Quantitative Proteomic Analysis of Primitive Neural Stem Cells from LRRK2 G2019S-Associated Parkinson’s Disease Patient-Derived iPSCs

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human-Induced Pluripotent Stem Cell (iPSC) Culture
2.2. Differentiation of iPSCs into pNSCs
2.3. Differentiation of pNSCs into Neuronal Cells
2.4. RNA Extraction and Quantitative Real-Time PCR (qRT-PCR)
2.5. Alkaline Phosphatase (AP) Staining
2.6. Immunocytochemistry
2.7. Flow Cytometry
2.8. Karyotyping
2.9. In-Solution Tryptic Digestion
2.10. LC-MS/MS Analysis and Database Search
2.11. Western Blotting
2.12. Statistical Analysis
3. Results
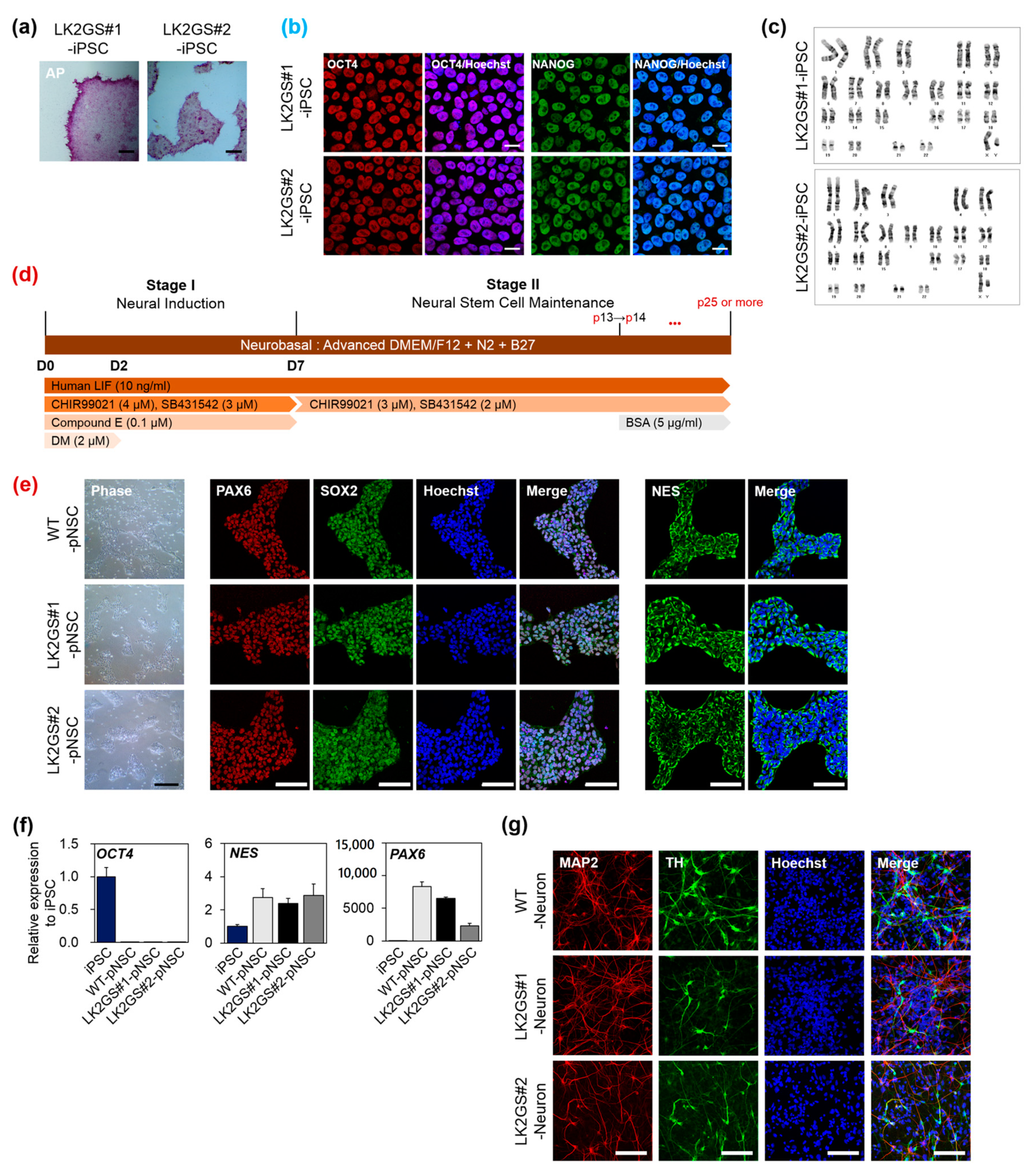
3.1. Generation and Characterization of pNSCs from PD Patients Cells Harboring LRRK2 G2019S
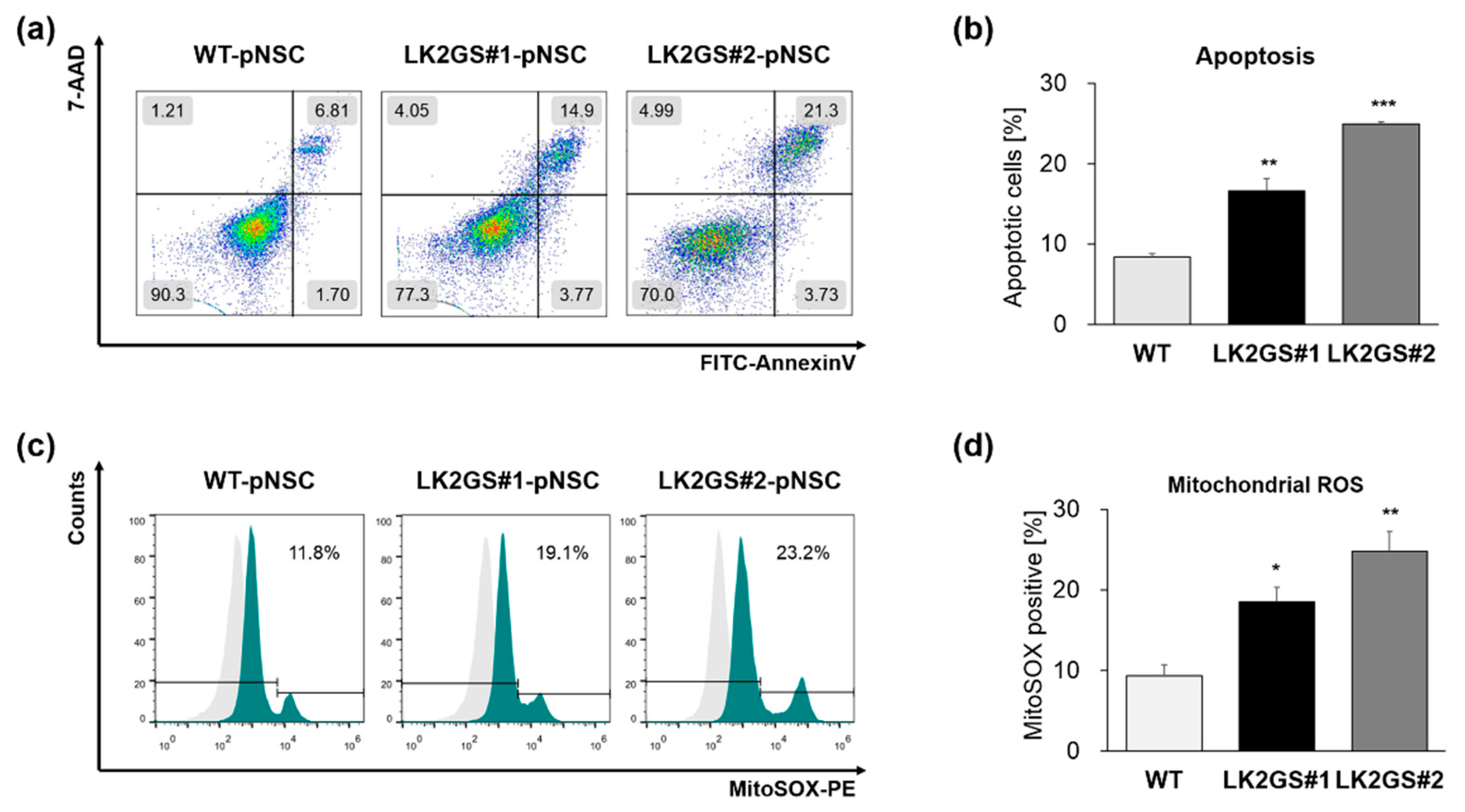
3.2. Functional Characerization for Apoptosis and Oxidative Stress in pNSCs
3.3. Ontological Classification of Differentially Regulated Proteins Between WT and LK2GS-pNSC
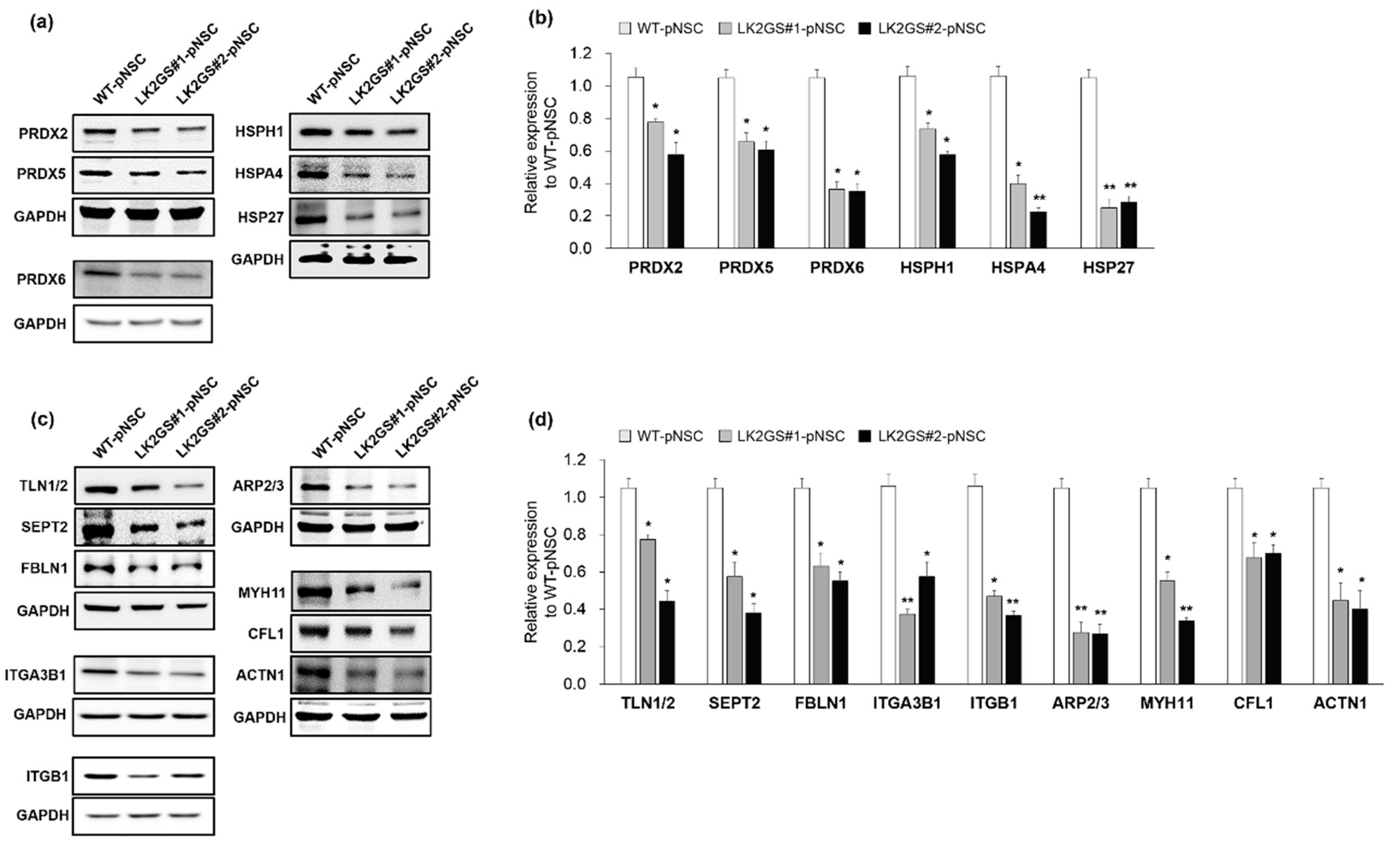
3.4. Regulation of Oxidative Stress, Cell Adhesion, Cytoskeleton-Related Proteins in LK2GS-pNSCs
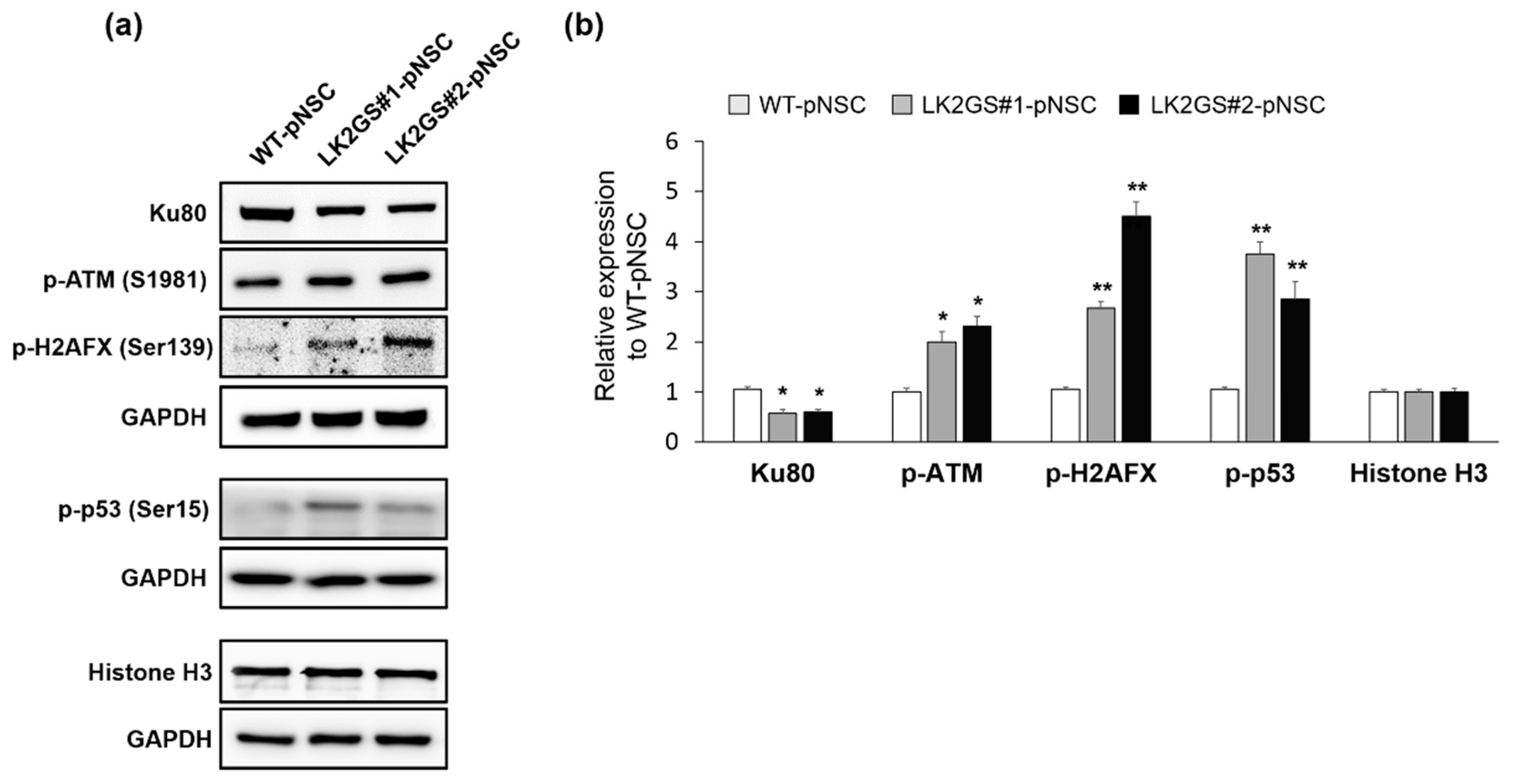
3.5. Induction of DNA Damage-Mediated Apoptosis in LK2GS-pNSCs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickson, D.W.; Fujishiro, H.; Orr, C.; DelleDonne, A.; Josephs, K.A.; Frigerio, R.; Burnett, M.; Parisi, J.E.; Klos, K.J.; Ahlskog, J.E. Neuropathology of non-motor features of Parkinson disease. Park. Relat. Disord. 2009, 15, S1–S5. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Prim. 2017, 3, 1–21. [Google Scholar] [CrossRef]
- Gasser, T. Mendelian forms of Parkinson’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2009, 1792, 587–596. [Google Scholar] [CrossRef] [Green Version]
- Pan-Montojo, F.; Reichmann, H. Considerations on the role of environmental toxins in idiopathic Parkinson’s disease pathophysiology. Transl. Neurodegener. 2014, 3, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Mata, I.F.; Wedemeyer, W.J.; Farrer, M.J.; Taylor, J.P.; Gallo, K.A. LRRK2 in Parkinson’s disease: Protein domains and functional insights. Trends Neurosci. 2006, 29, 286–293. [Google Scholar] [CrossRef]
- Jaleel, M.; Nichols, R.J.; Deak, M.; Campbell, D.G.; Gillardon, F.; Knebel, A.; Alessi, D.R. LRRK2 phosphorylates moesin at threonine-558: Characterization of how Parkinson’s disease mutants affect kinase activity. Biochem. J. 2007, 405, 307–317. [Google Scholar] [CrossRef]
