poe, B.T.; Misiti, V.; Cestelli Guidi, M. High-Temperature Evolution of Point Defect Equilibria in Hydrous Forsterite Synthesized at 1100 °C and up to 4 GPa. Minerals 2019, 9, 574. https://doi.org/10.3390/min9100574
Del Vecchio A, Poe BT, Misiti V, Cestelli Guidi M. High-Temperature Evolution of Point Defect Equilibria in Hydrous Forsterite Synthesized at 1100 °C and up to 4 GPa. Minerals. 2019; 9(10):574. https://doi.org/10.3390/min9100574
Chicago/Turabian StyleDel Vecchio, Alessandro, Brent T. Poe, Valeria Misiti, and Mariangela Cestelli Guidi. 2019. "High-Temperature Evolution of Point Defect Equilibria in Hydrous Forsterite Synthesized at 1100 °C and up to 4 GPa" Minerals 9, no. 10: 574. https://doi.org/10.3390/min9100574