- Daher, J.P.L.; Pletnikova, O.; Biskup, S.; Musso, A.; Gellhaar, S.; Galter, D.; Troncoso, J.C.; Lee, M.K.; Dawson, T.M.; Dawson, V.L.; et al. Neurodegenerative phenotypes in an A53T α-synuclein transgenic mouse model are independent of LRRK2. Hum. Mol. Genet. 2012, 21, 2420–2431. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, C.H.; Shaltouki, A.; Gonzalez, A.E.; Bettencourt da Cruz, A.; Burbulla, L.F.; St. Lawrence, E.; Schüle, B.; Krainc, D.; Palmer, T.D.; Wang, X. Functional Impairment in Miro Degradation and Mitophagy Is a Shared Feature in Familial and Sporadic Parkinson’s Disease. Cell Stem Cell 2016, 19, 709–724. [Google Scholar] [CrossRef] [Green Version]
- Manzoni, C.; Lewis, P.A. Dysfunction of the autophagy/lysosomal degradation pathway is a shared feature of the genetic synucleinopathies. FASEB J. 2013, 27, 3424–3429. [Google Scholar] [CrossRef] [Green Version]
- Chesselet, M.F.; Fleming, S.; Mortazavi, F.; Meurers, B. Strengths and limitations of genetic mouse models of Parkinson’s disease. Park. Relat. Disord. 2008, 14, 84–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giasson, B.I.; Duda, J.E.; Quinn, S.M.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron 2002, 34, 521–533. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.K.; Stirling, W.; Xu, Y.; Xu, E.; Qui, D.; Mandir, A.S.; Dawson, T.M.; Copeland, N.G.; Jenkins, N.A.; Price, D.L. Human α-synuclein-harboring familial Parkinson’s disease-linked Ala-53 → Thr mutation causes neurodegenerative disease with α-synuclein aggregation in transgenic mice. Proc. Natl. Acad. Sci. USA 2002, 99, 8968–8973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masliah, E.; Rockenstein, E.; Veinbergs, I.; Mallory, M.; Hashimoto, M.; Takeda, A.; Sagara, Y.; Sisk, A.; Mucke, L. Dopaminergic loss and inclusion body formation in α-synuclein mice: Implications for neurodegenerative disorders. Science 2000, 287, 1265–1269. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F. Flint Beal Experimental models of Parkinson disease. Nat. Rev. Neurosci. 2001, 2, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.Y.; Khurana, V.; Auluck, P.K.; Tardiff, D.F.; Mazzulli, J.R.; Soldner, F.; Baru, V.; Lou, Y.; Freyzon, Y.; Cho, S.; et al. Identification and Rescue of a-Synuclein Toxicity in Parkinson Patient-Derived Neurons. Science 2013, 342, 983–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.C. Neural subtype specification from embryonic stem cells. Brain Pathol. 2006, 16, 132–142. [Google Scholar] [CrossRef]
- Liu, G.H.; Qu, J.; Suzuki, K.; Nivet, E.; Li, M.; Montserrat, N.; Yi, F.; Xu, X.; Ruiz, S.; Zhang, W.; et al. Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature 2012, 491, 603–607. [Google Scholar] [CrossRef] [Green Version]
- Son, M.-Y.; Sim, H.; Son, Y.S.; Jung, K.B.; Lee, M.-O.; Oh, J.-H.; Chung, S.-K.; Jung, C.-R.; Kim, J. Distinctive genomic signature of neural and intestinal organoids from familial Parkinson’s disease patient-derived induced pluripotent stem cells. Neuropathol. Appl. Neurobiol. 2017, 43. [Google Scholar] [CrossRef]
- Lee, M.; Ha, J.; Son, Y.S.; Ahn, H.; Jung, K.B.; Son, M.Y.; Kim, J. Efficient exogenous DNA-free reprogramming with suicide gene vectors. Exp. Mol. Med. 2019, 51. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Sun, W.; Zhang, Y.; Wei, W.; Ambasudhan, R.; Xia, P.; Talantova, M.; Lin, T.; Kim, J.; Wang, X.; et al. Rapid induction and long-term self-renewal of primitive neural precursors from human embryonic stem cells by small molecule inhibitors. Proc. Natl. Acad. Sci. USA 2011, 108, 8299–8304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.; Sim, H.; Ahn, H.; Ha, J.; Baek, A.; Jeon, Y.-J.; Son, M.-Y.; Kim, J. Direct reprogramming to human induced neuronal progenitors from fibroblasts of familial and sporadic Parkinson’s disease patients. Int. J. Stem Cells 2019, 12, 474–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, X.; Echan, L.; Hembach, P.; Tang, H.Y.; Speicher, K.D.; Santoli, D.; Speicher, D.W. Towards global analysis of mammalian proteomes using sample prefractionation prior to narrow pH range two-dimensional gels and using one-dimensional gels for insoluble and large proteins. Electrophoresis 2001, 22, 1603–1615. [Google Scholar] [CrossRef]
- Niu, J.; Yu, M.; Wang, C.; Xu, Z. Leucine-rich repeat kinase 2 disturbs mitochondrial dynamics via dynamin-like protein. J. Neurochem. 2012, 122, 650–658. [Google Scholar] [CrossRef]
- Walter, J.; Bolognin, S.; Antony, P.M.A.; Nickels, S.L.; Poovathingal, S.K.; Salamanca, L.; Magni, S.; Perfeito, R.; Hoel, F.; Qing, X.; et al. Neural Stem Cells of Parkinson’s Disease Patients Exhibit Aberrant Mitochondrial Morphology and Functionality. Stem Cell Reports 2019, 12, 878–889. [Google Scholar] [CrossRef] [Green Version]
- Tolosa, E.; Vila, M.; Klein, C.; Rascol, O. LRRK2 in Parkinson disease: Challenges of clinical trials. Nat. Rev. Neurol. 2020, 16, 97–107. [Google Scholar] [CrossRef]
- Deng, X.; Dzamko, N.; Prescott, A.; Davies, P.; Liu, Q.; Yang, Q.; Lee, J.D.; Patricelli, M.P.; Nomanbhoy, T.K.; Alessi, D.R.; et al. Characterization of a selective inhibitor of the Parkinson’s disease kinase LRRK2. Nat. Chem. Biol. 2011, 7, 203–205. [Google Scholar] [CrossRef] [Green Version]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson’s disease. J. Parkinsons. Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef] [Green Version]
- Weng, M.; Xie, X.; Liu, C.; Lim, K.L.; Zhang, C.W.; Li, L. The Sources of Reactive Oxygen Species and Its Possible Role in the Pathogenesis of Parkinson’s Disease. Parkinsons. Dis. 2018, 2018. [Google Scholar] [CrossRef] [Green Version]
- Kish, S.J.; Morito, C.; Hornykiewicz, O. Glutathione peroxidase activity in Parkinson’s disease brain. Neurosci. Lett. 1985, 58, 343–346. [Google Scholar] [CrossRef]
- Keeney, P.M.; Xie, J.; Capaldi, R.A.; Bennett, J.P. Parkinson’s disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J. Neurosci. 2006, 26, 5256–5264. [Google Scholar] [CrossRef] [PubMed]
- De Simoni, S.; Goemaere, J.; Knoops, B. Silencing of peroxiredoxin 3 and peroxiredoxin 5 reveals the role of mitochondrial peroxiredoxins in the protection of human neuroblastoma SH-SY5Y cells toward MPP+. Neurosci. Lett. 2008, 433, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.; Kukkurainen, S.; Hytönen, V.P.; Wehrle-Haller, B. Cell adhesion by integrins. Physiol. Rev. 2019, 99, 1655–1699. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Reddy, D.S. Integrins as receptor targets for neurological disorders. Pharmacol. Ther. 2012, 134, 68–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izumi, Y.; Wakita, S.; Kanbara, C.; Nakai, T.; Akaike, A.; Kume, T. Integrin α5β1 expression on dopaminergic neurons is involved in dopaminergic neurite outgrowth on striatal neurons. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Schlie-Wolter, S.; Ngezahayo, A.; Chichkov, B.N. The selective role of ECM components on cell adhesion, morphology, proliferation and communication in vitro. Exp. Cell Res. 2013, 319, 1553–1561. [Google Scholar] [CrossRef]
- Mège, R.M.; Gavard, J.; Lambert, M. Regulation of cell-cell junctions by the cytoskeleton. Curr. Opin. Cell Biol. 2006, 18, 541–548. [Google Scholar] [CrossRef]
- Cartelli, D.; Goldwurm, S.; Casagrande, F.; Pezzoli, G.; Cappelletti, G. Microtubule destabilization is shared by genetic and idiopathic Parkinson’s disease patient fibroblasts. PLoS ONE 2012, 7, e37467. [Google Scholar] [CrossRef]
- Madabhushi, R.; Pan, L.; Tsai, L. Review DNA Damage and Its Links to Neurodegeneration. Neuron 2014, 83, 266–282. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Kozlov, S.; Lavin, M.F.; Person, M.D.; Paull, T.T. ATM Activation by Oxidative Stress. Science 2010, 330, 517–521. [Google Scholar] [CrossRef] [Green Version]
- Fell, V.L.; Schild-Poulter, C. The Ku heterodimer: Function in DNA repair and beyond. Mutat. Res. Rev. Mutat. Res. 2015, 763, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Tomimatsu, N.; Tahimic, C.G.T.; Otsuki, A.; Burma, S.; Fukuhara, A.; Sato, K.; Shiota, G.; Oshimura, M.; Chen, D.J.; Kurimasa, A. Ku70/80 modulates ATM and ATR signaling pathways in response to DNA double strand breaks. J. Biol. Chem. 2007, 282, 10138–10145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- So, S.; Davis, A.J.; Chen, D.J. Autophosphorylation at serine 1981 stabilizes ATM at DNA damage sites. J. Cell Biol. 2009, 187, 977–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kastan, M.B.; Lim, D. the Many Substrates and Functions of ATM. Nat. Rev. Mol. Cell Biol. 2000, 1. [Google Scholar] [CrossRef]
- Christine, K.; Ana, W. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef] [Green Version]
- Weykopf, B.; Haupt, S.; Jungverdorben, J.; Flitsch, L.J.; Hebisch, M.; Liu, G.H.; Suzuki, K.; Belmonte, J.C.I.; Peitz, M.; Blaess, S.; et al. Induced pluripotent stem cell-based modeling of mutant LRRK2-associated Parkinson’s disease. Eur. J. Neurosci. 2019, 49, 561–589. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.N.; Byers, B.; Cord, B.; Shcheglovitov, A.; Byrne, J.; Gujar, P.; Kee, K.; Schüle, B.; Dolmetsch, R.E.; Langston, W.; et al. LRRK2 mutant iPSC-derived da neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell 2011, 8, 267–280. [Google Scholar] [CrossRef] [Green Version]
- Delamarre, A.; Meissner, W.G. Épidémiologie, facteurs de risque environnementaux et génétiques de la maladie de Parkinson. Press. Med 2017, 46, 175–181. [Google Scholar] [CrossRef]
- James, A.M.; Cochemé, H.M.; Murphy, M.P. Mitochondria-targeted redox probes as tools in the study of oxidative damage and ageing. Mech. Ageing Dev. 2005, 126, 982–986. [Google Scholar] [CrossRef]
- Wood, Z.A.; Schröder, E.; Harris, J.R.; Poole, L.B. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem. Sci. 2003, 28, 32–40. [Google Scholar] [CrossRef]
- Concannon, C.G.; Gorman, A.M.; Samali, A. On the role of Hsp27 in regulating apoptosis. Apoptosis 2003, 8, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.R.; Udgata, A.; Tourlomousis, P.; Symmons, M.F.; Hopkins, L.J.; Bryant, C.E.; Gay, N.J. The Parkinson’s disease–associated kinase LRRK2 regulates genes required for cell adhesion, polarization, and chemotaxis in activated murine macrophages. J. Biol. Chem. 2020, 295, 10857–10867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, I.; Kim, B.; Byun, J.W.; Baik, S.H.; Huh, Y.H.; Kim, J.H.; Mook-Jung, I.; Song, W.K.; Shin, J.H.; Seo, H.; et al. LRRK2 G2019S mutation attenuates microglial motility by inhibiting focal adhesion kinase. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meberg, P.J.; Bamburg, J.R. Increase in neurite outgrowth mediated by overexpression of actin depolymerizing factor. J. Neurosci. 2000, 20, 2459–2469. [Google Scholar] [CrossRef] [Green Version]
- Goult, B.T.; Yan, J.; Schwartz, M.A. Talin as a mechanosensitive signaling hub. J. Cell Biol. 2018, 217, 3776–3784. [Google Scholar] [CrossRef] [Green Version]
- Gumbiner, B.M. Cell adhesion: The molecular basis of tissue architecture and morphogenesis. Cell 1996, 84, 345–357. [Google Scholar] [CrossRef] [Green Version]
- Neubauer, K.; Zieger, B. The Mammalian Septin Interactome. Front. Cell Dev. Biol. 2017, 5, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Hynes, R.O. Integrins: Bidirectional, Allosteric Signaling Machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef] [Green Version]
- Ross, R.S. Molecular and mechanical synergy: Cross-talk between integrins and growth factor receptors. Cardiovasc. Res. 2004, 63, 381–390. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, N.; Kostka, G.; Garbe, J.H.O.; Keene, D.R.; Bächinger, H.P.; Hanisch, F.G.; Markova, D.; Tsuda, T.; Timpl, R.; Chu, M.L.; et al. A comparative analysis of the fibulin protein family: Biochemical characterization, binding interactions, and tissue localization. J. Biol. Chem. 2007, 282, 11805–11816. [Google Scholar] [CrossRef] [Green Version]
- Twal, W.O.; Hammad, S.M.; Guffy, S.L.; Argraves, W.S. A novel intracellular fibulin-1D variant binds to the cytoplasmic domain of integrin beta 1 subunit. Matrix Biol. 2015, 43, 97–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero, S.; Le Clainche, C.; Gautreau, A.M. Actin polymerization downstream of integrins: Signaling pathways and mechanotransduction. Biochem. J. 2020, 477, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, T.B.; Meberg, P.J.; Brown, M.D.; Bernstein, B.W.; Minamide, L.S.; Jensen, J.R.; Okada, K.; Soda, E.A.; Bamburg, J.R. Regulating actin dynamics in neuronal growth cones by ADF/cofilin and Rho family GTPases. J. Neurobiol. 2000, 44, 126–144. [Google Scholar] [CrossRef]
- Kremer, B.E.; Adang, L.A.; Macara, I.G. Septins Regulate Actin Organization and Cell-Cycle Arrest through Nuclear Accumulation of NCK Mediated by SOCS7. Cell 2007, 130, 837–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiliotis, E.T.; Hunt, S.J.; Hu, Q.; Kinoshita, M.; Nelson, W.J. Epithelial polarity requires septin coupling of vesicle transport to polyglutamylated microtubules. J. Cell Biol. 2008, 180, 295–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Lasso, D.C.; Mollá, B.; Calap-Quintana, P.; García-Giménez, J.L.; Pallardo, F.V.; Palau, F.; Gonzalez-Cabo, P. Cofilin dysregulation alters actin turnover in frataxin-deficient neurons. Sci. Rep. 2020, 10, 5207. [Google Scholar] [CrossRef] [Green Version]
- Toshiyuki, M.; Reed, J.C. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995, 80, 293–299. [Google Scholar] [CrossRef] [Green Version]
- Baldereschi, M.; Di Carlo, A.; Rocca, W.A.; Vanni, P.; Maggi, S.; Perissinotto, E.; Grigoletto, F.; Amaducci, L.; Inzitari, D. Parkinson’s disease and parkinsonism in a longitudinal study: Two-fold higher incidence in men. ILSA Working Group. Italian Longitudinal Study on Aging. Neurology 2000, 55, 1358–1363. [Google Scholar] [CrossRef]
- Haaxma, C.A.; Bloem, B.R.; Borm, G.F.; Oyen, W.J.G.; Leenders, K.L.; Eshuis, S.; Booij, J.; Dluzen, D.E.; Horstink, M.W.I.M. Gender differences in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2007, 78, 819–824. [Google Scholar] [CrossRef] [Green Version]
- Moisan, F.; Kab, S.; Mohamed, F.; Canonico, M.; Le Guern, M.; Quintin, C.; Carcaillon, L.; Nicolau, J.; Duport, N.; Singh-Manoux, A.; et al. Parkinson disease male-to-female ratios increase with age: French nationwide study and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2016, 87, 952–957. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Acession no. | Protein Name | MW (kDa) | pI * Value | Coverage (%) † | Matched Peptides | Score | Fold Change (LK2GS/WT) | |
|---|---|---|---|---|---|---|---|---|
| WT | LK2GS | |||||||
| Oxidoreducates activity | ||||||||
| P32119 | Preoxiredoxin-2 (PRDX2) | 21.9 | 5.97 | 34.34 | 11 | 73.3 | 48.2 | 0.66 |
| P30044 | Peroxiredoxin-5 (PRDX5) | 22.1 | 8.7 | 25.7 | 3 | 8.38 | 3.92 | 0.47 |
| P30041 | Peroxiredoxin-6 (PRDX6) | 25 | 6.38 | 23.66 | 3 | 18.55 | 5.8 | 0.31 |
| Q92598 | Heat shock protein 105 (HSPH1) | 96.8 | 5.39 | 12.24 | 7 | 13.13 | 6.5 | 0.5 |
| P34932 | Heat shock 70 kDa protein 4 (HSPA4) | 94.3 | 5.19 | 12.98 | 7 | 24.6 | 14.56 | 0.6 |
| Q9BRX8 | Redox-regulatory protein FAM213A (F213A) | 25.7 | 8.84 | 6.55 | 1 | 4.65 | 0 | WT unique |
| Q9NRD8 | Dual oxidase 2 (DUOX2) | 175.3 | 7.85 | 1.03 | 1 | 0 | 3.4 | LK2GS unique |
| Q6DKJ4 | Nucleoredoxin (NXN) | 48.4 | 4.97 | 3.68 | 1 | 2.33 | 0 | WT unique |
| P21266 | Glutathione S-transferase Mu 3 (GSTM3) | 26.5 | 5.54 | 7.65 | 1 | 2.99 | 0 | WT unique |
| P10599 | Thioredoxin (TXN) | 11.7 | 4.9 | 8.57 | 1 | 1.71 | 0 | WT unique |
| Q96SL4 | Glutathione peroxidase 7 (GPX7) | 21 | 8.27 | 15.51 | 1 | 0 | 2.3 | LK2GS unique |
| Q8N543 | 2-oxoglutarate and iron-dependent oxygenase domain-containing protein 1 (OGFD1) | 63.2 | 5.11 | 4.06 | 1 | 2.41 | 0 | WT unique |
| Q9BUT1 | 3-hydroxybutyrate dehydrogenase type 2 (BDH2) | 26.7 | 7.65 | 19.59 | 3 | 4.1 | 6.3 | 1.5 |
| Q9C0B1 | Alpha-ketoglutarate-dependent dioxygenase FTO (FTO) | 58.2 | 5.22 | 6.35 | 2 | 10.3 | 3.9 | 0.37 |
| P31150 | Rab GDP dissociation inhibitor alpha (GDIA) | 50.6 | 5.14 | 21.03 | 8 | 88.02 | 35.94 | 0.41 |
| P50395 | Rab GDP dissociation inhibitor beta (GDIB) | 50.6 | 6.47 | 39.1 | 15 | 130.3 | 61.6 | 0.47 |
| Q15738 | Sterol-4-alpha-carboxylate 3-dehydrogenase, decarboxylating (NSDHL) | 41.9 | 8.06 | 6.43 | 1 | 2.6 | 0 | WT unique |
| Q8N183 | Mimitin, mitochondrial (MIMIT) | 19.8 | 8.98 | 3.55 | 1 | 1.93 | 0 | WT unique |
| P28331 | NADH-ubiquinone oxidoreductase 75 kDa subunit, mitochondrial (NDUS1) | 79.4 | 6.23 | 20.2 | 1 | 2.16 | 0 | WT unique |
| O60701 | UDP-glucose 6-dehydrogenas (UGDH) | 55 | 7.12 | 14.57 | 4 | 4.65 | 2.71 | 0.58 |
| P20674 | Cytochrome c oxidase subunit 5A, mitochondrial (COX5A) | 16.8 | 6.79 | 31.33 | 2 | 17.12 | 6.6 | 0.39 |
| Q9UDR5 | Alpha-aminoadipic semialdehyde synthase, mitochondrial (AASS) | 102.1 | 6.61 | 1.94 | 1 | 5.86 | 1.94 | 0.33 |
| P43304 | Glycerol-3-phosphate dehydrogenase, mitochondrial (GPDM) | 80.8 | 7.69 | 4.26 | 1 | 2.33 | 0 | WT unique |
| P40926 | Malate dehydrogenase, mitochondrial (MDHM) | 35.5 | 8.69 | 35.8 | 9 | 46.65 | 11.41 | 0.24 |
| Cell adhesion molecule binding | ||||||||
| Q9Y490 | Talin-1 (TLN1) | 269.6 | 6.07 | 5.51 | 5 | 26.58 | 14.37 | 0.54 |
| Q9Y4G6 | Talin-2 (TLN2) | 271.4 | 5.57 | 1.06 | 1 | 2.24 | 0 | WT unique |
| P23528 | Cofilin-1 (CFL1) | 18.5 | 8.09 | 44.58 | 6 | 95.82 | 46.26 | 0.48 |
| Q92616 | Translational activator GCN1 (GCN1L) | 292.6 | 7.46 | 2.28 | 3 | 10.01 | 3.84 | 0.38 |
| P04075 | Fructose-bisphosphate aldolase A (ALDOA) | 39.4 | 8.09 | 26.1 | 8 | 39.43 | 15.43 | 0.39 |
| P06733 | Alpha-enolase (ENOA) | 434 | 7.39 | 48.62 | 17 | 232.4 | 106.1 | 0.47 |
| P26038 | Moesin (MOES) | 67.8 | 6.4 | 13 | 8 | 45 | 22.64 | 0.5 |
| Q13813 | Spectrin alpha chain, non-erythrocytic 1 (SPTN1) | 284.4 | 5.35 | 1.94 | 3 | 1.53 | 0 | WT unique |
| Cytoskeleton molecules | ||||||||
| P35749 | Myosin-11 (MYH11) | 227.2 | 5.5 | 1.67 | 3 | 6.65 | 1.81 | 0.27 |
| Q15019 | Septin-2 (SEPT2) | 41.5 | 6.6 | 20.78 | 4 | 20.12 | 7.3 | 0.36 |
| P12814 | Alpha-actinin-1 (ACTN1) | 103 | 5.41 | 2.47 | 2 | 9.02 | 0 | WT unique |
| P62873 | Guanine nucleotide-binding protein G(I)/G(S)/G(T) subunit beta-1 (GBB1) | 37.4 | 6 | 6.18 | 2 | 5.76 | 3.73 | 0.65 |
| P62158 | Calmodulin (CALM) | 16.8 | 4.22 | 46.98 | 3 | 26.26 | 14.79 | 0.56 |
| P37231 | Peroxisome proliferator-activated receptor gamma (PPARG) | 57.6 | 5.94 | 3.37 | 1 | 1.88 | 0 | WT unique |
| DNA damage response | ||||||||
| P13010 | 86 kDa subunit of Ku antigen (XRCC5) | 82.7 | 5.81 | 19.67 | 10 | 69.55 | 31.34 | 0.45 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sim, H.; Seo, J.-H.; Kim, J.; Oh, M.; Lee, J.-E.; Baek, A.; Lee, S.-Y.; Chung, S.-K.; Son, M.-Y.; Chae, J.-I.; et al. Quantitative Proteomic Analysis of Primitive Neural Stem Cells from LRRK2 G2019S-Associated Parkinson’s Disease Patient-Derived iPSCs. Life 2020, 10, 331. https://doi.org/10.3390/life10120331
Sim H, Seo J-H, Kim J, Oh M, Lee J-E, Baek A, Lee S-Y, Chung S-K, Son M-Y, Chae J-I, et al. Quantitative Proteomic Analysis of Primitive Neural Stem Cells from LRRK2 G2019S-Associated Parkinson’s Disease Patient-Derived iPSCs. Life. 2020; 10(12):331. https://doi.org/10.3390/life10120331
Chicago/Turabian StyleSim, Hyuna, Ji-Hye Seo, Jumi Kim, Minyoung Oh, Joo-Eun Lee, Areum Baek, Seo-Young Lee, Sun-Ku Chung, Mi-Young Son, Jung-Il Chae, and et al. 2020. "Quantitative Proteomic Analysis of Primitive Neural Stem Cells from LRRK2 G2019S-Associated Parkinson’s Disease Patient-Derived iPSCs" Life 10, no. 12: 331. https://doi.org/10.3390/life10120331
APA StyleSim, H., Seo, J.-H., Kim, J., Oh, M., Lee, J.-E., Baek, A., Lee, S.-Y., Chung, S.-K., Son, M.-Y., Chae, J.-I., Jeon, Y.-J., & Kim, J. (2020). Quantitative Proteomic Analysis of Primitive Neural Stem Cells from LRRK2 G2019S-Associated Parkinson’s Disease Patient-Derived iPSCs. Life, 10(12), 331. https://doi.org/10.3390/life10120331